Identification of a Common Different Gene Expression Signature in Ischemic Cardiomyopathy

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Differential Expression Analysis
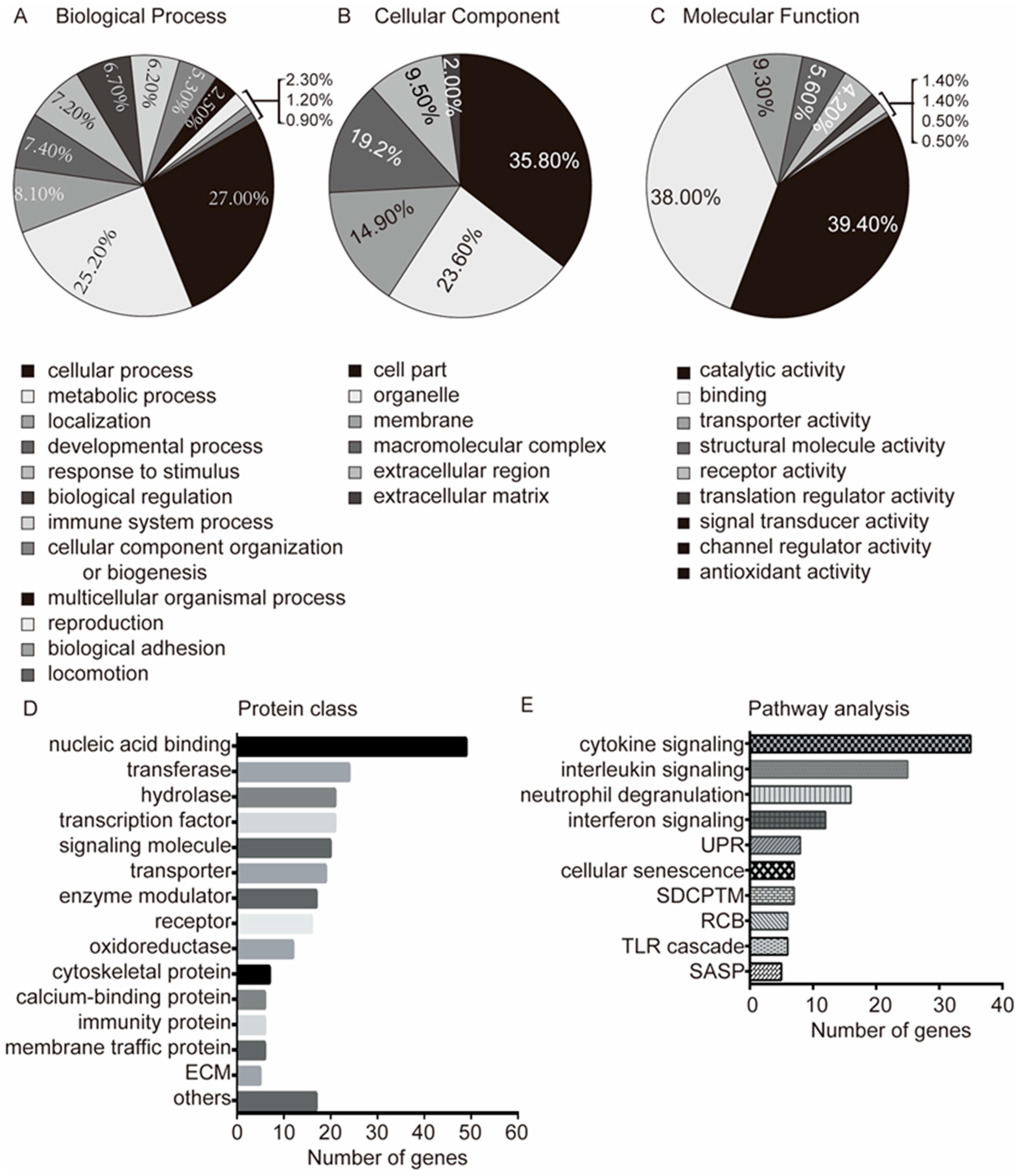
2.3. Gene Ontology Analyses
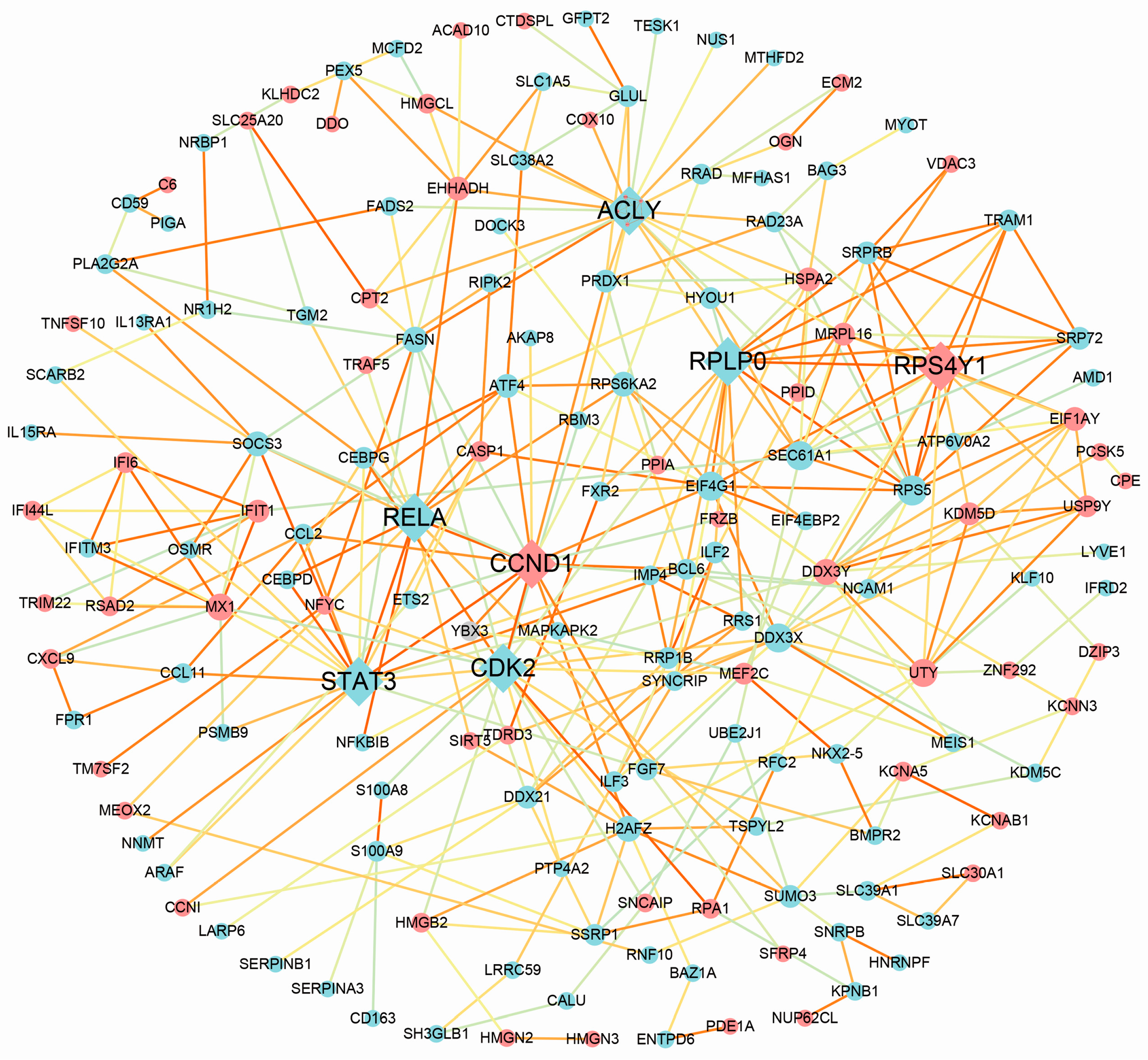
2.4. Protein–Protein Interaction Network Construction Analysis
2.5. Sample Collection
2.6. RNA Isolation and Real-Time Polymerase Chain Reaction
2.7. Statistical Analysis
3. Results
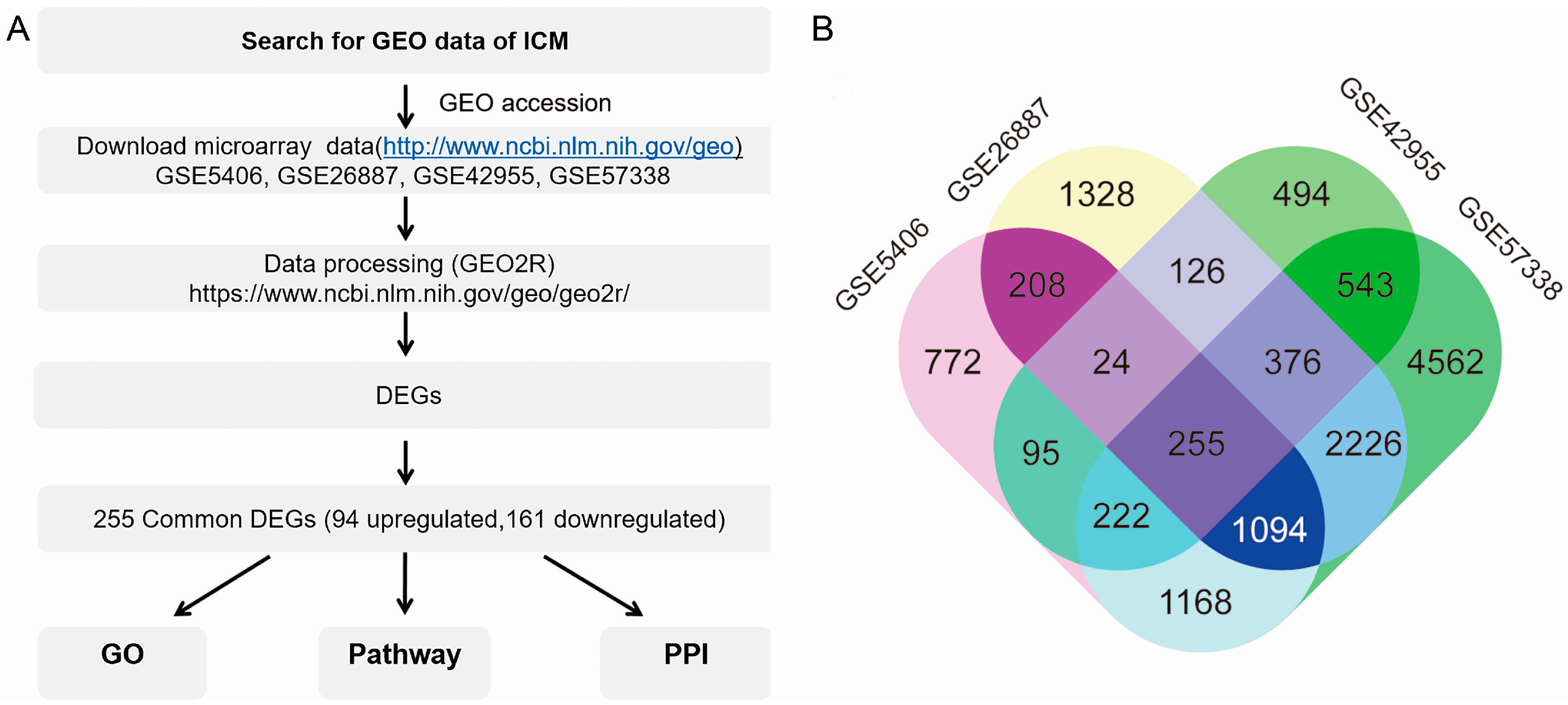
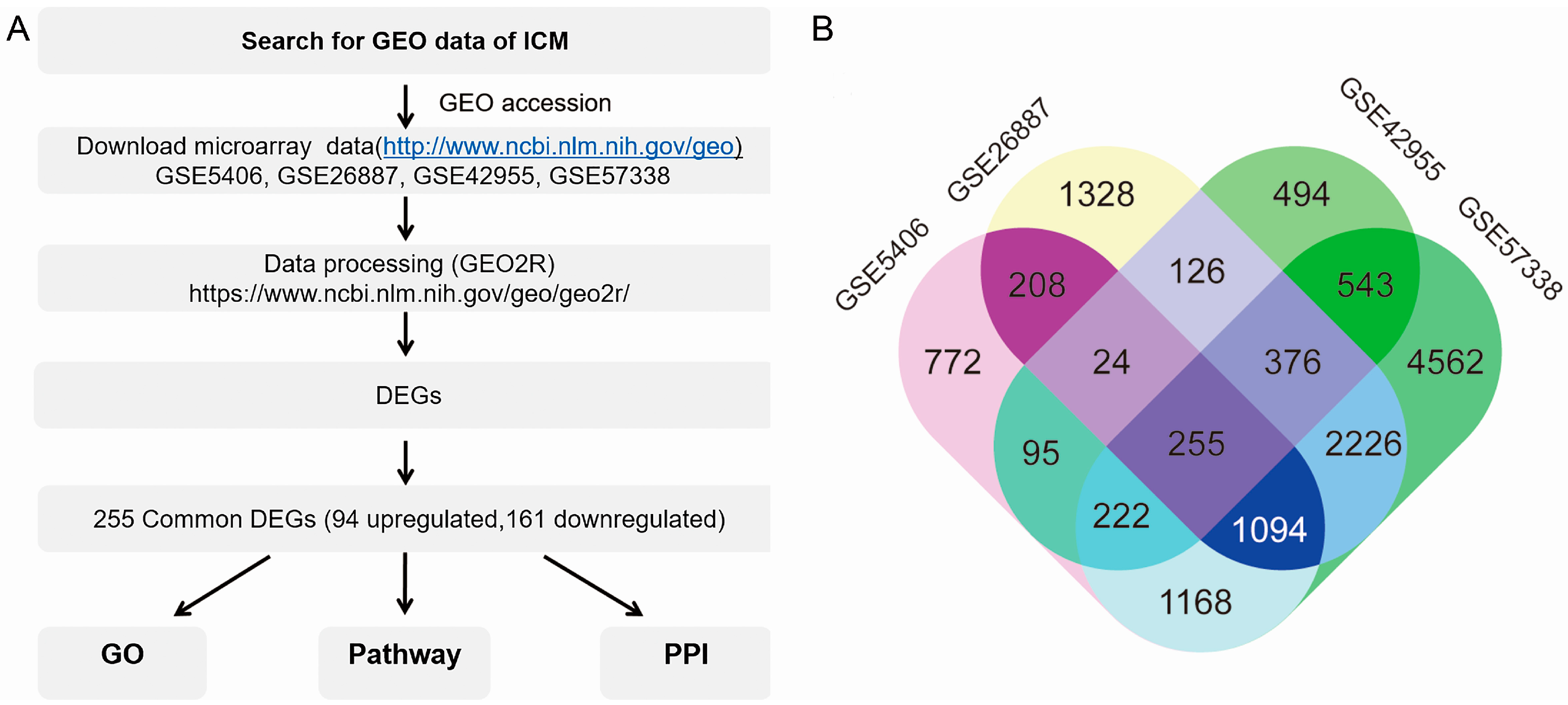
3.1. Screening Common Differentially Expressed Genes from Four Gene Espression Omnibus Series
3.2. Functional Enrichment and Integrated Analysis of the Differentially Expressed Genes
3.3. Pathway Analysis
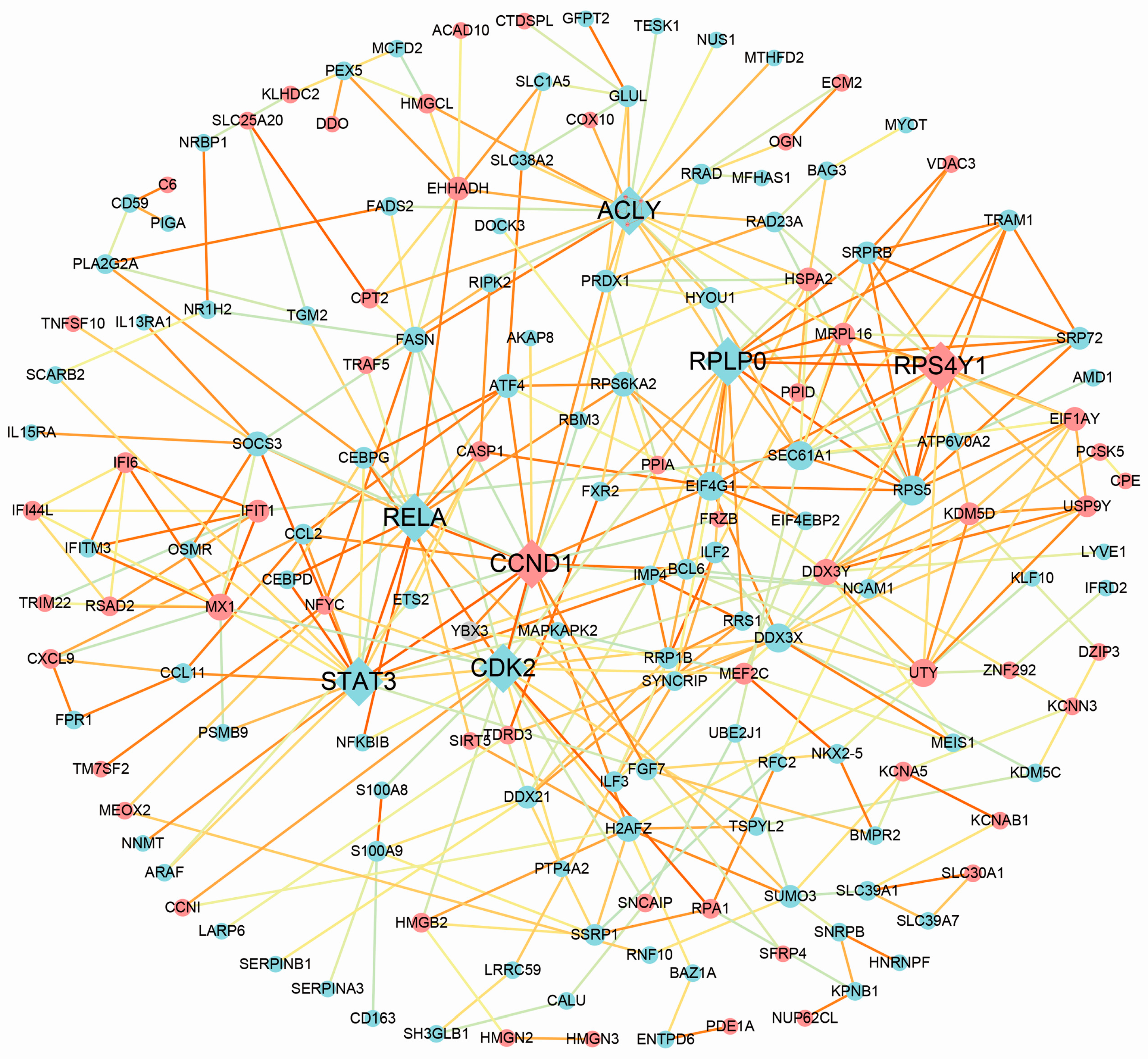
3.4. Protein–Protein Interaction Analysis
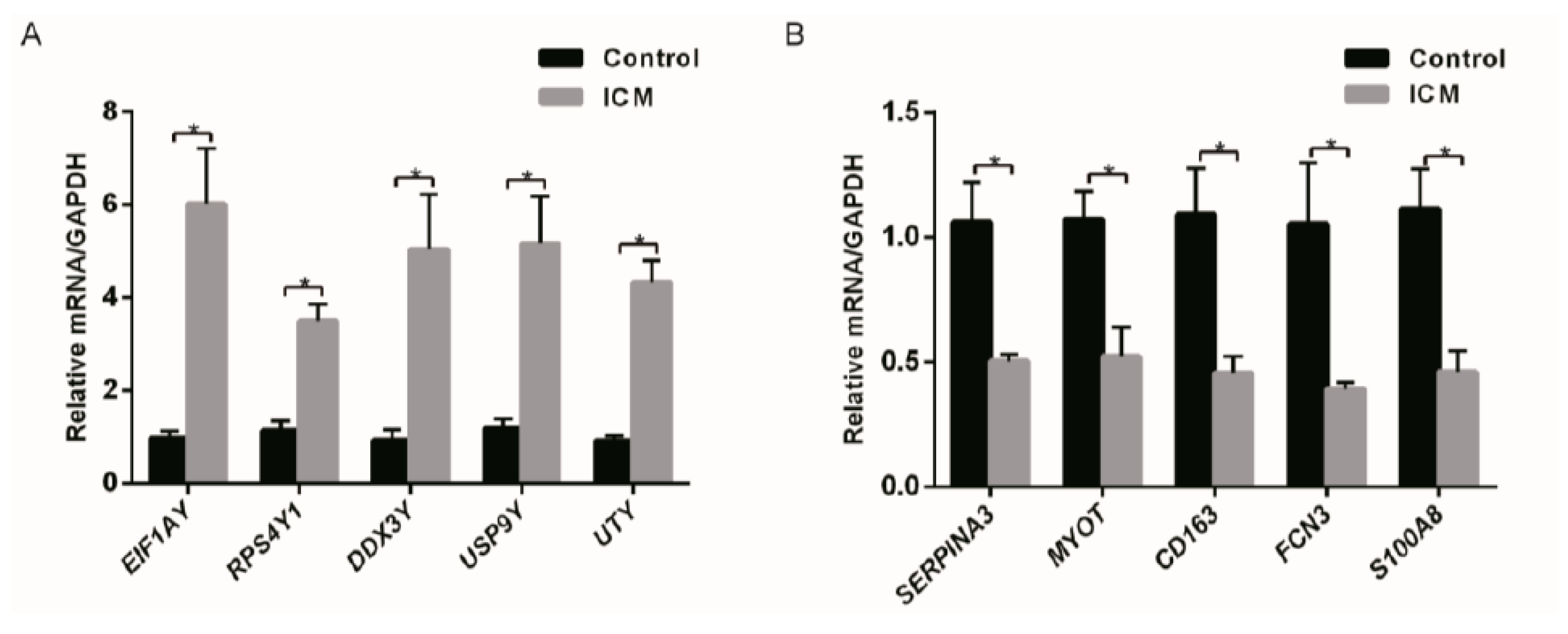
3.5. Real-Time Polymerase Chain Reaction Validation
4. Discussion
4.1. Inflammation and the Immune System Are Critical in the Development of Ischemic Cardiomyopathy
4.2. The Improvement of Lipid Metabolism Could Be a Potential Target for Therapy of Ischemic Cardiomyopathy
4.3. Particular Genes on Chromosome Y May Be Risk Factors for Coronary Heart Disease and Ischemic Cardiomyopathy
4.4. Could Heart Failure Induce Myocardial Regeneration?
4.5. Limitations of the Study
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; Gonzalez-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. Rev. Esp. Cardiol. 2016, 69, 1167. [Google Scholar] [PubMed]
- Felker, G.M.; Shaw, L.K.; O’Connor, C.M. A standardized definition of ischemic cardiomyopathy for use in clinical research. J. Am. Coll. Cardiol. 2002, 39, 210–218. [Google Scholar] [CrossRef]
- Sutton, M.G.; Sharpe, N. Left ventricular remodeling after myocardial infarction: Pathophysiology and therapy. Circulation 2000, 101, 2981–2988. [Google Scholar] [CrossRef] [PubMed]
