Genome-Wide Identification of circRNAs in Pathogenic Basidiomycetous Yeast Cryptococcus neoformans Suggests Conserved circRNA Host Genes over Kingdoms


Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Media
2.2. RNA Isolation and Quality Control
2.3. Deep RNA Sequencing and In Silico Discovery of Circular RNAs
2.4. Gene Ontology Category and Kyoto Encyclopedia of Genes and Genomes Pathway Analysis
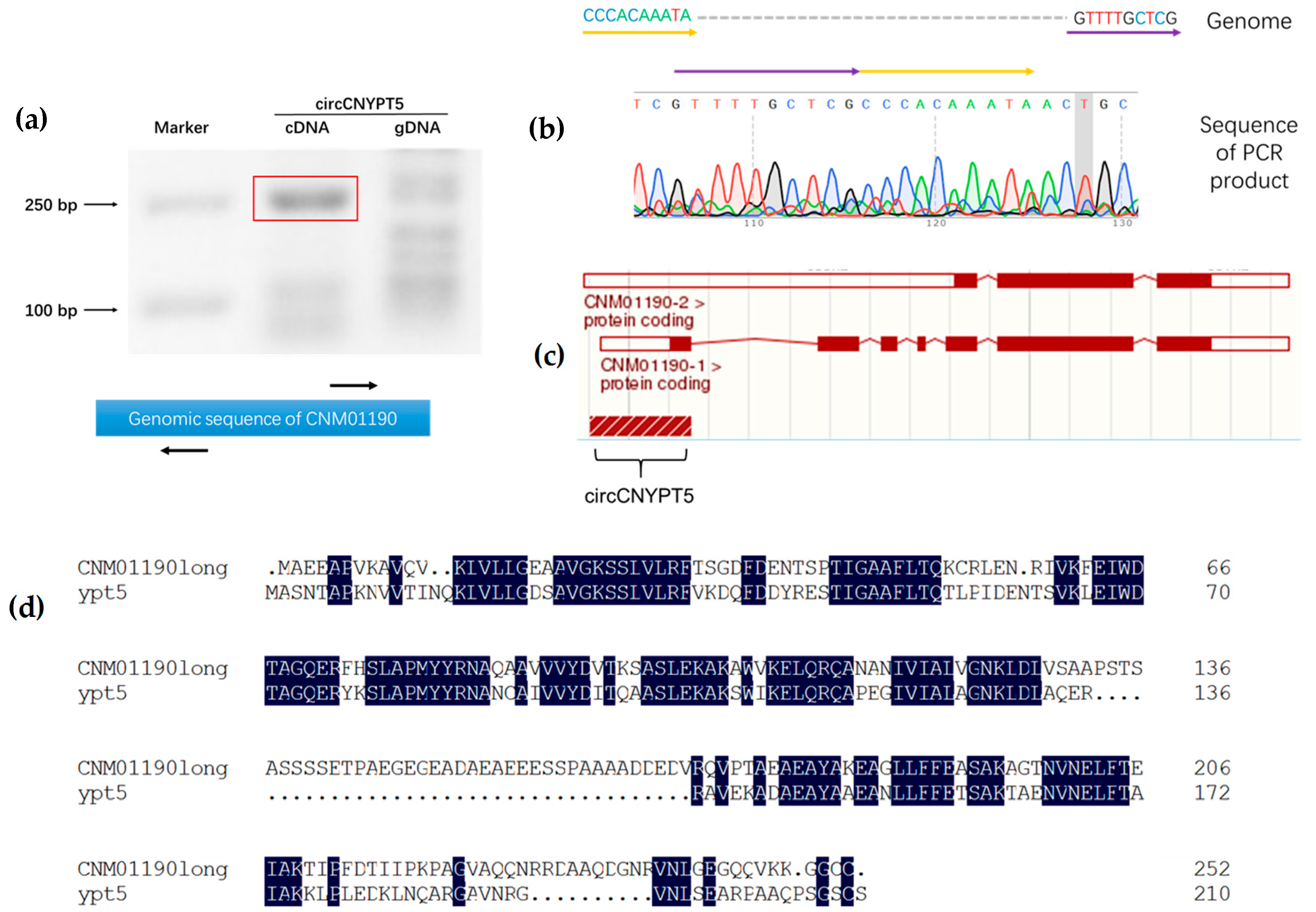
2.5. Validation of Circular RNAs
2.6. Other Online Database and Software
2.7. Construction of DBR1 Gene Overexpression Vector
2.8. Quantitative and Semiquantitative Reverse Transcription Polymerase Chain Reaction
3. Results
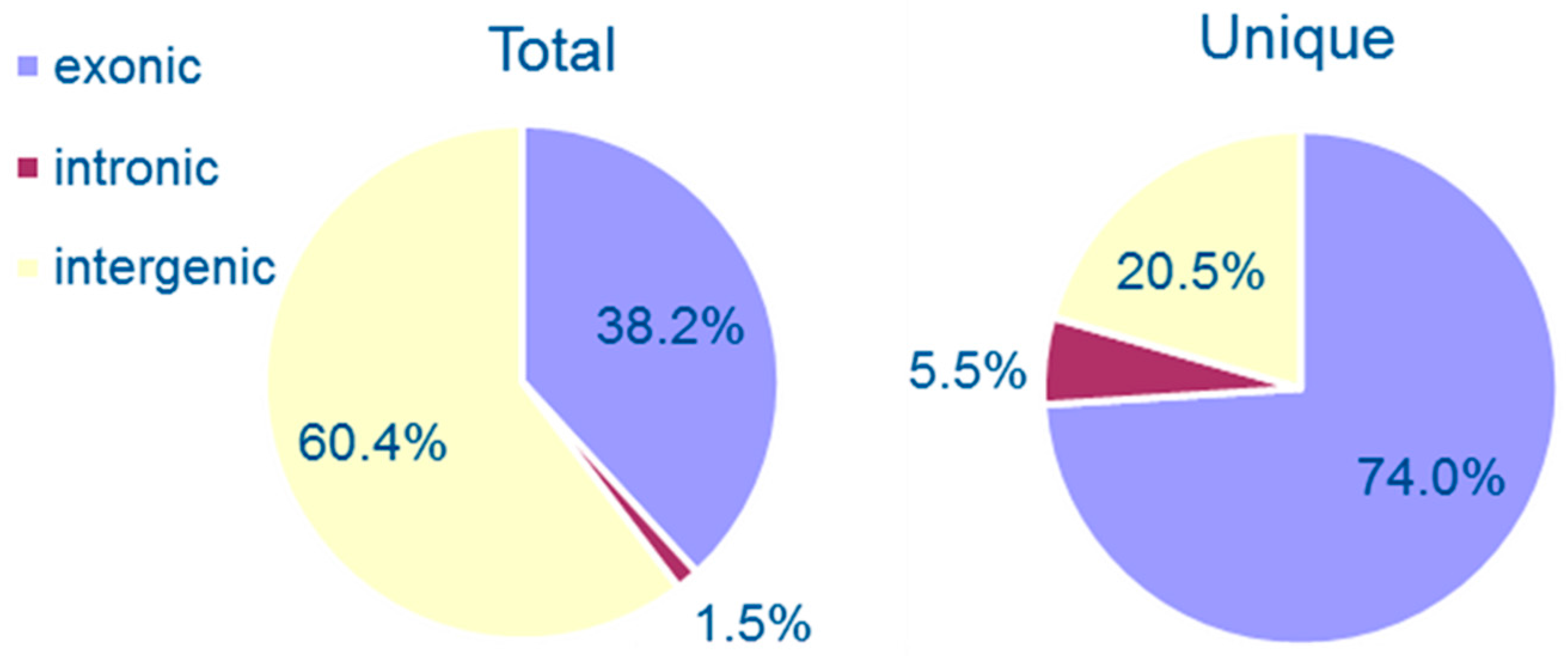
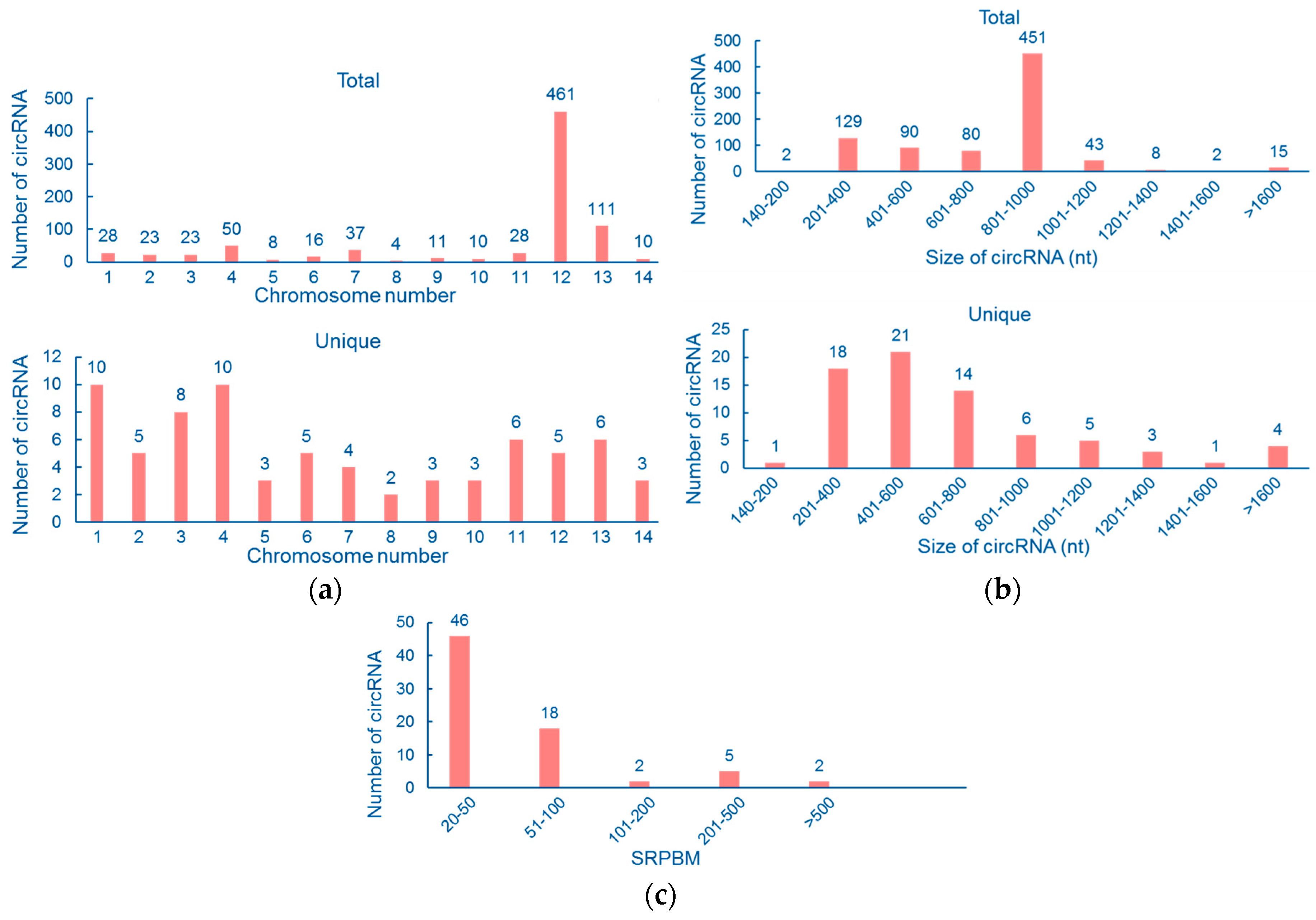
3.1. Genomewide Identification of Circular RNAs
3.2. Properties of Cryptococcal Circular RNAs
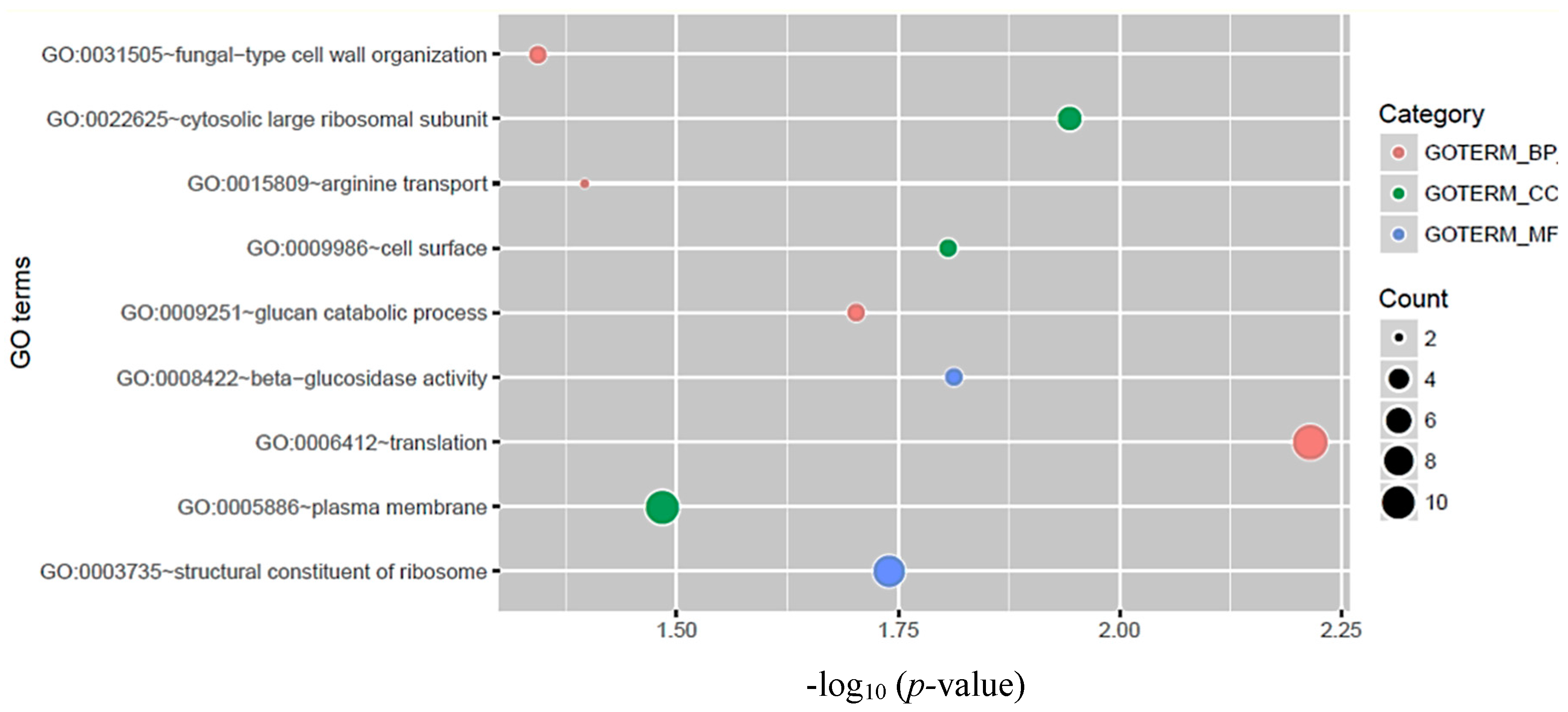
3.3. Functional Analysis of circRNA Host Genes
3.4. Small Guanosine Triphosphatase-Encoding Orthologs Are Conserved circRNA Hosts
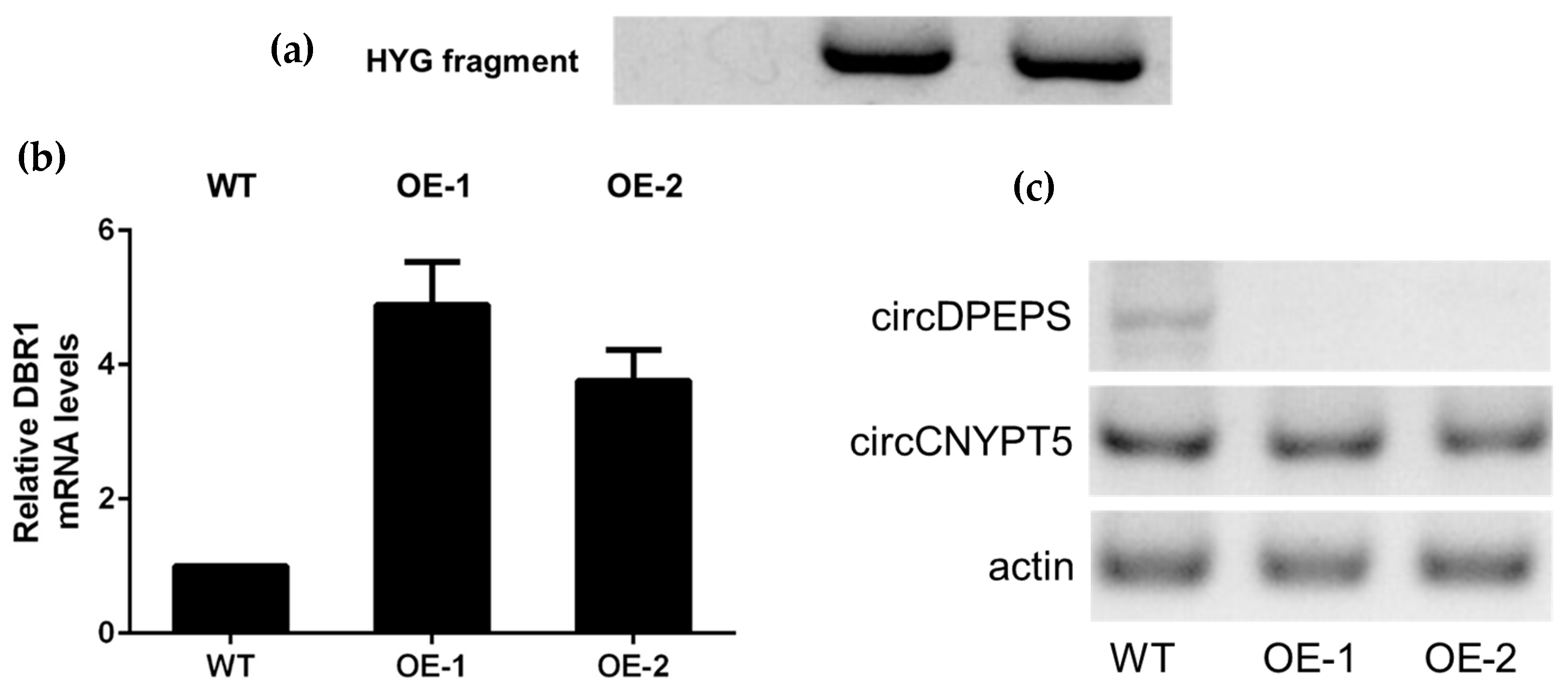
3.5. DBR1 Expression Level Is Negatively Associated with circDPEPS but not circCNYPT5
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Salzman, J. Circular RNA expression: Its potential regulation and function. Trends. Genet. 