
Maternal Factors that Induce Epigenetic Changes Contribute to Neurological Disorders in Offspring

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
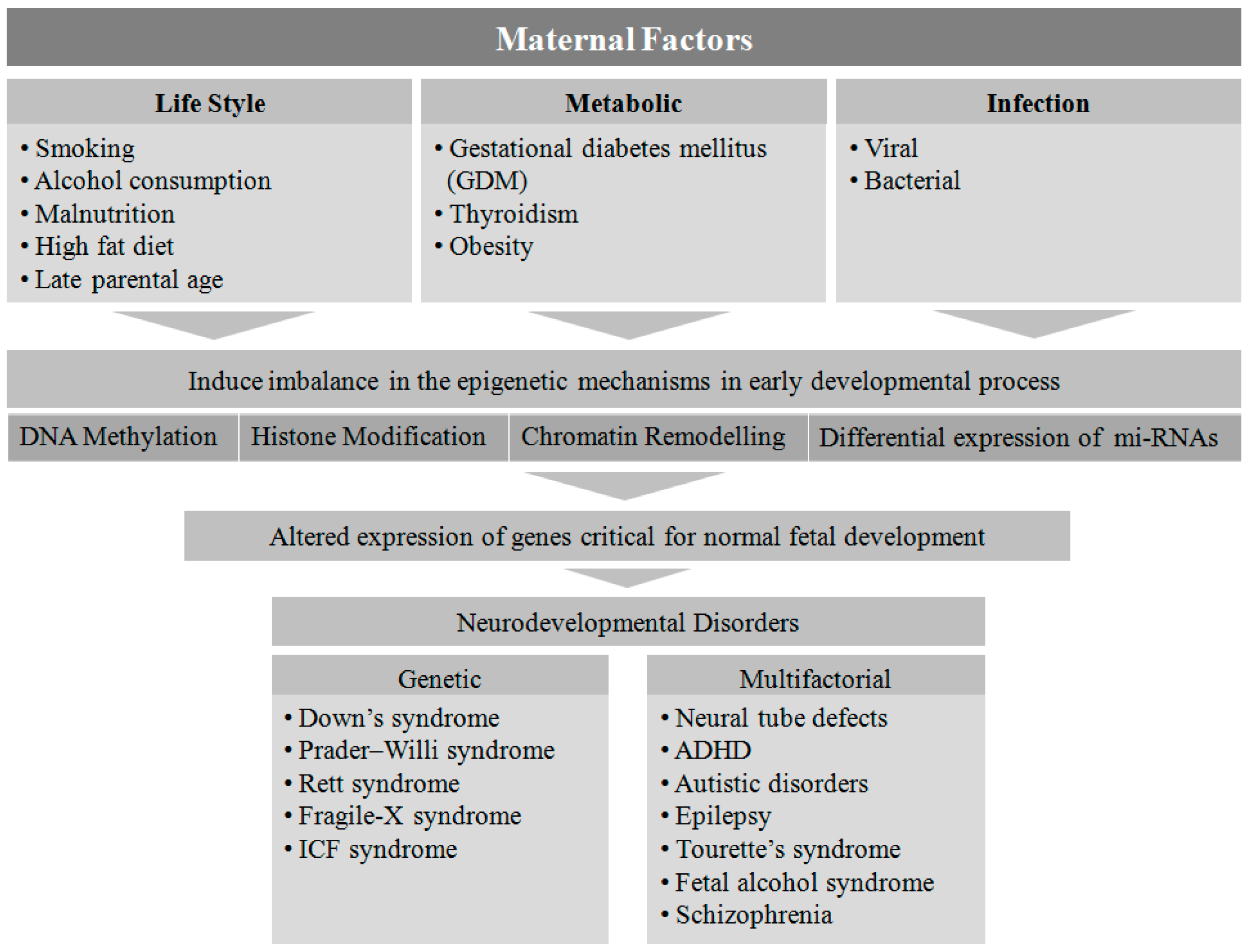
2. Maternal Factors Contributing to Neurodevelopmental Disorders in Offspring
3. Epigenetic Association in Pathophysiology of NDs
4. Maternal Lifestyle
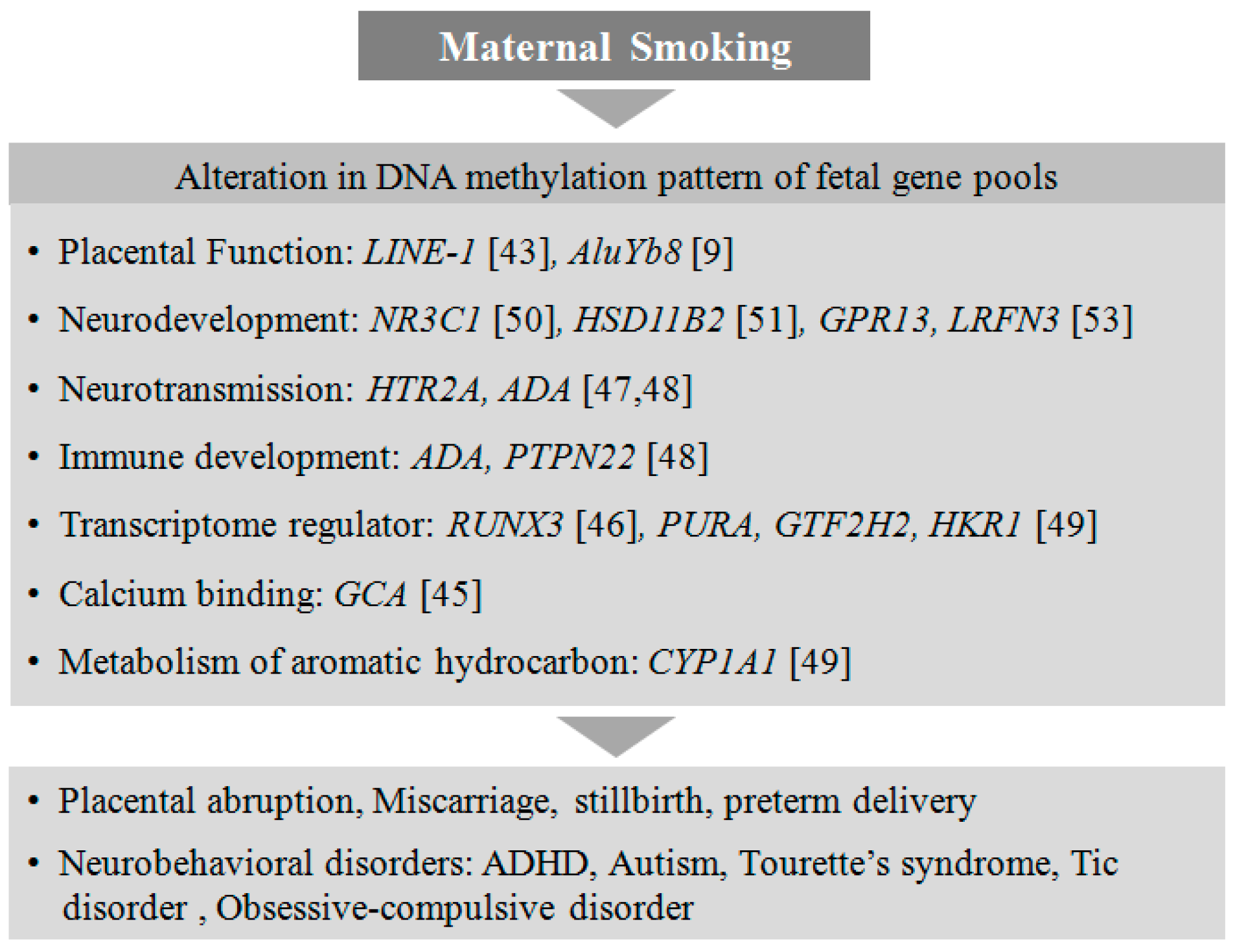
4.1. Smoking
4.2. Alcohol Consumption
4.3. Malnutrition
4.4. Late Maternal Age and Assisted Reproductive Procedures
5. Maternal Metabolic Disorders
5.1. Gestational Diabetes Mellitus (GDM)
5.2. Obesity
5.3. Hypothyroidism
6. Infection
7. Genetic and Epigenetic Regulation of Neurodevelopmental Disorders
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van Loo, K.M.; Martens, G.J. Genetic and environmental factors in complex neurodevelopmental disorders. Curr. Genom. 2007, 8, 429–444. [Google Scholar]
- Pierre, G. Neurodegenerative disordersand metabolic disease. Arch. Dis. Child 2013, 98, 618–624. [Google Scholar] [CrossRef] [PubMed]
- Thapar, A.; Cooper, M.; Rutter, M. Neurodevelopmental disorders. Lancet Psychiatry 2016. [Google Scholar] [CrossRef]
- Polanczyk, G.; de Lima, M.S.; Horta, B.L.; Biederman, J.; Rohde, L.A. The worldwide prevalence of ADHD: A systematic review and metaregression analysis. Am. J. Psychiatry 2007, 164, 942–948. [Google Scholar] [CrossRef] [PubMed]
- Visser, S.N.; Danielson, M.L.; Bitsko, R.H.; Holbrook, J.R.; Kogan, M.D.; Ghandour, R.M.; Perou, R.; Blumberg, S.J. Trends in the parent-report of health care provider-diagnosed and medicated attention-deficit/hyperactivity disorder: United States, 2003–2011. J. Am. Acad. Child Adolesc. Psychiatry 2014, 53, 34–46. [Google Scholar] [CrossRef]
- Polanczyk, G.V.; Willcutt, E.G.; Salum, G.A.; Kieling, C.; Rohde, L.A. ADHD prevalence estimates across three decades: An updated systematic review and meta-regression analysis. Int. J. Epidemiol. 2014, 43, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Kiser, D.P.; Rivero, O.; Lesch, K.P. Annual researchreview: The (epi)genetics ofneurodevelopmental disordersin the era of whole-genome sequencing--unveiling the dark matter. J. Child Psychol. Psychiatry 2015, 56, 278–295. [Google Scholar] [CrossRef] [PubMed]
- Salilew-Wondim, D.; Tesfaye, D.; Hoelker, M.; Schellander, K. Embryo transcriptome response to environmental factors: Implication for its survival under suboptimal conditions. Anim. Reprod. Sci. 2014, 149, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm-Benartzi, C.S.; Houseman, E.A.; Maccani, M.A.; Poage, G.M.; Koestler, D.C.; Langevin, S.M.; Gagne, L.A.; Banister, C.E.; Padbury, J.F.; Marsit, C.J. In utero exposures, infant growth, and DNA methylation of repetitive elements and developmentally related genes in human placenta. Environ. Health Perspect. 2012, 120, 296. [Google Scholar] [CrossRef] [PubMed]
- Dhobale, M.V.; Pisal, H.R.; Mehendale, S.S.; Joshi, S.R. Differential expression of humanplacentalneurotrophic factors in preterm and term deliveries. Int. J. Dev. Neurosci. 2013, 31, 719–723. [Google Scholar] [CrossRef]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [PubMed]
- Abel, T.; Zukin, R.S. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr. Opin. Pharmacol. 2008, 8, 57–64. [Google Scholar] [PubMed]
- Urdinguio, R.G.; Sanchez-Mut, J.V.; Esteller, M. Epigenetic mechanisms in neurological diseases: Genes, syndromes, and therapies. Lancet Neurol. 2009, 8, 1056–1072. [Google Scholar] [PubMed]
- Kubota, T.; Miyake, K.; Hirasawa, T. Epigenetics in neurodevelopmental and mental disorders. Med. Epigenet. 2013, 1, 52–59. [Google Scholar]
- Simpkin, A.J.; Hemani, G.; Suderman, M.; Gaunt, T.R.; Lyttleton, O.; Mcardle, W.L.; Ring, S.M.; Sharp, G.C.; Tilling, K.; Horvath, S. Prenatal and early life influences on epigenetic age in children: A study of mother-offspring pairs from two cohort studies. Hum. Mol. Genet. 2015, 25, 191–201. [Google Scholar] [PubMed]
- Lillycrop, K.A.; Burdge, G.C. The effect of nutrition during early life on the epigenetic regulation of transcription and implications for human diseases. J. Nutrigenet. Nutrigenom. 2012, 4, 248–260. [Google Scholar]
- Chango, A.; Pogribny, I.P. Considering maternal dietary modulators for epigenetic regulation and programming of the fetal epigenome. Nutrients 2015, 7, 2748–2770. [Google Scholar] [PubMed]
- Ornoy, A.; Reece, E.A.; Pavlinkova, G.; Kappen, C.; Miller, R.K. Effect of maternal diabetes on the embryo, fetus, and children: Congenital anomalies, genetic and epigenetic changes and developmental outcomes. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 53–72. [Google Scholar]
- Schroeder, S.R. Mental retardation and developmental disabilities influenced by environmental neurotoxic insults. Environ. Health Perspect. 2000, 108, 395. [Google Scholar] [PubMed]
- Pina-Camacho, L.; Jensen, S.; Gaysina, D.; Barker, E. Maternal depression symptoms, unhealthy diet and child emotional-behavioural dysregulation. Psychol. Med. 2015, 45, 1851–1860. [Google Scholar] [PubMed]
- Tang, B.; Jia, H.; Kast, R.J.; Thomas, E.A. Epigenetic changes at gene promoters in response to immune activation in utero. Brain Behav. Immun. 2013, 30, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Panchenko, P.E.; Voisin, S.; Jouin, M.; Jouneau, L.; Prézelin, A.; Lecoutre, S.; Breton, C.; Jammes, H.; Junien, C.; Gabory, A. Expression of epigenetic machinery genes is sensitive to maternal obesity and weight loss in relation to fetal growth in mice. Clin. Epigenet. 2016, 8, 22. [Google Scholar] [CrossRef] [PubMed]
- Huizink, A.C.; Mulder, E.J. Maternal smoking, drinking or cannabis use during pregnancy and neurobehavioral and cognitive functioning in human offspring. Neurosci. Biobehav. Rev. 2006, 30, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Hiramatsu, Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology 2012, 153, 2823–2830. [Google Scholar] [CrossRef] [PubMed]
- Sandin, S.; Hultman, C.M.; Kolevzon, A.; Gross, R.; MacCabe, J.H.; Reichenberg, A. Advancing maternal age is associated with increasing risk for autism: A review and meta-analysis. J. Am. Acad. Child Adolesc. Psychiatry 2012, 51, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Feil, R.; Fraga, M.F. Epigeneticsand the environment: Emerging patterns and implications. Nat. Rev. Genet. 2012, 13, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.W.; Huang, K.; Yang, C.; Kang, C.S. Non-coding RNAs as regulators inepigenetics (Review). Oncol. Rep. 2017, 37, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Wolffe, A.P.; Matzke, M.A. Epigenetics: Regulation through repression. Science 1999, 286, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Bergman, Y. Epigenetics of haematopoietic cell development. Nat. Rev. Immunol. 2011, 11, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Resendiz, M.; Mason, S.; Lo, C.-L.; Zhou, F.C. Epigenetic regulation of the neural transcriptome and alcohol interference during development. Front. Genet. 2014, 5, 285. [Google Scholar] [CrossRef] [PubMed]