- Rouleau, J.L.; de Champlain, J.; Klein, M.; Bichet, D.; Moye, L.; Packer, M.; Dagenais, G.R.; Sussex, B.; Arnold, J.M.; Sestier, F.; et al. Activation of neurohumoral systems in postinfarction left ventricular dysfunction. J. Am. Coll. Cardiol. 1993, 22, 390–398. [Google Scholar] [CrossRef]
- Ripatti, S.; Tikkanen, E.; Orho-Melander, M.; Havulinna, A.S.; Silander, K.; Sharma, A.; Guiducci, C.; Perola, M.; Jula, A.; Sinisalo, J.; et al. A multilocus genetic risk score for coronary heart disease: Case-control and prospective cohort analyses. Lancet 2010, 376, 1393–1400. [Google Scholar] [CrossRef]
- Yamazaki, T.; Komuro, I.; Kudoh, S.; Zou, Y.; Shiojima, I.; Mizuno, T.; Takano, H.; Hiroi, Y.; Ueki, K.; Tobe, K.; et al. Angiotensin II partly mediates mechanical stress-induced cardiac hypertrophy. Circ. Res. 1995, 77, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H2181–H2190. [Google Scholar] [CrossRef] [PubMed]
- Bogoyevitch, M.A.; Glennon, P.E.; Andersson, M.B.; Clerk, A.; Lazou, A.; Marshall, C.J.; Parker, P.J.; Sugden, P.H. Endothelin-1 and fibroblast growth factors stimulate the mitogen-activated protein kinase signaling cascade in cardiac myocytes. The potential role of the cascade in the integration of two signaling pathways leading to myocyte hypertrophy. J. Biol. Chem. 1994, 269, 1110–1119. [Google Scholar] [PubMed]
- Zhao, W.; Zhao, T.; Huang, V.; Chen, Y.; Ahokas, R.A.; Sun, Y. Platelet-derived growth factor involvement in myocardial remodeling following infarction. J. Mol. Cell. Cardiol. 2011, 51, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Rainer, P.P.; Hao, S.; Vanhoutte, D.; Lee, D.I.; Koitabashi, N.; Molkentin, J.D.; Kass, D.A. Cardiomyocyte-specific transforming growth factor beta suppression blocks neutrophil infiltration, augments multiple cytoprotective cascades, and reduces early mortality after myocardial infarction. Circ. Res. 2014, 114, 1246–1257. [Google Scholar] [CrossRef] [PubMed]
