De Novo Transcriptome Assembly and Characterization of the Synthesis Genes of Bioactive Constituents in Abelmoschus esculentus (L.) Moench



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA Isolation and Library Construction
2.3. Sequencing, Quality Control, and De Novo Assembly
2.4. Calculation of the Expression Level of the Transcripts and Analysis of Differentially Expressed Genes
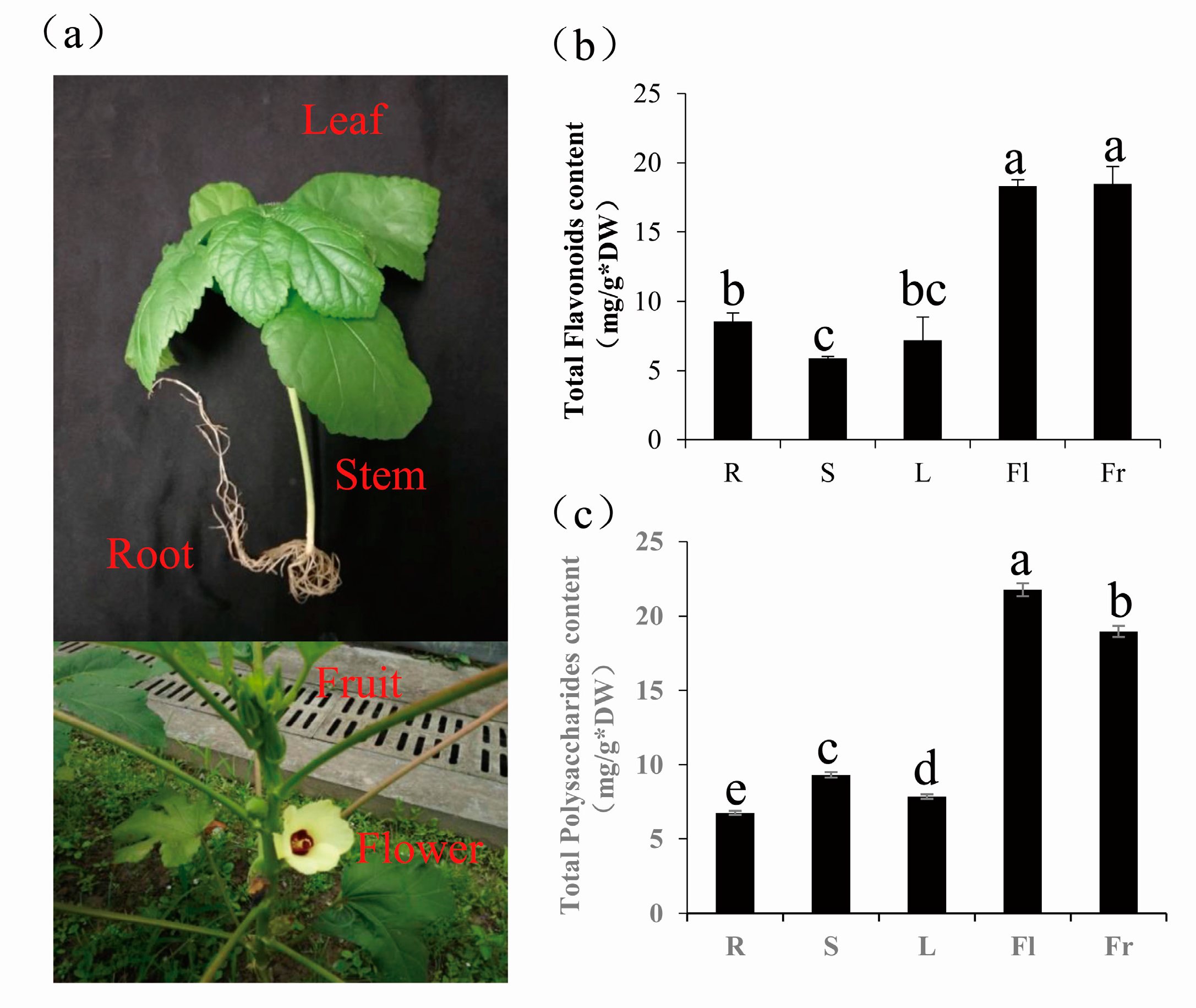
2.5. Determination of Total