- Reik, W.; Romer, I.; Barton, S.C.; Surani, M.A.; Howlett, S.K.; Klose, J. Adult phenotype in the mouse can be affected by epigenetic events in the early embryo. Development 1993, 119, 933–942. [Google Scholar] [PubMed]
- Rideout, W.M.; Eggan, K.; Jaenisch, R. Nuclear cloning and epigenetic reprogramming of the genome. Science 2001, 293, 1093–1098. [Google Scholar] [CrossRef] [PubMed]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in x-linked MeCP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [PubMed]
- Xu, G.L.; Bestor, T.H.; Bourc’his, D.; Hsieh, C.L.; Tommerup, N.; Bugge, M.; Hulten, M.; Qu, X.; Russo, J.J.; Viegas-Péquignot, E. Chromosome instability and immunodeficiency syndrome caused by mutations in a DNA methyltransferase gene. Nature 1999, 402, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Jin, P.; Warren, S.T. Understanding the molecular basis of fragile x syndrome. Hum. Mol. Genet. 2000, 9, 901–908. [Google Scholar] [CrossRef] [PubMed]
- Cone-Wesson, B. Prenatal alcohol and cocaine exposure: Influences on cognition, speech, language, and hearing. J. Commun. Disord. 2005, 38, 279–302. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.M.; Ou, J.J.; Liu, L.; Zhang, D.; Zhao, J.-P.; Tang, S.-Y. Association between maternal obesity and autism spectrum disorder in offspring: A meta-analysis. J. Autism Dev. Disord. 2016, 46, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Joehanes, R.; Just, A.C.; Marioni, R.E.; Pilling, L.C.; Reynolds, L.M.; Mandaviya, P.R.; Guan, W.; Xu, T.; Elks, C.E.; Aslibekyan, S. Epigenetic signatures of cigarette smoking. Circ. Cardiovasc. Genet. 2016, 9, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Stroud, L.R.; Papandonatos, G.D.; Rodriguez, D.; McCallum, M.; Salisbury, A.L.; Phipps, M.G.; Lester, B.; Huestis, M.A.; Niaura, R.; Padbury, J.F. Maternal smoking during pregnancy and infant stress response: Test of a prenatal programming hypothesis. Psychoneuroendocrinology 2014, 48, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Linnet, K.M.; Dalsgaard, S.; Obel, C.; Wisborg, K.; Henriksen, T.B.; Rodriguez, A.; Kotimaa, A.; Moilanen, I.; Thomsen, P.H.; Olsen, J. Maternal lifestyle factors in pregnancy risk of attention deficit hyperactivity disorder and associated behaviors: Review of the current evidence. Am. J. Psychiatry 2003, 160, 1028–1040. [Google Scholar] [CrossRef] [PubMed]
- Motlagh, M.G.; Sukhodolsky, D.G.; Landeros-Weisenberger, A.; Katsovich, L.; Thompson, N.; Scahill, L.; King, R.A.; Peterson, B.S.; Schultz, R.T.; Leckman, J.F. Adverse effects of heavy prenatal maternal smoking on attentional control in children with ADHD. J. Atten. Disord. 2011, 15, 593–603. [Google Scholar] [CrossRef] [PubMed]
- Mathews, C.A.; Bimson, B.; Lowe, T.L.; Herrera, L.D.; Budman, C.L.; Erenberg, G.; Naarden, A.; Bruun, R.D.; Freimer, N.B.; Reus, V.I. Association between maternal smoking and increased symptom severity in tourette’s syndrome. Am. J. Psychiatry 2006, 163, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Burris, H.H.; Rifas-Shiman, S.L.; Baccarelli, A.; Tarantini, L.; Boeke, C.E.; Kleinman, K.; Litonjua, A.A.; Rich-Edwards, J.W.; Gillman, M.W. Associations of line-1 DNA methylation with preterm birth in a prospective cohort study. J. Dev. Orig. Health Dis. 2012, 3, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Delpisheh, A.; Brabin, L.; Brabin, B.J. Pregnancy, smoking and birth outcomes. Womens Health 2006, 2, 389–403. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.; Ma, J.; Harris, A.S.; Patterson, L.; Brown, K.A.; Shope, C.; Showalter, L.; Abramovici, A.; Aagaard-Tillery, K.M. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics 2011, 6, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Maccani, J.Z.; Koestler, D.C.; Houseman, E.A.; Marsit, C.J.; Kelsey, K.T. Placental DNA methylation alterations associated with maternal tobacco smoking at the RUNX3 gene are also associated with gestational age. Epigenomics 2013, 5, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Paquette, A.G.; Lesseur, C.; Armstrong, D.A.; Koestler, D.C.; Appleton, A.A.; Lester, B.M.; Marsit, C.J. Placental HTR2A methylation is associated with infant neurobehavioral outcomes. Epigenetics 2013, 8, 796–801. [Google Scholar] [CrossRef] [PubMed]
- Sato, K. Placenta-derived hypo-serotonin situations in the developing forebrain cause autism. Med. Hypotheses 2013, 80, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Suter, M.; Abramovici, A.; Showalter, L.; Hu, M.; Do Shope, C.; Varner, M.; Aagaard-Tillery, K. In utero tobacco exposure epigenetically modifies placental CYP1A1 expression. Metabolism 2010, 59, 1481–1490. [Google Scholar] [CrossRef] [PubMed]
- Stroud, L.R.; Paster, R.L.; Goodwin, M.S.; Shenassa, E.; Buka, S.; Niaura, R.; Rosenblith, J.F.; Lipsitt, L.P. Maternal smoking during pregnancy and neonatal behavior: A large-scale community study. Pediatrics 2009, 123, e842–e848. [Google Scholar] [CrossRef] [PubMed]
- Appleton, A.A.; Armstrong, D.A.; Lesseur, C.; Lee, J.; Padbury, J.F.; Lester, B.M.; Marsit, C.J. Patterning in placental 11-b hydroxysteroid dehydrogenase methylation according to prenatal socioeconomic adversity. PLoS ONE 2013, 8, e74691. [Google Scholar] [CrossRef] [PubMed]
- Conradt, E.; Lester, B.M.; Appleton, A.A.; Armstrong, D.A.; Marsit, C.J. The roles of DNA methylation of NR3C1 and 11β-HSD2 and exposure to maternal mood disorder in utero on newborn neurobehavior. Epigenetics 2013, 8, 1321–1329. [Google Scholar] [CrossRef] [PubMed]
- Maccani, M.A.; Marsit, C.J. Review article: Epigenetics in the placenta. Am. J. Reprod. Immun. 2009, 62, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.; Smith, D.; Ulleland, C.; Streissguth, A. Pattern of malformation in offspring of chronic alcoholic mothers. Lancet 1973, 301, 1267–1271. [Google Scholar] [CrossRef]
- Spohr, H.-L.; Willms, J.; Steinhausen, H.-C. Prenatal alcohol exposure and long-term developmental consequences. Lancet 1993, 341, 907–910. [Google Scholar] [CrossRef]
- Riley, E.P.; McGee, C.L. Fetal alcohol spectrum disorders: An overview with emphasis on changes in brain and behavior. Exp. Biol. Med. 2005, 230, 357–365. [Google Scholar] [CrossRef]
- Church, M.; Kaltenbach, J. Effects of fetal alcohol exposure on the auditory and vestibular systems. Alcohol. Clin. Exp. Res. 1997, 21, 495–512. [Google Scholar] [PubMed]
- Steinhausen, H.-C.; Nestler, V.; Spohr, H.-L. Development and psychopathology of children with the fetal alcohol syndrome. J. Dev. Behav. Pediatr. 1982, 3, 49–54. [Google Scholar] [CrossRef] [PubMed]