- Desmoulière, A.; Geinoz, A.; Gabbiani, F.; Gabbiani, G. Transforming growth factor-β1 induces α-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J. Cell. Biol. 1993, 122, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Guarda, E.; Katwa, L.C.; Myers, P.R.; Tyagi, S.C.; Weber, K.T. Effects of endothelins on collagen turnover in cardiac fibroblasts. Cardiovasc. Res. 1993, 27, 2130–2134. [Google Scholar] [CrossRef] [PubMed]
- Hannenhalli, S.; Putt, M.E.; Gilmore, J.M.; Wang, J.; Parmacek, M.S.; Epstein, J.A.; Morrisey, E.E.; Margulies, K.B.; Cappola, T.P. Transcriptional genomics associates FOX transcription factors with human heart failure. Circulation 2006, 114, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Kittleson, M.M.; Ye, S.Q.; Irizarry, R.A.; Minhas, K.M.; Edness, G.; Conte, J.V.; Parmigiani, G.; Miller, L.W.; Chen, Y.; Hall, J.L.; et al. Identification of a gene expression profile that differentiates between ischemic and nonischemic cardiomyopathy. Circulation 2004, 110, 3444–3451. [Google Scholar] [CrossRef] [PubMed]
- Kittleson, M.M.; Minhas, K.M.; Irizarry, R.A.; Ye, S.Q.; Edness, G.; Breton, E.; Conte, J.V.; Tomaselli, G.; Garcia, J.G.; Hare, J.M. Gene expression analysis of ischemic and nonischemic cardiomyopathy: Shared and distinct genes in the development of heart failure. Physiol. Genom. 2005, 21, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Qiao, A.; Zhao, Z.; Zhang, H.; Sun, Z.; Cui, X. Gene expression profiling reveals genes and transcription factors associated with dilated and ischemic cardiomyopathies. Pathol. Res. Pract. 2017, 213, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- Jupe, S.; Fabregat, A.; Hermjakob, H. Expression data analysis with Reactome. Curr. Protoc. Bioinform. 2015, 49, 8–20. [Google Scholar]