Flavonoids and Polysaccharides in Okra
3. Results
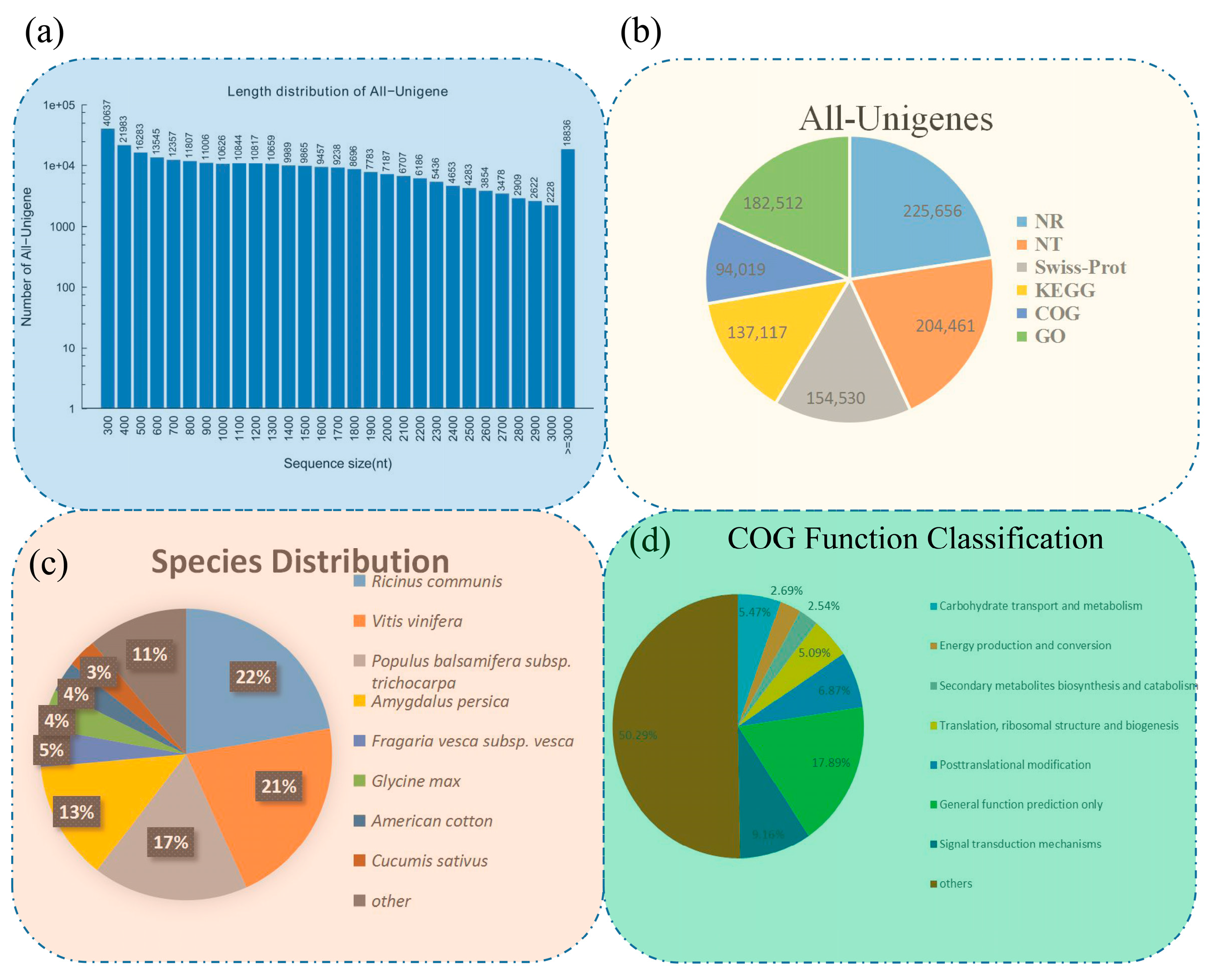
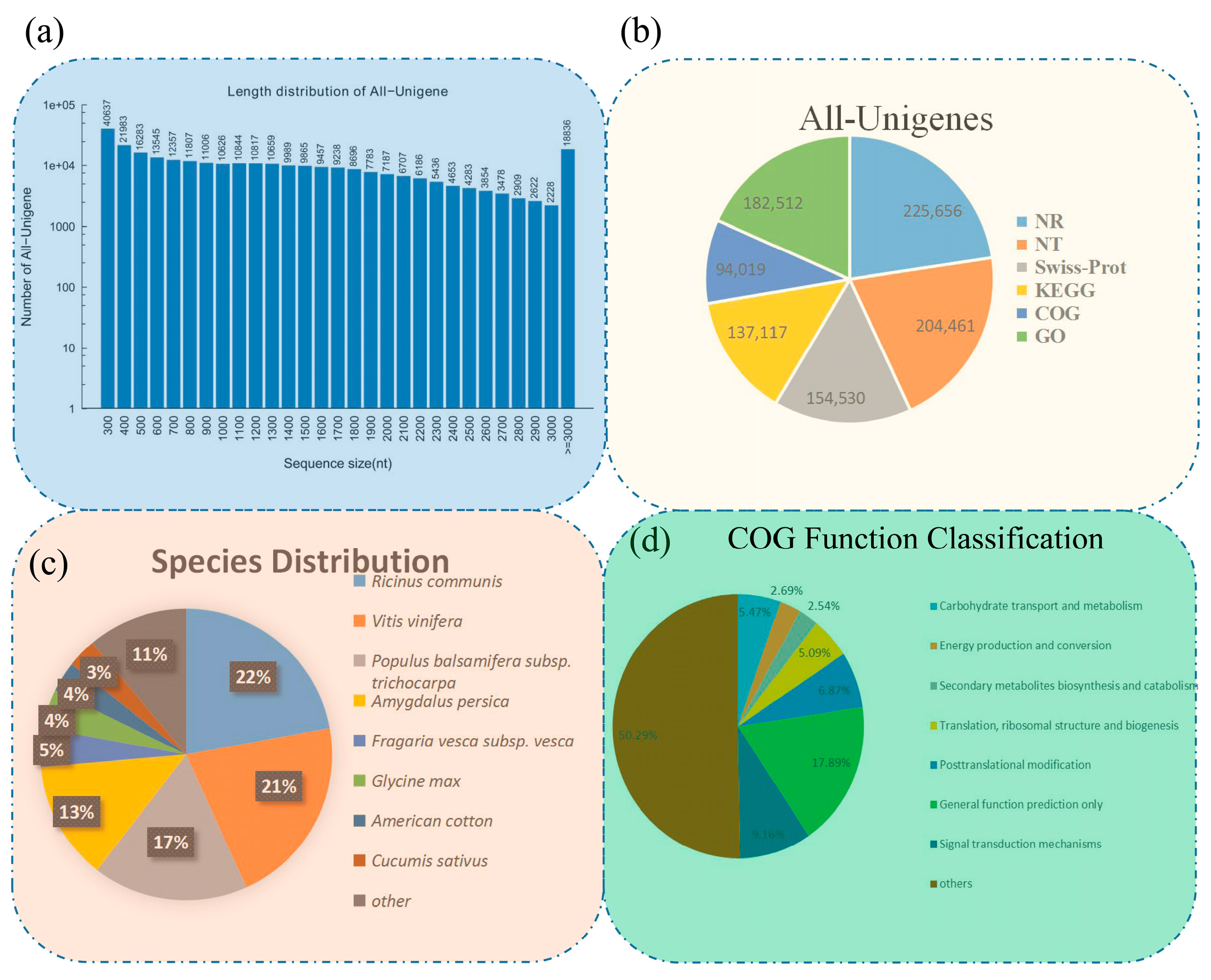