2016, 32, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Yang, L. Regulation of circRNA biogenesis. RNA Biol. 2015, 12, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Zuo, Y.; Wang, J.; Zhang, M.Q.; Malhotra, A.; Mayeda, A. Characterization of RNase R-digested cellular RNA source that consists of lariat and circular RNAs from pre-mRNA splicing. Nucleic Acids Res. 2006, 34, e63. [Google Scholar] [CrossRef] [PubMed]
- Sanger, H.L.; Klotz, G.; Riesner, D.; Gross, H.J.; Kleinschmidt, A.K. Viroids are single-stranded covalently closed circular RNA molecules existing as highly base-paired rod-like structures. Proc. Natl. Acad. Sci. USA 1976, 73, 3852–3856. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, T.; Wang, X.; He, A. Circles reshaping the RNA world: From waste to treasure. Mol. Cancer 2017, 16, 58. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, J.; Zhao, F. Ciri: An efficient and unbiased algorithm for de novo circular RNA identification. Genome Biol. 2015, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.O.; Wang, H.B.; Zhang, Y.; Lu, X.; Chen, L.L.; Yang, L. Complementary sequence-mediated exon circularization. Cell 2014, 159, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Szabo, L.; Morey, R.; Palpant, N.J.; Wang, P.L.; Afari, N.; Jiang, C.; Parast, M.M.; Murry, C.E.; Laurent, L.C.; Salzman, J. Statistically based splicing detection reveals neural enrichment and tissue-specific induction of circular RNA during human fetal development. Genome Biol. 2015, 16, 126. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Alexandrov, P.N.; Jaber, V.; Lukiw, W.J. Deficiency in the ubiquitin conjugating enzyme UBE2A in Alzheimer’s Disease (AD) is linked to deficits in a natural circular miRNA-7 sponge (circRNA; ciRS-7). Genes 2016, 7, 116. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell Biol. 2016, 17, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Lasda, E.; Parker, R. Circular RNAs: Diversity of form and function. RNA 2014, 20, 1829–1842. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.; Wilusz, J.E. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014, 28, 2233–2247. [Google Scholar] [CrossRef] [PubMed]
- Conn, S.J.; Pillman, K.A.; Toubia, J.; Conn, V.M.; Salmanidis, M.; Phillips, C.A.; Roslan, S.; Schreiber, A.W.; Gregory, P.A.; Goodall, G.J. The RNA binding protein quaking regulates formation of circRNAs. Cell 2015, 160, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Barrett, S.P.; Wang, P.L.; Salzman, J. Circular RNA biosynthesis can proceed through an exon-containing lariat precursor. eLife 2015, 4, e07540. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Yang, B.; Chen, B.J.; Bliim, N.; Ueberham, U.; Arendt, T.; Janitz, M. The emerging role of circular RNAs in transcriptome regulation. Genomics 2017, 109, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Granados-Riveron, J.T.; Aquino-Jarquin, G. The complexity of the translation