- Szklarczyk, D.; Morris, J.H.; Cook, H.; Kuhn, M.; Wyder, S.; Simonovic, M.; Santos, A.; Doncheva, N.T.; Roth, A.; Bork, P.; et al. The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017, 45, D362–D368. [Google Scholar] [CrossRef] [PubMed]
- Doncheva, N.T.; Assenov, Y.; Domingues, F.S.; Albrecht, M. Topological analysis and interactive visualization of biological networks and protein structures. Nat Protoc. 2012, 7, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Torre-Amione, G. Immune activation in chronic heart failure. Am. J. Cardiol. 2005, 95, 3C–8C. [Google Scholar] [CrossRef] [PubMed]
- Flores-Arredondo, J.H.; Garcia-Rivas, G.; Torre-Amione, G. Immune modulation in heart failure: Past challenges and future hopes. Curr. Heart Fail. Rep. 2011, 8, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Gao, W.; Yuan, J.; Wu, C.; Yao, K.; Zhang, L.; Ma, L.; Zhu, J.; Zou, Y.; Ge, J. Exosomes derived from dendritic cells improve cardiac function via activation of CD4+ T lymphocytes after myocardial infarction. J. Mol. Cell. Cardiol. 2016, 91, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S.; et al. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012, 125, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Dalod, M.; Chelbi, R.; Malissen, B.; Lawrence, T. Dendritic cell maturation: Functional specialization through signaling specificity and transcriptional programming. EMBO J. 2014, 33, 1104–1116. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Trujillo, L.; Vazquez-Garza, E.; Castillo, E.C.; Garcia-Rivas, G.; Torre-Amione, G. Role of adaptive immunity in the development and progression of heart failure: New evidence. Arch. Med. Res. 2017, 48, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Diamantopoulos, A.P.; Larsen, A.I.; Omdal, R. Is it safe to use TNF-α blockers for systemic inflammatory disease in patients with heart failure? Importance of dosage and receptor specificity. Int. J. Cardiol. 