- May, P.A.; Gossage, J.P. Estimating the prevalence of fetal alcohol syndrome: A summary. Alcohol Res. Health 2001, 25, 159–167. [Google Scholar] [PubMed]
- Zhou, F.C.; Kane, C.J.; Smith, S.M. Proceedings of the 2009 annual meeting of the fetal alcohol spectrum disorders study group. Alcohol 2012, 46, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Junaid, M.A. Lifestyle, pregnancy and epigenetic effects. Epigenomics 2015, 7, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, V.R.; Curtis, K.; Zachariah, R.M.; Chudley, A.E.; Rastegar, M. Overview of the genetic basis andepigeneticmechanisms that contribute to FASD pathobiology. Curr. Top. Med. Chem. 2017, 17, 808–828. [Google Scholar] [CrossRef] [PubMed]
- Lussier, A.A.; Weinberg, J.; Kobor, M.S. Epigenetics studies of fetalalcoholspectrumdisorder: Where are we now? Epigenomics 2017, 9, 291–311. [Google Scholar] [CrossRef] [PubMed]
- Portales-Casamar, E.; Lussier, A.A.; Jones, M.J.; MacIsaac, J.L.; Edgar, R.D.; Mah, S.M.; Barhdadi, A.; Provost, S.; Lemieux-Perreault, L.P.; Cynader, M.S.; et al. DNA methylation signature of human fetalalcoholspectrumdisorder. Epigenet. Chromatin 2016, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Kleiber, M.L.; Mantha, K.; Stringer, R.L.; Singh, S.M. Neurodevelopmental alcohol exposure elicits long-term changes to gene expression that alter distinct molecular pathways dependent on timing of exposure. J. Neurodev. Disord. 2013, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Marquardt, K.; Brigman, J.L. The impact of prenatalalcoholexposureon social, cognitive and affective behavioral domains: Insights from rodent models. Alcohol 2016, 51, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Perkins, A.; Lehmann, C.; Lawrence, R.C.; Kelly, S.J. Alcohol exposure during development: Impact on the epigenome. Int. J. Dev. Neurosci. 2013, 31, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Da Lee, R.; Rhee, G.S.; An, S.M.; Kim, S.S.; Kwack, S.J.; Seok, J.H.; Chae, S.Y.; Park, C.H.; Yoon, H.J.; Cho, D.H.; et al. Differential gene profiles in developing embryo and fetus after in utero exposure to ethanol. J. Toxicol. Environ. Health A 2004, 67, 2073–2084. [Google Scholar] [CrossRef] [PubMed]
- Houlé, K.; Abdi, M.; Clabough, E.B.D. Acute ethanol exposure during late mouse neurodevelopment results in long-term deficits in memory retrieval, but not in social responsiveness. Brain Behav. 2017, 7, e00636. [Google Scholar] [CrossRef] [PubMed]
- Przybycien-Szymanska, M.M.; Rao, Y.S.; Prins, S.A.; Pak, T.R. Parental binge alcohol abuse alters F1 generation hypothalamic gene expression in the absence of direct fetal alcohol exposure. PLoS ONE 2014, 9, e89320. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Choi, C.S.; Park, J.H.; Joo, S.H.; Kim, S.Y.; Ko, H.M.; Kim, K.C.; Jeon, S.J.; Park, S.H.; Han, S.H.; et al. Chronic exposure to ethanol of male mice before mating produces attention deficit hyperactivity disorder-like phenotype along with epigenetic dysregulation of dopamine transporter expression in mouse offspring. J. Neurosci. Res. 2014, 92, 658–670. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Alvarez, R.; Gayen, S.; Vadigepalli, R.; Anni, H. Ethanol diverts early neuronal differentiation trajectory of embryonic stem cells by disrupting the balance of lineage specifiers. PLoS ONE 2013, 8, e63794. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Balaraman, Y.; Wang, G.; Nephew, K.P.; Zhou, F.C. Alcohol exposure alters DNA methylation profiles in mouse embryos at early neurulation. Epigenetics 2009, 4, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Hicks, S.D.; Middleton, F.A.; Miller, M.W. Ethanol-induced methylation of cell cycle genes in neural stem cells. J. Neurochem. 2010, 114, 1767–1780. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.C.; Balaraman, Y.; Teng, M.; Liu, Y.; Singh, R.P.; Nephew, K.P. Alcohol alters DNA methylation patterns and inhibits neural stem cell differentiation. Alcohol. Clin. Exp. Res. 2011, 35, 735–746. [Google Scholar] [CrossRef] [PubMed]
- Ouko, L.A.; Shantikumar, K.; Knezovich, J.; Haycock, P.; Schnugh, D.J.; Ramsay, M. Effect of alcohol consumption on CpG methylation in the differentially methylated regions of H19 and IG-DMR in male gametes—Implications for fetal alcohol spectrum disorders. Alcohol. Clin. Exp. Res. 2009, 33, 1615–1627. [Google Scholar] [CrossRef] [PubMed]
- Haycock, P.C. Fetal alcohol spectrum disorders: The epigenetic perspective 1. Biol. Reprod. 2009, 81, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Marutha Ravindran, C.R.; Ticku, M.K. Changes in methylation pattern of NMDA receptorNR2B gene in cortical neurons after chronic ethanol treatment in mice. Brain Res. Mol. Brain Res. 2004, 121, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Laufer, B.I.; Mantha, K.; Kleiber, M.L.; Diehl, E.J.; Addison, S.M.; Singh, S.M. Long-lasting alterations to DNA methylation and ncRNAs could underlie the effects of fetal alcohol exposure in mice. Dis. Models Mech. 2013, 6, 977–992. [Google Scholar] [CrossRef] [PubMed]
- Govorko, D.; Bekdash, R.A.; Zhang, C.; Sarkar, D.K. Male germline transmits fetal alcohol adverse effect on hypothalamic proopiomelanocortin gene across generations. Biol. Psychiatry 2012, 72, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Shukla, S.D. Acute in vivo effect of ethanol (binge drinking) on histone h3 modifications in rat tissues. Alcohol Alcohol. 2006, 41, 126–132. [Google Scholar] [CrossRef] [PubMed]
- D’Addario, C.; Caputi, F.F.; Ekström, T.J.; Di Benedetto, M.; Maccarrone, M.; Romualdi, P.; Candeletti, S. Ethanol induces epigenetic modulation of prodynorphin and pronociceptin gene expression in the rat amygdala complex. J. Mol. Neurosci. 2013, 49, 312–319. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Crossey, E.L.; Zhang, L.; Zucca, S.; George, O.L.; Valenzuela, C.F.; Zhao, X. Alcohol exposure decreases CREB binding protein expression and histone acetylation in the developing cerebellum. PLoS ONE 2011, 6, e19351. [Google Scholar] [CrossRef] [PubMed]
- Veazey, K.J.; Parnell, S.E.; Miranda, R.C.; Golding, M.C. Dose dependent alcohol-induced alterations in chromatin structure persist beyond the window of exposure and correlate with fetal alcohol syndrome birth defects. Epigenet. Chromatin 2015, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Miranda, R. MicroRNAs and fetal brain development: Implications for ethanol teratology during the second trimester period of neurogenesis. Front. Genet. 2012, 3, 77. [Google Scholar] [CrossRef] [PubMed]