ability of circRNAs. Biochim. Biophys. Acta 2016, 1859, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- Loftus, B.J.; Fung, E.; Roncaglia, P.; Rowley, D.; Amedeo, P.; Bruno, D.; Vamathevan, J.; Miranda, M.; Anderson, I.J.; Fraser, J.A.; et al. The genome of the basidiomycetous yeast and human pathogen Cryptococcus neoformans. Science 2005, 307, 1321–1324. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.K.; Jain, M. NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zheng, Q.; Bao, C.; Li, S.; Guo, W.; Zhao, J.; Chen, D.; Gu, J.; He, X.; Huang, S. Circular RNA is enriched and stable in exosomes: A promising biomarker for cancer diagnosis. Cell Res. 2015, 25, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOABS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Guo, X.; Wang, W. Identification and characterization of circular RNAs in zebrafish. FEBS Lett. 2017, 591, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Cheng, Y.; Zhang, C.; You, Q.; Shen, X.; Guo, W.; Jiao, Y. Genome-wide identification and characterization of circular RNAs by high throughput sequencing in soybean. Sci. Rep. 2017, 7, 5636. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.L.; Bao, Y.; Yee, M.C.; Barrett, S.P.; Hogan, G.J.; Olsen, M.N.; Dinneny, J.R.; Brown, P.O.; Salzman, J. Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE 2014, 9, e90859. [Google Scholar] [CrossRef] [PubMed]