2013, 167, 1719–1723. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L.; McMurray, J.J.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Coats, A.J. How to RECOVER from RENAISSANCE? The significance of the results of RECOVER, RENAISSANCE, RENEWAL and ATTACH. Int. J. Cardiol. 2002, 86, 123–130. [Google Scholar] [CrossRef]
- Azevedo, P.S.; Minicucci, M.F.; Santos, P.P.; Paiva, S.A.; Zornoff, L.A. Energy metabolism in cardiac remodeling and heart failure. Cardiol. Rev. 2013, 21, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Hatzivassiliou, G.; Zhao, F.; Bauer, D.E.; Andreadis, C.; Shaw, A.N.; Dhanak, D.; Hingorani, S.R.; Tuveson, D.A.; Thompson, C.B. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 2005, 8, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Morvan, F.; Jourde, B.; Meier, V.; Kahle, P.; Brebbia, P.; Toussaint, G.; Glass, D.J.; Fornaro, M. ATP citrate lyase improves mitochondrial function in skeletal muscle. Cell Metab. 2015, 21, 868–876. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.Y.; Jeon, H.K.; Choi, J.S.; Kim, Y.J. Reduced expression of FASN through SREBP-1 down-regulation is responsible for hypoxic cell death in HepG2 cells. J. Cell Biochem. 2012, 113, 3730–3739. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Jin, G.; Mi, R.; Zhang, J.; Zhang, J.; Xu, H.; Cheng, S.; Zhang, Y.; Song, W.; Liu, F. Inhibition of fatty acid synthase suppresses neovascularization via regulating the expression of VEGF-A in glioma. J. Cancer Res. Clin. Oncol. 2016, 142, 2447–2459. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Zhang, H.; Schulze, P.C.; Schilling, J.D.; Verbsky, J.; Lodhi, I.J.; Topkara, V.K.; Feng, C.; Coleman, T.; Kovacs, A.; et al. Fatty acid synthase modulates homeostatic responses to myocardial stress. J. Biol. Chem. 2011, 286, 30949–30961. [Google Scholar] [CrossRef] [PubMed]