3.1. Results of De Novo Assembly and Unigene Annotation
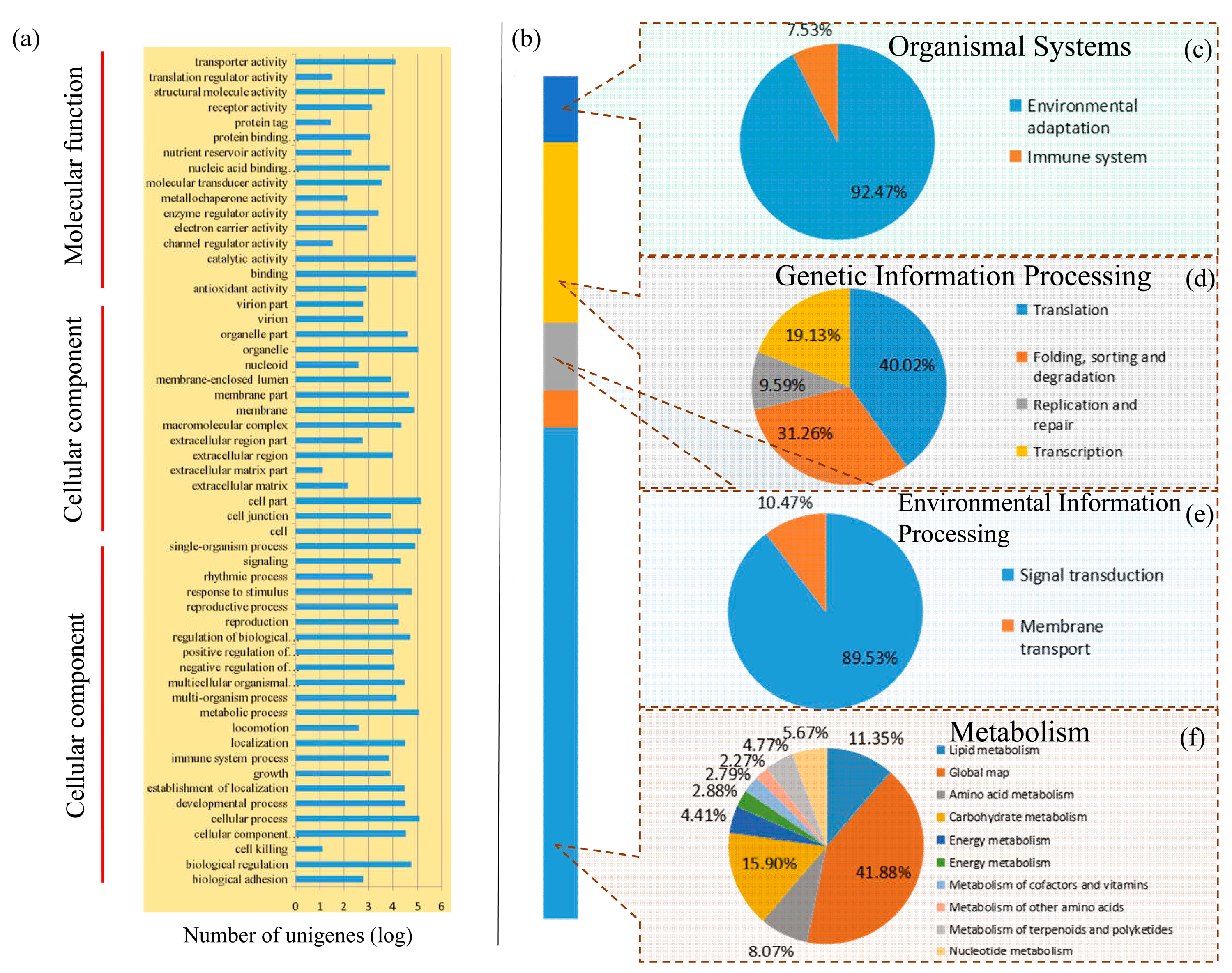