- Bitton, D.A.; Atkinson, S.R.; Rallis, C.; Smith, G.C.; Ellis, D.A.; Chen, Y.Y.; Malecki, M.; Codlin, S.; Lemay, J.F.; Cotobal, C.; et al. Widespread exon skipping triggers degradation by nuclear RNA surveillance in fission yeast. Genome Res. 2015, 25, 884–896. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wei, D.; Zhu, X.; Pan, J.; Zhang, P.; Huo, L.; Zhu, X. A ‘suicide’ CRISPR-Cas9 system to promote gene deletion and restoration by electroporation in Cryptococcus neoformans. Sci. Rep. 2016, 6, 31145. [Google Scholar] [CrossRef] [PubMed]
- Rahim, K.; Huo, L.; Li, C.; Zhang, P.; Basit, A.; Xiang, B.; Ting, B.; Hao, X.; Zhu, X. Identification of a basidiomycete-specific Vilse-like GTPase activating proteins (GAPs) and its roles in the production of virulence factors in Cryptococcus neoformans. FEMS Yeast Res. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Cui, L.; Zhou, Y.; Zhu, C.; Fan, D.; Gong, H.; Zhao, Q.; Zhou, C.; Zhao, Y.; Lu, D.; et al. Transcriptome-wide investigation of circular RNAs in rice. RNA 2015, 21, 2076–2087. [Google Scholar] [CrossRef] [PubMed]
- You, X.; Vlatkovic, I.; Babic, A.; Will, T.; Epstein, I.; Tushev, G.; Akbalik, G.; Wang, M.; Glock, C.; Quedenau, C.; et al. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat. Neurosci. 