- Gusarova, V.; Brodsky, J.L.; Fisher, E.A. Apolipoprotein B100 exit from the endoplasmic reticulum (ER) is COPII-dependent, and its lipidation to very low density lipoprotein occurs post-ER. J. Biol. Chem. 2003, 278, 48051–48058. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.S.; Goldstein, J.L. Sterol regulatory element binding proteins (SREBPs): Controllers of lipid synthesis and cellular uptake. Nutr. Rev. 1998, 56, S1–S3. [Google Scholar] [CrossRef] [PubMed]
- Schiavoni, G.; Bennati, A.M.; Castelli, M.; Della, F.M.; Beccari, T.; Servillo, G.; Roberti, R. Activation of TM7SF2 promoter by SREBP-2 depends on a new sterol regulatory element, a GC-box, and an inverted CCAAT-box. Biochim. Biophys. Acta. 2010, 1801, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Sato, R. SREBPs: Protein interaction and SREBPs. FEBS J. 2009, 276, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Charchar, F.J.; Bloomer, L.D.; Barnes, T.A.; Cowley, M.J.; Nelson, C.P.; Wang, Y.; Denniff, M.; Debiec, R.; Christofidou, P.; Nankervis, S.; et al. Inheritance of coronary artery disease in men: An analysis of the role of the Y chromosome. Lancet 2012, 379, 915–922. [Google Scholar] [CrossRef]
- Bloomer, L.D.; Nelson, C.P.; Eales, J.; Denniff, M.; Christofidou, P.; Debiec, R.; Moore, J.; Zukowska-Szczechowska, E.; Goodall, A.H.; Thompson, J.; et al. Male-specific region of the Y chromosome and cardiovascular risk: Phylogenetic analysis and gene expression studies. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1722–1727. [Google Scholar] [CrossRef] [PubMed]