- Sathyan, P.; Golden, H.B.; Miranda, R.C. Competing interactions between micro-RNAs determine neural progenitor survival and proliferation after ethanol exposure: Evidence from an ex vivo model of the fetal cerebral cortical neuroepithelium. J. Neurosci. 2007, 27, 8546–8557. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Zhang, Z.; Li, Q.; Yang, R.; Pei, X.; Xu, Y.; Wang, J.; Zhou, S.-F.; Li, Y. Ethanol exposure induces differential microRNA and target gene expression and teratogenic effects which can be suppressed by folic acid supplementation. Hum. Reprod. 2009, 24, 562–579. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.K.; Gupta, V.K.; Shirasaka, T. An update on fetal alcohol syndrome-pathogenesis, risks, and treatment. Alcohol. Clin. Exp. Res. 2016, 40, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.J.; Van Der Meulen, J.H.; Ravelli, A.C.; Osmond, C.; Barker, D.J.; Bleker, O.P. Effects of prenatal exposure to the dutch famine on adult disease in later life: An overview. Mol. Cell. Endocrinol. 2001, 185, 93–98. [Google Scholar] [CrossRef]
- Bygren, L.O.; Edvinsson, S.; Broström, G. Change in food availability during pregnancy: Is it related to adult sudden death from cerebro- and cardiovascular disease in offspring? Am. J. Hum. Biol. 2000, 12, 447–453. [Google Scholar] [CrossRef]
- Brown, A.S.; Susser, E.S. Sex differences in prevalence of congenital neural defects after periconceptional famine exposure. Epidemiology 1997, 8, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Susser, E.; Neugebauer, R.; Hoek, H.W.; Brown, A.S.; Lin, S.; Labovitz, D.; Gorman, J.M. Schizophrenia after prenatal famine: Further evidence. Arch. Gen. Psychiatry 1996, 53, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Begum, G.; Stevens, A.; Smith, E.B.; Connor, K.; Challis, J.R.; Bloomfield, F.; White, A. Epigenetic changes in fetal hypothalamic energy regulating pathways are associated with maternal undernutrition and twinning. FASEB J. 2012, 26, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.; Murani, E.; Schwerin, M.; Metges, C.C.; Wimmers, K.; Ponsuksili, S. Maternal dietary protein restriction and excess affects offspring gene expression and methylation of non-smc subunits of condensin i in liver and skeletal muscle. Epigenetics 2012, 7, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Takaya, J.; Iharada, A.; Okihana, H.; Kaneko, K. Magnesium deficiency in pregnant rats alters methylation of specific cytosines in the hepatic hydroxysteroid dehydrogenase-2 promoter of the offspring. Epigenetics 2011, 6, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Mathers, J. Early nutrition: Impact on epigenetics. In Nutrigenomics-Opportunities in Asia; Karger Publishers: Basel, Switzerland, 2007; Volume 60, pp. 42–48. [Google Scholar]
- Szyf, M. The early life environment and the epigenome. Biochim. Biophys. Acta BBA Gen. Subj. 2009, 1790, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Waterland, R.A. Assessing the effects of high methionine intake on DNA methylation. J. Nutr. 2006, 136, 1706S–1710S. [Google Scholar] [PubMed]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar] [PubMed]
- Waterland, R.; Travisano, M.; Tahiliani, K.; Rached, M.; Mirza, S. Methyl donor supplementation prevents transgenerational amplification of obesity. Int. J. Obes. 2008, 32, 1373–1379. [Google Scholar] [CrossRef] [PubMed]
- Elango, N.; Hunt, B.G.; Goodisman, M.A.; Soojin, V.Y. DNA methylation is widespread and associated with differential gene expression in castes of the honeybee, Apis mellifera. Proc. Natl. Acad. Sci. USA 2009, 106, 11206–11211. [Google Scholar] [CrossRef] [PubMed]
- Hunt, B.G.; Brisson, J.A.; Soojin, V.Y.; Goodisman, M.A. Functional conservation of DNA methylation in the pea aphid and the honeybee. Genome Biol. Evol. 2010, 2, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Yajnik, C.S.; Deshmukh, U.S. Fetal programming: Maternal nutrition and role of one-carbon metabolism. Rev. Endocr. Metab. Disord. 2012, 13, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, N.; Jia, D.; Geary-Joo, C.; Wu, X.; Ferguson-Smith, A.C.; Fung, E.; Bieda, M.C.; Snyder, F.F.; Gravel, R.A.; Cross, J.C. Mutation in folate metabolism causes epigenetic instability and transgenerational effects on development. Cell 2013, 155, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Sable, P.; Kale, A.; Joshi, A.; Joshi, S. Maternal micronutrient imbalance alters gene expression of BDNF, NGF, TRKB and CREB in the offspring brain at an adult age. Int. J. Dev. Neurosci. 2014, 34, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Yan, J.; West, A.A.; Perry, C.A.; Malysheva, O.V.; Devapatla, S.; Pressman, E.; Vermeylen, F.; Caudill, M.A. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J. 2012, 26, 3563–3574. [Google Scholar] [CrossRef] [PubMed]
- Oliva, J.; Bardag-Gorce, F.; Li, J.; French, B.A.; Nguyen, S.K.; Lu, S.C.; French, S.W. Betaine prevents mallory-denk body formation in drug-primed mice by epigenetic mechanisms. Exp. Mol. Pathol. 2009, 86, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.N. Non-canonical activity of retinoic acid in epigenetic control of embryonic stem cell. Transcription 2013, 4, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Loewy, A.D.; Niles, K.M.; Anastasio, N.; Watkins, D.; Lavoie, J.; Lerner-Ellis, J.P.; Pastinen, T.; Trasler, J.M.; Rosenblatt, D.S. Epigenetic modification of the gene for the vitamin B12 chaperone mmachc can result in increased tumorigenicity and methionine dependence. Mol. Genet. Metab. 2009, 96, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Karlic, H.; Varga, F. Impact of vitamin d metabolism on clinical epigenetics. Clin. Epigenet. 2011, 2, 55. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; McIntosh, G.H.; Le Leu, R.K.; Nyskohus, L.S.; Woodman, R.J.; Young, G.P. Combination of selenium and green tea improves the efficacy of chemoprevention in a rat colorectal cancer model by modulating genetic and epigenetic biomarkers. PLoS ONE 2013, 8, e64362. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.; Diaz, F. Acute dietary zinc deficiency before conception compromises oocyte epigenetic programming and disrupts embryonic development. Dev. Biol. 2013, 376, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.; Kumar, D.; Srivastava, R.K. Epigenetic modifications by dietary phytochemicals: Implications for personalized nutrition. Pharmacol. Ther. 2013, 138, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Pogribny, I.P.; Tryndyak, V.P.; Muskhelishvili, L.; Rusyn, I.; Ross, S.A. Methyl deficiency, alterations in global histone modifications, and carcinogenesis. J. Nutr. 2007, 137, 216S–222S. [Google Scholar] [PubMed]
- Khorram, O.; Han, G.; Bagherpour, R.; Magee, T.R.; Desai, M.; Ross, M.G.; Chaudhri, A.A.; Toloubeydokhti, T.; Pearce, W.J. Effect of maternal undernutrition on vascular expression of micro and messenger RNA in newborn and aging offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1366–R1374. [Google Scholar] [CrossRef] [PubMed]
- Stone, N.; Pangilinan, F.; Molloy, A.M.; Shane, B.; Scott, J.M.; Ueland, P.M.; Mills, J.L.; Kirke, P.N.; Sethupathy, P.; Brody, L.C. Bioinformatic and genetic association analysis of microRNA target sites in one-carbon metabolism genes. PLoS ONE 2011, 6, e21851. [Google Scholar] [CrossRef] [PubMed]