2015, 18, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. CircRNA biosynthesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.O.; Chen, T.; Xiang, J.F.; Yin, Q.F.; Xing, Y.H.; Zhu, S.; Yang, L.; Chen, L.L. Circular intronic long noncoding RNAs. Mol. Cell 2013, 51, 792–806. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.Y.; Chen, L.; Liu, C.; Zhu, Q.H.; Fan, L. Widespread noncoding circular RNAs in plants. New Phytol. 2015, 208, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Zhou, Z.; Niu, Y.; Sun, X.; Deng, Z. Identification and functional characterization of tomato circRNAs derived from genes involved in fruit pigment accumulation. Sci. Rep. 2017, 7, 8594. [Google Scholar] [CrossRef] [PubMed]
- Siede, D.; Rapti, K.; Gorska, A.A.; Katus, H.A.; Altmuller, J.; Boeckel, J.N.; Meder, B.; Maack, C.; Volkers, M.; Muller, O.J.; et al. Identification of circular RNAs with host gene-independent expression in human model systems for cardiac differentiation and disease. J. Mol. Cell Cardiol. 2017, 109, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Ballou, E.R.; Selvig, K.; Narloch, J.L.; Nichols, C.B.; Alspaugh, J.A. Two Rac paralogs regulate polarized growth in the human fungal pathogen Cryptococcus neoformans. Fungal Genet. Biol. 2013, 57, 58–75. [Google Scholar] [CrossRef] [PubMed]
- Ballou, E.R.; Nichols, C.B.; Miglia, K.J.; Kozubowski, L.; Alspaugh, J.A. Two CDC42 paralogues modulate Cryptococcus neoformans thermotolerance and morphogenesis under host physiological conditions. Mol. Microbiol. 2010, 75, 763–780. [Google Scholar] [CrossRef] [PubMed]