- Heidecker, B.; Lamirault, G.; Kasper, E.K.; Wittstein, I.S.; Champion, H.C.; Breton, E.; Russell, S.D.; Hall, J.; Kittleson, M.M.; Baughman, K.L.; et al. The gene expression profile of patients with new-onset heart failure reveals important gender-specific differences. Eur. Heart J. 2010, 31, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, O.; Bhardwaj, R.D.; Bernard, S.; Zdunek, S.; Barnabe-Heider, F.; Walsh, S.; Zupicich, J.; Alkass, K.; Buchholz, B.A.; Druid, H.; et al. Evidence for cardiomyocyte renewal in humans. Science 2009, 324, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Nakada, Y.; Canseco, D.C.; Thet, S.; Abdisalaam, S.; Asaithamby, A.; Santos, C.X.; Shah, A.M.; Zhang, H.; Faber, J.E.; Kinter, M.T.; et al. Hypoxia induces heart regeneration in adult mice. Nature 2017, 541, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Olson, E.N.; Bassel-Duby, R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Mankoo, B.S.; Collins, N.S.; Ashby, P.; Grigorieva, E.; Pevny, L.H.; Candia, A.; Wright, C.V.; Rigby, P.W.; Pachnis, V. Mox2 is a component of the genetic hierarchy controlling limb muscle development. Nature 1999, 400, 69–73. [Google Scholar] [PubMed]
- Coppiello, G.; Collantes, M.; Sirerol-Piquer, M.S.; Vandenwijngaert, S.; Schoors, S.; Swinnen, M.; Vandersmissen, I.; Herijgers, P.; Topal, B.; van Loon, J.; et al. Meox2/Tcf15 heterodimers program the heart capillary endothelium for cardiac fatty acid uptake. Circulation 2015, 131, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Kumarswamy, R.; Thum, T. Non-coding RNAs in cardiac remodeling and heart failure. Circ. Res. 2013, 113, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Boheler, K.R.; Volkova, M.; Morrell, C.; Garg, R.; Zhu, Y.; Margulies, K.; Seymour, A.M.; Lakatta, E.G. Sex- and age-dependent human transcriptome variability: Implications for chronic heart failure. Proc. Natl. Acad. Sci. USA 2003, 100, 2754–2759. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene symbol | Log(FCA) | Biological Function |