- Franchina, T.; Amodeo, V.; Bronte, G.; Savio, G.; Ricciardi, G.R.; Picciotto, M.; Russo, A.; Giordano, A.; Adamo, V. Circulating mir-22, mir-24 and mir-34a as novel predictive biomarkers to pemetrexed-based chemotherapy in advanced non-small cell lung cancer. J. Cell. Physiol. 2014, 229, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Koturbash, I.; Melnyk, S.; James, S.J.; Beland, F.A.; Pogribny, I.P. Role of epigenetic and miR-22 and miR-29b alterations in the downregulation of Mat1a and Mthfr genes in early preneoplastic livers in rats induced by 2-acetylaminofluorene. Mol. Carcinog. 2013, 52, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Kenny, L.C.; Lavender, T.; McNamee, R.; O’Neill, S.M.; Mills, T.; Khashan, A.S. Advanced maternal age and adverse pregnancy outcome: Evidence from a large contemporary cohort. PLoS ONE 2013, 8, e56583. [Google Scholar] [CrossRef] [PubMed]
- Jolly, M.; Sebire, N.; Harris, J.; Robinson, S.; Regan, L. The risks associated with pregnancy in women aged 35 years or older. Hum. Reprod. 2000, 15, 2433–2437. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.; Syngelaki, A.; Maiz, N.; Zinevich, Y.; Nicolaides, K.H. Maternal age and adverse pregnancy outcome: A cohort study. Ultrasound Obstet. Gynecol. 2013, 42, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Seoud, M.A.-F.; Nassar, A.H.; Usta, I.M.; Melhem, Z.; Kazma, A.; Khalil, A.M. Impact of advanced maternal age on pregnancy outcome. Am. J. Perinatol. 2002, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Novakovic, B.; Yuen, R.K.; Gordon, L.; Penaherrera, M.S.; Sharkey, A.; Moffett, A.; Craig, J.M.; Robinson, W.P.; Saffery, R. Evidence for widespread changes in promoter methylation profile in human placenta in response to increasing gestational age and environmental/stochastic factors. BMC Genom. 2011, 12, 529. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Taylor, J.A. Genome-wide age-related DNA methylation changes in blood and other tissues relate to histone modification, expression and cancer. Carcinogenesis 2014, 35, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Smallwood, S.A.; Kelsey, G. De novo DNA methylation: A germ cell perspective. Trends Genet. 2012, 28, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Yue, M.X.; Fu, X.W.; Zhou, G.B.; Hou, Y.P.; Ming, D.; Wang, L.; Zhu, S.E. Abnormal DNA methylation in oocytes could be associated with a decrease in reproductive potential in old mice. J. Assist. Reprod. Genet. 2012, 29, 643–650. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, T.G.; Aston, K.I.; Pflueger, C.; Cairns, B.R.; Carrell, D.T. Age-associated sperm DNA methylation alterations: Possible implications in offspring disease susceptibility. PLoS Genet. 2014, 10, e1004458. [Google Scholar] [CrossRef] [PubMed]
- Adkins, R.M.; Thomas, F.; Tylavsky, F.A.; Krushkal, J. Parental ages and levels of DNA methylation in the newborn are correlated. BMC Med. Genet. 2011, 12, 47. [Google Scholar] [CrossRef] [PubMed]
- Grether, J.K.; Anderson, M.C.; Croen, L.A.; Smith, D.; Windham, G.C. Risk of autism and increasing maternal and paternal age in a large north American population. Am. J. Epidemiol. 2009, 170, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Shelton, J.F.; Tancredi, D.J.; Hertz-Picciotto, I. Independent and dependent contributions of advanced maternal and paternal ages to autism risk. Autism Res. 2010, 3, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Bolton, P.F.; Murphy, M.; Macdonald, H.; Whitlock, B.; Pickles, A.; Rutter, M. Obstetric complications in autism: Consequences or causes of the condition? J. Am. Acad. Child Adolesc. Psychiatry 1997, 36, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Kamel, R.M. Assisted reproductive technology after the birth of Louise Brown. J. Reprod. Infertil. 2013, 14, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Bonduelle, M.; Wennerholm, U.B.; Loft, A.; Tarlatzis, B.C.; Peters, C.; Henriet, S.; Mau, C.; Victorin-Cederquist, A.; Van Steirteghem, A.; Balaska, A.; et al. A multi-centre cohort study of the physical health of 5-year-old children conceived after intracytoplasmic sperm injection, in vitro fertilization and natural conception. Hum. Reprod. 2005, 20, 413–419. [Google Scholar] [CrossRef] [PubMed]
- Ponjaert-Kristoffersen, I.; Bonduelle, M.; Barnes, J.; Nekkebroeck, J.; Loft, A.; Wennerholm, U.B.; Tarlatzis, B.C.; Peters, C.; Hagberg, B.S.; Berner, A.; et al. International collaborative study of intracytoplasmic sperm injection–conceived, in vitro fertilization–conceived, and naturally conceived 5-year-old child outcomes: Cognitive and motor assessments. Pediatrics 2005, 115, e283–e289. [Google Scholar] [CrossRef] [PubMed]
- Ramoğlu, M.; Kavuncuoğlu, S.; Aldemir, E.; Yarar, C.; Eras, Z. Neurodevelopment of preterm infants born after in vitro fertilization and spontaneous multiple pregnancy. Pediatr. Int. 2016, 58, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Mainigi, M.; Rosenzweig, J.M.; Lei, J.; Mensah, V.; Thomaier, L.; Talbot, C.C., Jr.; Olalere, D.; Ord, T.; Rozzah, R.; Johnston, M.V.; et al. Peri-implantation hormonal milieu: Elucidating mechanisms of adverse neurodevelopmental outcomes. Reprod. Sci. 2016, 23, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Krakowiak, P.; Walker, C.K.; Bremer, A.A.; Baker, A.S.; Ozonoff, S.; Hansen, R.L.; Hertz-Picciotto, I. Maternal metabolic conditions and risk for autism and other neurodevelopmental disorders. Pediatrics 2012, 129, e1121–e1128. [Google Scholar] [CrossRef] [PubMed]
- Van Lieshout, R.; Taylor, V.; Boyle, M. Pre-pregnancy and pregnancy obesity and neurodevelopmental outcomes in offspring: A systematic review. Obes. Rev. 2011, 12, e548–e559. [Google Scholar] [CrossRef] [PubMed]
- O’reilly, J.R.; Reynolds, R.M. The risk of maternal obesity to the long-term health of the offspring. Clin. Endocrinol. 2013, 78, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ruchat, S.M.; Houde, A.A.; Voisin, G.; St-Pierre, J.; Perron, P.; Baillargeon, J.P.; Gaudet, D.; Hivert, M.F.; Brisson, D.; Bouchard, L. Gestational diabetes mellitus epigenetically affects genes predominantly involved in metabolic diseases. Epigenetics 2013, 8, 935–943. [Google Scholar] [CrossRef] [PubMed]
- West, N.A.; Kechris, K.; Dabelea, D. Exposure to maternal diabetes in utero and DNA methylation patterns in the offspring. Immunometabolism 2013, 1, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, M.C.; Adrover, E.; Pallarés, M.E.; Antonelli, M.C.; Frasch, A.C.; Brocco, M.A. Prenatal stress changes the glycoprotein gpm6a gene expression and induces epigenetic changes in rat offspring brain. Epigenetics 2014, 9, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Lester, B.M.; Conradt, E.; Marsit, C.J. Epigenetic basis for the development of depression in children. Clin. Obstet. Gynecol. 2013, 56, 556. [Google Scholar] [CrossRef] [PubMed]