- Ballou, E.R.; Kozubowski, L.; Nichols, C.B.; Alspaugh, J.A. Ras1 acts through duplicated Cdc42 and Rac proteins to regulate morphogenesis and pathogenesis in the human fungal pathogen Cryptococcus neoformans. PLoS Genet. 2013, 9, e1003687. [Google Scholar] [CrossRef] [PubMed]
- Dumesic, P.A.; Natarajan, P.; Chen, C.; Drinnenberg, I.A.; Schiller, B.J.; Thompson, J.; Moresco, J.J.; Yates, J.R., 3rd; Bartel, D.P.; Madhani, H.D. Stalled spliceosomes are a signal for RNAi-mediated genome defense. Cell 2013, 152, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Raw Reads | Filtered Reads | Raw Base | Filtered Base | Q20 (%) 1 |
|---|---|---|---|---|---|
| Cn JEC21 | 41,703,834 | 39,280,024 | 6.26 Gnt | 6.16 Gnt | 98.52 |
| circRNA ID | Chr | RNA Size | circRNA Start Loci | CircRNA End Loci | Junction Reads 1 |
|---|---|---|---|---|---|
| 12:174494-175325 | 12 | 831 | 174494 | 175325 | 410 |
| 13:359406-359654 | 13 | 248 | 359406 | 359654 | 67 |
| 7:1027082-1027487 | 7 | 405 | 1027082 | 1027487 | 29 |
| 13:603431-604144 | 13 | 713 | 603431 | 604144 | 28 |
| 4:1303545-1304160 | 4 | 615 | 1303545 | 1304160 | 24 |
| 12:174265-175325 | 12 | 1060 | 174265 | 175325 | 23 |
| 12:174461-175325 | 12 | 864 | 174461 | 175325 | 22 |
| 11:62574-63095 | 11 | 521 | 62574 | 63095 | 11 |
| 13:89597-90783 | 13 | 1186 | 89597 | 90783 | 8 |