|---|---|---|
| EIF1AY | 3.44 | Chromatin organization |
| UTY | 2.28 | Chromatin organization |
| USP9Y | 2.12 | Ubiquitin-Proteasome Dependent Proteolysis |
| RPS4Y1 | 1.95 | Chromatin organization |
| DDX3Y | 1.78 | Translational initiation |
| SERPINA3 | −3.04 | Immune Response to elevated platelet cytosolic Ca2+ |
| MYOT | −2.27 | Structural constituent of muscle |
| S100A8 | −2.03 | Immune System (Activated TLR4 signaling) |
| FCN3 | −1.96 | Immune System (Lectin pathway of complement activation) |
| CD163 | −1.86 | Scavenger Receptors signaling |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Jiang, Q.; Ding, Z.; Liu, G.; Yu, P.; Jiang, G.; Yu, Z.; Yang, C.; Qian, J.; Jiang, H.; et al. Identification of a Common Different Gene Expression Signature in Ischemic Cardiomyopathy. Genes 2018, 9, 56. https://doi.org/10.3390/genes9010056
Li Y, Jiang Q, Ding Z, Liu G, Yu P, Jiang G, Yu Z, Yang C, Qian J, Jiang H, et al. Identification of a Common Different Gene Expression Signature in Ischemic Cardiomyopathy. Genes. 2018; 9(1):56. https://doi.org/10.3390/genes9010056
Chicago/Turabian StyleLi, Yana, Qiu Jiang, Zhiwen Ding, Guijian Liu, Peng Yu, Guoliang Jiang, Ziqing Yu, Chunjie Yang, Juying Qian, Hong Jiang, and et al. 2018. "Identification of a Common Different Gene Expression Signature in Ischemic Cardiomyopathy" Genes 9, no. 1: 56. https://doi.org/10.3390/genes9010056
APA StyleLi, Y., Jiang, Q., Ding, Z., Liu, G., Yu, P., Jiang, G., Yu, Z., Yang, C., Qian, J., Jiang, H., & Zou, Y. (2018). Identification of a Common Different Gene Expression Signature in Ischemic Cardiomyopathy. Genes, 9(1), 56. https://doi.org/10.3390/genes9010056