- Wild, S.; Roglic, G.; Green, A.; Sicree, R.; King, H. Global prevalence of diabetes: Estimates for the year 2000 and projections for 2030. Diabetes Care 2004, 27, 1047–1053. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.J.; Schuller, K.L. The increasing prevalence of diabetes in pregnancy. Obstet. Gynecol. Clin. N. Am. 2007, 34, 173–199. [Google Scholar] [CrossRef] [PubMed]
- Perera, F.; Herbstman, J. Prenatal environmental exposures, epigenetics, and disease. Reprod. Toxicol. 2011, 31, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Miyake, K.; Hariya, N.; Mochizuki, K. Understanding the epigenetics of neurodevelopmental disorders and dohad. J. Dev. Orig. Health Dis. 2015, 6, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chase, M.; Jung, S.-K.; Smith, P.J.; Loeken, M.R. Hypoxic stress in diabetic pregnancy contributes to impaired embryo gene expression and defective development by inducing oxidative stress. Am. J. Physiol. Endocrinol. Metab. 2005, 289, E591–E599. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Schoneveld, O.; Georgakilas, A.G.; Panayiotidis, M.I. Oxidative stress, DNA methylation and carcinogenesis. Cancer Lett. 2008, 266, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Kumar, S.D.; Loh, W.T.; Manikandan, J.; Ling, E.A.; Tay, S.S.; Dheen, S.T. Global gene expression analysis of cranial neural tubes in embryos of diabetic mice. J. Neurosci. Res. 2008, 86, 3481–3493. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Tay, S.; Ling, E.; Dheen, S. High glucose alters the expression of genes involved in proliferation and cell-fate specification of embryonic neural stem cells. Diabetologia 2006, 49, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Shyamasundar, S.; Jadhav, S.P.; Bay, B.H.; Tay, S.S.W.; Kumar, S.D.; Rangasamy, D.; Dheen, S.T. Analysis of epigenetic factors in mouse embryonic neural stem cells exposed to hyperglycemia. PLoS ONE 2013, 8, e65945. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.; Fleming, T.; Robinson, M.; Thomson, B.; Graetz, N.; Margono, C.; Mullany, E.C.; Biryukov, S.; Abbafati, C.; Abera, S.F. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: A systematic analysis for the global burden of disease study 2013. Lancet 2014, 384, 766–781. [Google Scholar] [CrossRef]
- Rivera, H.M.; Christiansen, K.J.; Sullivan, E.L. The role of maternal obesity in the risk of neuropsychiatric disorders. Front. Neurosci. 2015, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.L.; Grayson, B.; Takahashi, D.; Robertson, N.; Maier, A.; Bethea, C.L.; Smith, M.S.; Coleman, K.; Grove, K.L. Chronic consumption of a high-fat diet during pregnancy causes perturbations in the serotonergic system and increased anxiety-like behavior in nonhuman primate offspring. J. Neurosci. 2010, 30, 3826–3830. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Bilbo, S.D. Developmental programming of brain and behavior by perinatal diet: Focus on inflammatory mechanisms. Dialogues Clin. Neurosci. 2014, 16, 307. [Google Scholar] [PubMed]
- Salbaum, J.M.; Kappen, C. Responses of the embryonic epigenome to maternal diabetes. Birth Defects Res. Part A Clin. Mol. Teratol. 2012, 94, 770–781. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, R.L.; Hsieh, J.; Barbosa, A.C.; Richardson, J.A.; Olson, E.N. Histone deacetylases 1 and 2 control the progression of neural precursors to neurons during brain development. Proc. Natl. Acad. Sci. USA 2009, 106, 7876–7881. [Google Scholar] [CrossRef] [PubMed]
- Aagaard-Tillery, K.M.; Grove, K.; Bishop, J.; Ke, X.; Fu, Q.; McKnight, R.; Lane, R.H. Developmental origins of disease and determinants of chromatin structure: Maternal diet modifies the primate fetal epigenome. J. Mol. Endocrinol. 2008, 41, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Salas, P.; Moore, S.E.; Baker, M.S.; Bergen, A.W.; Cox, S.E.; Dyer, R.A.; Fulford, A.J.; Guan, Y.; Laritsky, E.; Silver, M.J. Maternal nutrition at conception modulates DNA methylation of human metastable epialleles. Nat. Commun. 2014, 5, 3746. [Google Scholar] [CrossRef] [PubMed]
- Laker, R.C.; Wlodek, M.E.; Connelly, J.J.; Yan, Z. Epigenetic origins of metabolic disease: The impact of the maternal condition to the offspring epigenome and later health consequences. Food Sci. Hum. Wellness 2013, 2, 1–11. [Google Scholar] [CrossRef]
- Law, W.; Bradley, D.; Lazarus, J.; John, R.; Gregory, J. Congenital hypothyroidism in Wales (1982–1993): Demographic features, clinical presentation and effects on early neurodevelopment. Clin. Endocrinol. 1998, 48, 201–207. [Google Scholar] [CrossRef]
- Navarro, D.; Alvarado, M.; Morte, B.; Berbel, D.; Sesma, J.; Pacheco, P.; de Escobar, G.M.; Bernal, J.; Berbel, P. Late maternal hypothyroidism alters the expression of camk4 in neocortical subplate neurons: A comparison with nurr1 labeling. Cereb. Cortex 2013, 24, 2694–2706. [Google Scholar] [CrossRef] [PubMed]
- Pinazo-Durán, M.D.; Pons-Vázquez, S.; Gallego-Pinazo, R.; Estrada, C.G.; Zanón-Moreno, V.; Bou, V.V.; Solana, P.S. Thyroid hormone deficiency disrupts rat eye neurodevelopment. Brain Res. 2011, 1392, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.E.; Hedge, J.M.; Valentín-Blasini, L.; Blount, B.C.; Kannan, K.; Tietge, J.; Zoeller, R.T.; Crofton, K.M.; Jarrett, J.M.; Fisher, J.W. An animal model of marginal iodine deficiency during development: The thyroid axis and neurodevelopmental outcome. Toxicol. Sci. 2013, 132, 177–195. [Google Scholar] [CrossRef] [PubMed]
- Rovet, J.F.; Hepworth, S. Attention problems in adolescents with congenital hypothyroidism: A multicomponential analysis. J. Int. Neuropsychol. Soc. 2001, 7, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Moog, N.; Entringer, S.; Heim, C.; Wadhwa, P.; Kathmann, N.; Buss, C. Influence of maternal thyroid hormones during gestation on fetal brain development. Neuroscience 2017, 342, 68–100. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Shan, Z.; Teng, W.; Yu, X.; Li, Y.; Fan, C.; Teng, X.; Guo, R.; Wang, H.; Li, J. Abnormalities of maternal thyroid function during pregnancy affect neuropsychological development of their children at 25–30 months. Clin. Endocrinol. 2010, 72, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Teng, W.; Shan, Z.; Yu, X.; Gao, Y.; Wang, S.; Fan, C.; Wang, H.; Zhang, H. The effect of maternal subclinical hypothyroidism during pregnancy on brain development in rat offspring. Thyroid 2010, 20, 909–915. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Mohan, V.; Sinha, R.A.; Chagtoo, M.; Godbole, M.M. Histone deacetylase inhibition reduces hypothyroidism-induced neurodevelopmental defects in rats. J. Endocrinol. 2015, 227, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Romanet, P.; Osei, L.; Netchine, I.; Pertuit, M.; Enjalbert, A.; Reynaud, R.; Barlier, A. Case report of gnas epigenetic defect revealed by a congenital hypothyroidism. Pediatrics 2015, 135, e1079–e1083. [Google Scholar] [CrossRef] [PubMed]