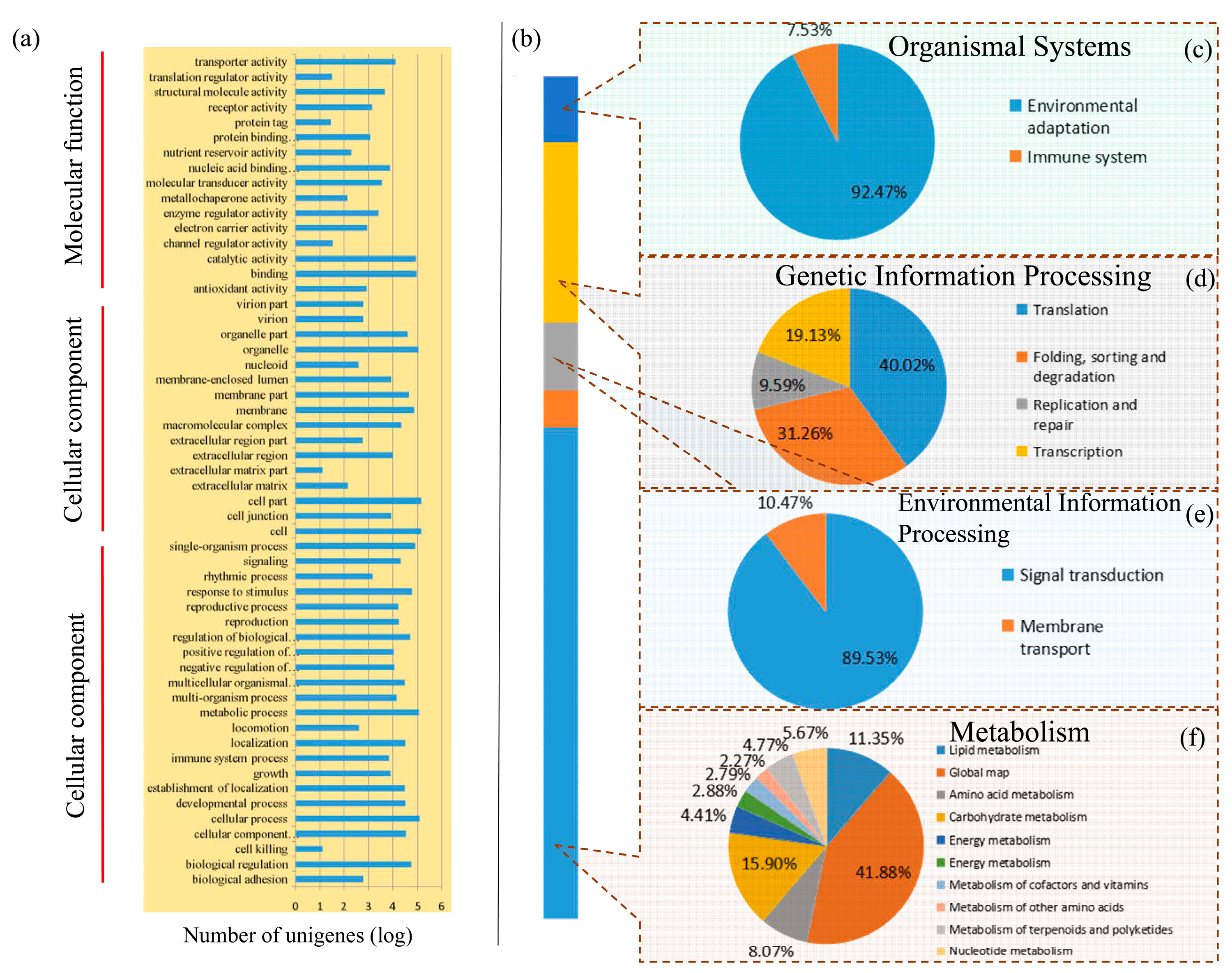
3.2. Classification of Enriched Gene Ontology Terms and KEGG Pathways