| 2:661847-663003 | 2 | 1156 | 661847 | 663003 | 7 |
| Pathway ID 1 | Description | Gene Count | p-Value |
|---|---|---|---|
| cne03010 | Ribosome | 6 | 0.023 |
| cne00500 | Starch and sucrose metabolism | 3 | 0.031 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huo, L.; Zhang, P.; Li, C.; Rahim, K.; Hao, X.; Xiang, B.; Zhu, X. Genome-Wide Identification of circRNAs in Pathogenic Basidiomycetous Yeast Cryptococcus neoformans Suggests Conserved circRNA Host Genes over Kingdoms. Genes 2018, 9, 118. https://doi.org/10.3390/genes9030118
Huo L, Zhang P, Li C, Rahim K, Hao X, Xiang B, Zhu X. Genome-Wide Identification of circRNAs in Pathogenic Basidiomycetous Yeast Cryptococcus neoformans Suggests Conserved circRNA Host Genes over Kingdoms. Genes. 2018; 9(3):118. https://doi.org/10.3390/genes9030118
Chicago/Turabian StyleHuo, Liang, Ping Zhang, Chenxi Li, Kashif Rahim, Xiaoran Hao, Biyun Xiang, and Xudong Zhu. 2018. "Genome-Wide Identification of circRNAs in Pathogenic Basidiomycetous Yeast Cryptococcus neoformans Suggests Conserved circRNA Host Genes over Kingdoms" Genes 9, no. 3: 118. https://doi.org/10.3390/genes9030118
APA StyleHuo, L., Zhang, P., Li, C., Rahim, K., Hao, X., Xiang, B., & Zhu, X. (2018). Genome-Wide Identification of circRNAs in Pathogenic Basidiomycetous Yeast Cryptococcus neoformans Suggests Conserved circRNA Host Genes over Kingdoms. Genes, 9(3), 118. https://doi.org/10.3390/genes9030118