- Ratnayake, U.; Quinn, T.; Walker, D.; Dickinson, H. Cytokines and the neurodevelopmental basis of mental illness. Front. Neurosci. 2013, 7, 180. [Google Scholar] [CrossRef] [PubMed]
- Burton, A.; Kizhner, O.; Brown, M.B.; Peltier, M.R. Effect of experimental genital mycoplasmosis on gene expression in the fetal brain. J. Reprod. Immunol. 2012, 93, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Mazina, V.; Gerdts, J.; Trinh, S.; Ankenman, K.; Ward, T.; Dennis, M.Y.; Girirajan, S.; Eichler, E.E.; Bernier, R. Interactive effects of copy number variation and maternal infection on autism impairment. J. Dev. Behav. Pediatr. JDBP 2015, 36, 61. [Google Scholar] [CrossRef] [PubMed]
- Khandaker, G.; Zimbron, J.; Lewis, G.; Jones, P. Prenatal maternal infection, neurodevelopment and adult schizophrenia: A systematic review of population-based studies. Psychol. Med. 2013, 43, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Goeden, N.; Velasquez, J.; Arnold, K.A.; Chan, Y.; Lund, B.T.; Anderson, G.M.; Bonnin, A. Maternal inflammation disrupts fetal neurodevelopment via increased placental output of serotonin to the fetal brain. J. Neurosci. 2016, 36, 6041–6049. [Google Scholar] [CrossRef] [PubMed]
- Ohkawara, T.; Katsuyama, T.; Ida-Eto, M.; Narita, N.; Narita, M. Maternal viral infection during pregnancy impairs development of fetal serotonergic neurons. Brain Dev. 2015, 37, 88–93. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, J.; Yaddanapudi, K.; Hornig, M.; Villar, G.; Serge, R.; Lipkin, W.I. Induction of toll-like receptor 3-mediated immunity during gestation inhibits cortical neurogenesis and causes behavioral disturbances. MBio 2010, 1, e00176-10. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.R.; Falleiros-Arlant, L.H.; Dueñas, L.; Pleitez-Navarrete, J.; Salgado, D.M.; Brea-Del Castillo, J. Congenital and perinatal complications of chikungunya fever: A Latin American experience. Int. J. Infect. Dis. 2016, 51, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Villamil-Gómez, W.; Alba-Silvera, L.; Menco-Ramos, A.; Gonzalez-Vergara, A.; Molinares-Palacios, T.; Barrios-Corrales, M.; Rodríguez-Morales, A.J. Congenital chikungunya virus infection in Sincelejo, Colombia: A case series. J. Trop. Pediatr. 2015, 61, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Gérardin, P.; Barau, G.; Michault, A.; Bintner, M.; Randrianaivo, H.; Choker, G.; Lenglet, Y.; Touret, Y.; Bouveret, A.; Grivard, P. Multidisciplinary prospective study of mother-to-child chikungunya virus infections on the island of La Réunion. PLoS Med. 2008, 5, e60. [Google Scholar] [CrossRef] [PubMed]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.; Sampaio, S.A.; de Filippis, I.; Fabri, A.; Araujo, E.S.; de Sequeira, P.C.; de Mendonça, M.C.; de Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T. Zika virus disrupts phospho-tbk1 localization and mitosis in human neuroepithelial stem cells and radial glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, F.T.; Tesla, B.; Simchick, G.; Zhao, Q.; Hodge, T.; Brindley, M.A.; Stice, S.L. Zika virus induced mortality and microcephaly in chicken embryos. Stem Cells Dev. 2016, 25, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Cauchemez, S.; Besnard, M.; Bompard, P.; Dub, T.; Guillemette-Artur, P.; Eyrolle-Guignot, D.; Salje, H.; van Kerkhove, M.D.; Abadie, V.; Garel, C. Association between zika virus and microcephaly in french polynesia, 2013–15: A retrospective study. Lancet 2016, 387, 2125–2132. [Google Scholar] [CrossRef]
- Belman, A.L.; Muenz, L.R.; Marcus, J.C.; Goedert, J.J.; Landesman, S.; Rubinstein, A.; Goodwin, S.; Durako, S.; Willoughby, A. Neurologic status of human immunodeficiency virus 1-infected infants and their controls: A prospective study from birth to 2 years. Pediatrics 1996, 98, 1109–1118. [Google Scholar] [PubMed]
- Tran, L.T.; Roos, A.; Fouche, J.-P.; Koen, N.; Woods, R.P.; Zar, H.J.; Narr, K.L.; Stein, D.J.; Donald, K.A. White matter microstructural integrity and neurobehavioral outcome of hiv-exposed uninfected neonates. Medicine 2016, 95, e2577. [Google Scholar] [CrossRef] [PubMed]
- Kandawasvika, G.Q.; Ogundipe, E.; Gumbo, F.Z.; Kurewa, E.N.; Mapingure, M.P.; Stray-Pedersen, B. Neurodevelopmental impairment among infants born to mothers infected with human immunodeficiency virus and uninfected mothers from three peri-urban primary care clinics in harare, zimbabwe. Dev. Med. Child Neurol. 2011, 53, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Drotar, D.; Olness, K.; Wiznitzer, M.; Guay, L.; Marum, L.; Svilar, G.; Hom, D.; Fagan, J.F.; Ndugwa, C.; Kiziri-Mayengo, R. Neurodevelopmental outcomes of ugandan infants with human immunodeficiency virus type 1 infection. Pediatrics 1997, 100, e5. [Google Scholar] [CrossRef] [PubMed]
- Salemi, J.; Whiteman, V.; August, E.; Chandler, K.; Mbah, A.; Salihu, H. Maternal hepatitis B and hepatitis C infection and neonatal neurological outcomes. J. Viral Hepat. 2014, 21, e144–e153. [Google Scholar] [CrossRef] [PubMed]
- Euscher, E.; Davis, J.; Holzman, I.; Nuovo, G.J. Coxsackie virus infection of the placenta associated with neurodevelopmental delays in the newborn. Obstet. Gynecol. 2001, 98, 1019–1026. [Google Scholar] [CrossRef] [PubMed]
- Cheslack-Postava, K.; Brown, A.S.; Chudal, R.; Suominen, A.; Huttunen, J.; Surcel, H.-M.; Sourander, A. Maternal exposure to sexually transmitted infections and schizophrenia among offspring. Schizophr. Res. 2015, 166, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Basil, P.; Li, Q.; Dempster, E.; Mill, J.; Sham, P.; Wong, C.; McAlonan, G. Prenatal maternal immune activation causes epigenetic differences in adolescent mouse brain. Trans. Psychiatry 2014, 4, e434. [Google Scholar] [CrossRef] [PubMed]
- Labouesse, M.A.; Dong, E.; Grayson, D.R.; Guidotti, A.; Meyer, U. Maternal immune activation induces gad1 and gad2 promoter remodeling in the offspring prefrontal cortex. Epigenetics 2015, 10, 1143–1155. [Google Scholar] [CrossRef] [PubMed]
- Poletaev, A.B.; Poletaeva, A.A.; Pukhalenko, A.I.; Zamaleeva, R.S.; Cherepanova, N.A.; Frizin, D.V. Adaptive maternal immune deviations as a ground for autism spectrum disorders development in children. Folia Med. 2014, 56, 73–80. [Google Scholar] [CrossRef]
- Wang, H.; Cao, Q.; Ge, J.; Liu, C.; Ma, Y.; Meng, Y.; Wang, Y.; Zhao, X.; Liu, R.; Li, C. LncRNA-regulated infection and inflammation pathways associated with pregnancy loss: Genome wide differential expression of lncRNAs in early spontaneous abortion. Am. J. Reprod. Immunol. 2014, 72, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Shi, Q.; Gu, Y.; Pan, J.; Hua, M.; Liu, M.; Dong, Z.; Zhang, M.; Wang, L.; Gu, Y. LncRNA pathway involved in premature preterm rupture of membrane (PPROM): An epigenomic approach to study the pathogenesis of reproductive disorders. PLoS ONE 2013, 8, e79897. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Sameshima, H.; Minematsu, T.; Kusumoto, K.; Yamauchi, A.; Ikenoue, T. Maternal IgG avidity, IgM and ultrasound abnormalities: Combined method to detect congenital cytomegalovirus infection with sequelae. J. Perinatol. 2013, 33, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Corey, L.; Wald, A. Maternal and neonatal herpes simplex virus infections. N. Engl. J. Med. 2009, 361, 1376–1385. [Google Scholar] [CrossRef] [PubMed]
- Buiting, K.; Gross, S.; Lich, C.; Gillessen-Kaesbach, G.; el-Maarri, O.; Horsthemke, B. Epimutations in Prader-Willi and Angelman syndromes: A molecular study of 136 patients with an imprinting defect. Am. J. Hum. Genet. 2003, 72, 571–577. [Google Scholar] [CrossRef] [PubMed]
- El-Maarri, O.; Buiting, K.; Peery, E.G.; Kroisel, P.M.; Balaban, B.; Wagner, K.; Urman, B.; Heyd, J.; Lich, C.; Brannan, C.I.; et al. Maternal methylation imprints on human chromosome 15 are established during or after fertilization. Nat. Genet. 2001, 27, 341–344. [Google Scholar] [CrossRef] [PubMed]
- Vieira, J.P.; Lopes, F.; Silva-Fernandes, A.; Sousa, M.V.; Moura, S.; Sousa, S.; Costa, B.M.; Barbosa, M.; Ylstra, B.; Temudo, T.; et al. Variant Rett syndrome in a girl with a pericentric X-chromosome inversion leading to epigenetic changes and overexpression of the MeCP2 gene. Int. J. Dev. Neurosci. 2015, 46, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Brasa, S.; Mueller, A.; Jacquemont, S.; Hahne, F.; Rozenberg, I.; Peters, T.; He, Y.; McCormack, C.; Gasparini, F.; Chibout, S.D.; et al. Reciprocal changes in DNA methylation and hydroxymethylation and a broad repressiveepigenetic switch characterize FMR1 transcriptional silencing infragile X syndrome. Clin. Epigenet. 2016, 8, 15. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Salvioli, S.; Gentilini, D.; Di Blasio, A.M.; Giuliani, C.; Tung, S.; Vinters, H.V.; et al. Accelerated epigenetic aging in Down syndrome. Aging Cell 2015, 14, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Bürger, J.; Buiting, K.; Dittrich, B.; Gross, S.; Lich, C.; Sperling, K.; Horsthemke, B.; Reis, A. Different mechanisms and recurrence risks of imprinting defects in Angelman syndrome. Am. J. Hum. Genet. 1997, 61, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kucukkal, T.G.; Li, J.; Alexov, E.; Cao, W. Binding analysis of methyl-CpG binding domain of MeCP2 and Rett syndrome mutations. ACS Chem. Biol. 2016, 11, 2706–2715. [Google Scholar] [CrossRef] [PubMed]
- Petazzi, P.; Sandoval, J.; Szczesna, K.; Jorge, O.C.; Roa, L.; Sayols, S.; Gomez, A.; Huertas, D.; Esteller, M. Dysregulation of the long non-coding RNA transcriptome in a Rett syndrome mouse model. RNA Biol. 2013, 10, 1197–1203. [Google Scholar] [CrossRef] [PubMed]
- Hunihan, L.; Brown, J.; Cacace, A.; Fernandes, A.; Weston, A. Generation of a clonal induced pluripotent stem cell (iPSC) line expressing the mutant MeCP2 allele from a Rett Syndrome patient fibroblast line. Stem Cell Res. 2017, 20, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Hagerman, R.J.; Berry-Kravis, E.; Kaufmann, W.E.; Ono, M.Y.; Tartaglia, N.; Lachiewicz, A.; Kronk, R.; Delahunty, C.; Hessl, D.; Visootsak, J.; et al. Advances in the treatment of fragile X syndrome. Pediatrics 2009, 123, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Chiurazzi, P. Epigenetics, fragile X syndromeand transcriptional therapy. Am. J. Med. Genet. A 2013, 161A, 2797–2808. [Google Scholar] [CrossRef] [PubMed]
- Resendiz, M.; Chen, Y.; Öztürk, N.C.; Zhou, F.C. Epigenetic medicine and fetal alcohol spectrum disorders. Epigenomics 2013, 5, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Miyake, K.; Hariya, N.; Mochizuki, K. Epigenetics as a basis for diagnosis of neurodevelopmental disorders: Challenges and opportunities. Expert Rev. Mol. Diagn. 2014, 14, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Agudelo, M.; Gandhi, N.; Saiyed, Z.; Pichili, V.; Thangavel, S.; Khatavkar, P.; Yndart-Arias, A.; Nair, M. Effects of alcohol on histone deacetylase 2 (HDAC2) and the neuroprotective role of trichostatin a (TSA). Alcohol. Clin. Exp. Res. 2011, 35, 1550–1556. [Google Scholar] [CrossRef] [PubMed]
- Takuma, K.; Hara, Y.; Kataoka, S.; Kawanai, T.; Maeda, Y.; Watanabe, R.; Takano, E.; Hayata-Takano, A.; Hashimoto, H.; Ago, Y. Chronic treatment with valproic acid or sodium butyrate attenuates novel object recognition deficits and hippocampal dendritic spine loss in a mouse model of autism. Pharmacol. Biochem. Behav. 2014, 126, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Sodium Butyrate for Improving Cognitive Function in Schizophrenia. Available online: https://clinicaltrials.gov/ct2/show/NCT02654405 (accessed on 15 March 2017).
- Treatment of Children with Autism Spectrum Disorders and Epileptiform EEG with Divalproex Sodium. Available online: https://clinicaltrials.gov/ct2/show/NCT02094651 (accessed on 15 March 2017).
- Jacka, F.N.; Ystrom, E.; Brantsaeter, A.L.; Karevold, E.; Roth, C.; Haugen, M.; Meltzer, H.M.; Schjolberg, S.; Berk, M. Maternal and early postnatal nutrition and mental health of offspring by age 5 years: A prospective cohort study. J. Am. Acad. Child Adolesc. Psychiatry 2013, 52, 1038–1047. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, A.; Itsiopoulos, C.; Skouteris, H.; Opie, R.S.; McPhie, S.; Hill, B.; Jacka, F.N. Preventing mental health problems in offspring by targeting dietary intake of pregnant women. BMC Med. 2014, 12, 208. [Google Scholar] [CrossRef] [PubMed]




© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banik, A.; Kandilya, D.; Ramya, S.; Stünkel, W.; Chong, Y.S.; Dheen, S.T. Maternal Factors that Induce Epigenetic Changes Contribute to Neurological Disorders in Offspring. Genes 2017, 8, 150. https://doi.org/10.3390/genes8060150
Banik A, Kandilya D, Ramya S, Stünkel W, Chong YS, Dheen ST. Maternal Factors that Induce Epigenetic Changes Contribute to Neurological Disorders in Offspring. Genes. 2017; 8(6):150. https://doi.org/10.3390/genes8060150
Chicago/Turabian StyleBanik, Avijit, Deepika Kandilya, Seshadri Ramya, Walter Stünkel, Yap Seng Chong, and S. Thameem Dheen. 2017. "Maternal Factors that Induce Epigenetic Changes Contribute to Neurological Disorders in Offspring" Genes 8, no. 6: 150. https://doi.org/10.3390/genes8060150
APA StyleBanik, A., Kandilya, D., Ramya, S., Stünkel, W., Chong, Y. S., & Dheen, S. T. (2017). Maternal Factors that Induce Epigenetic Changes Contribute to Neurological Disorders in Offspring. Genes, 8(6), 150. https://doi.org/10.3390/genes8060150