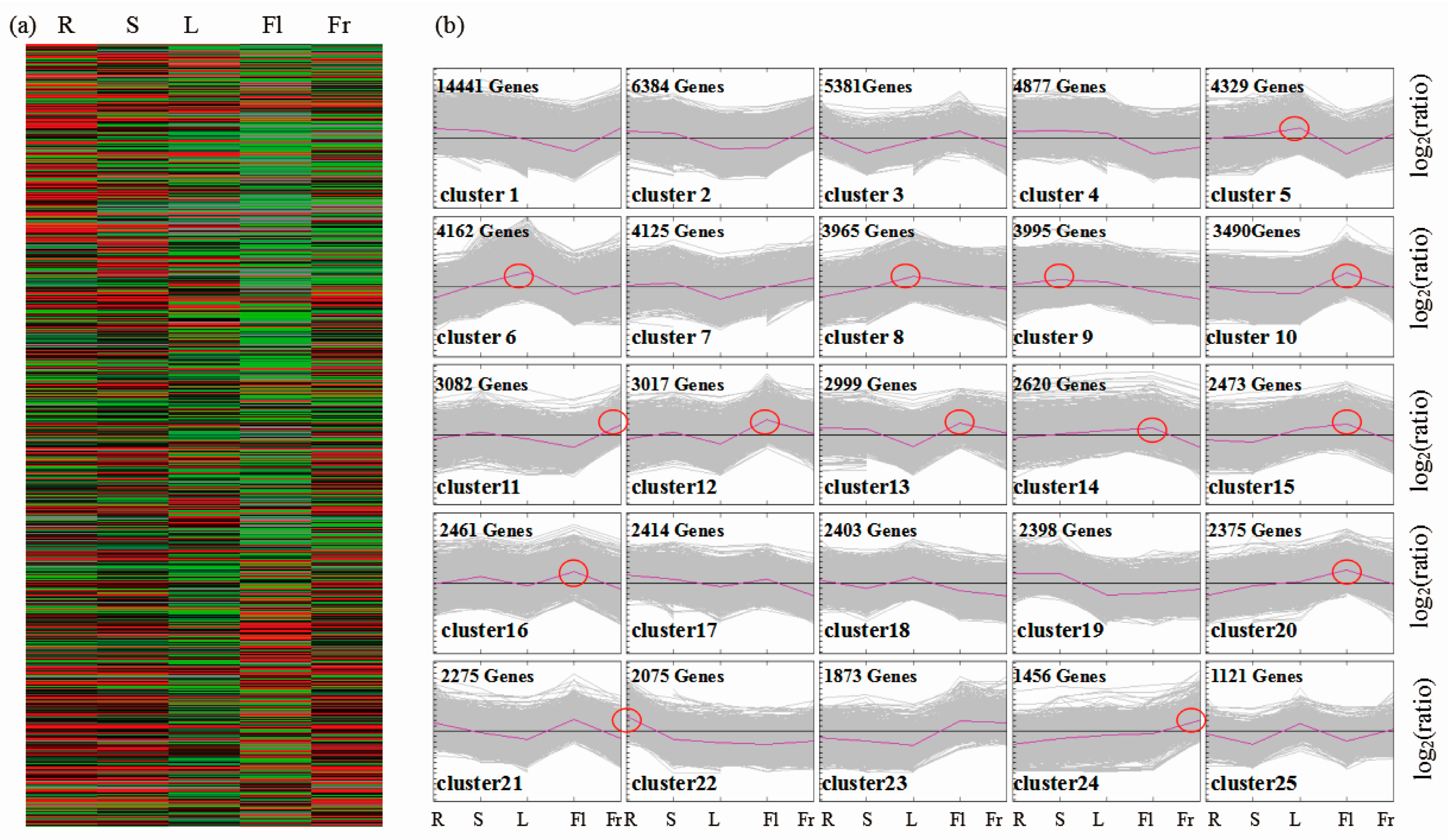
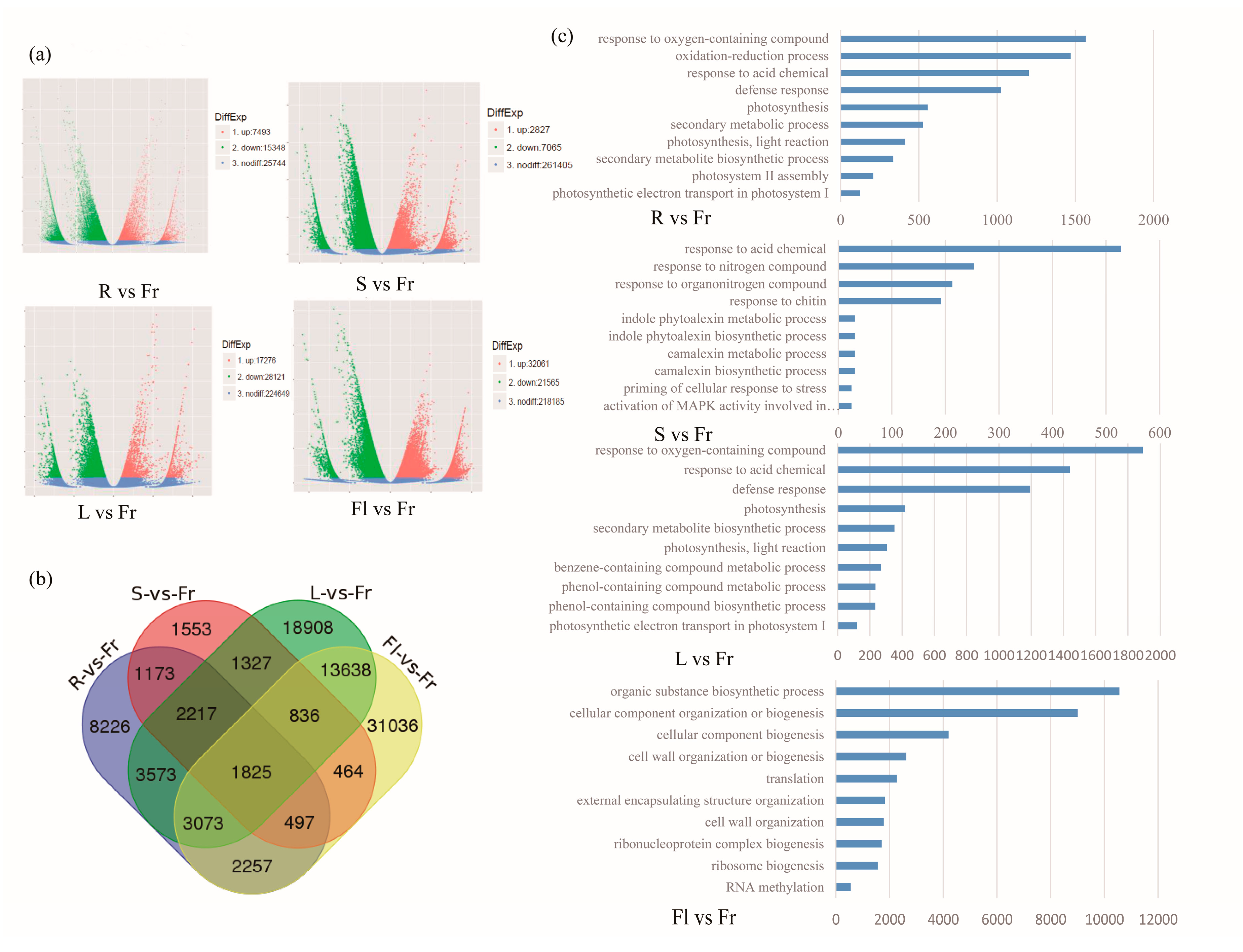
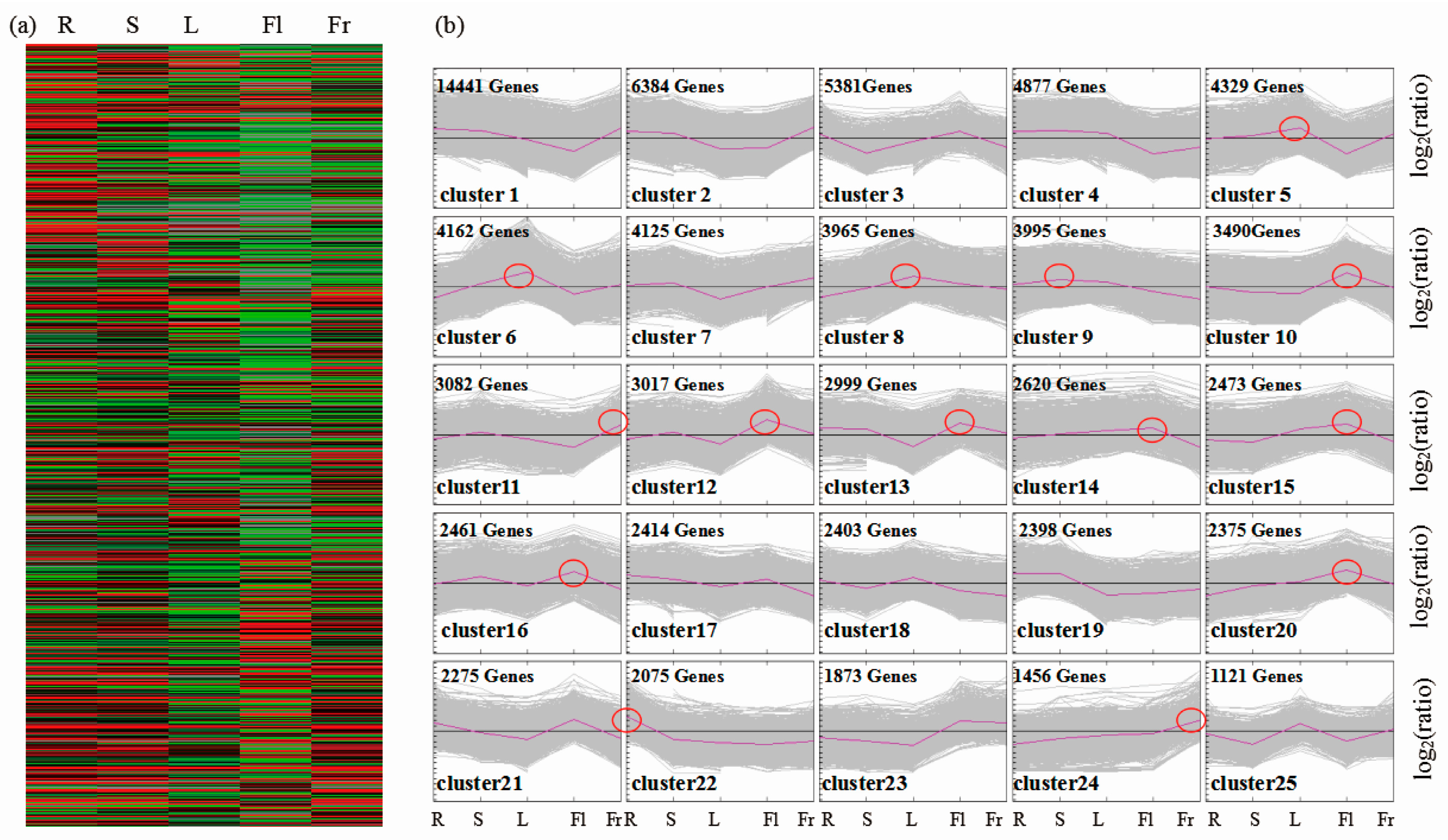
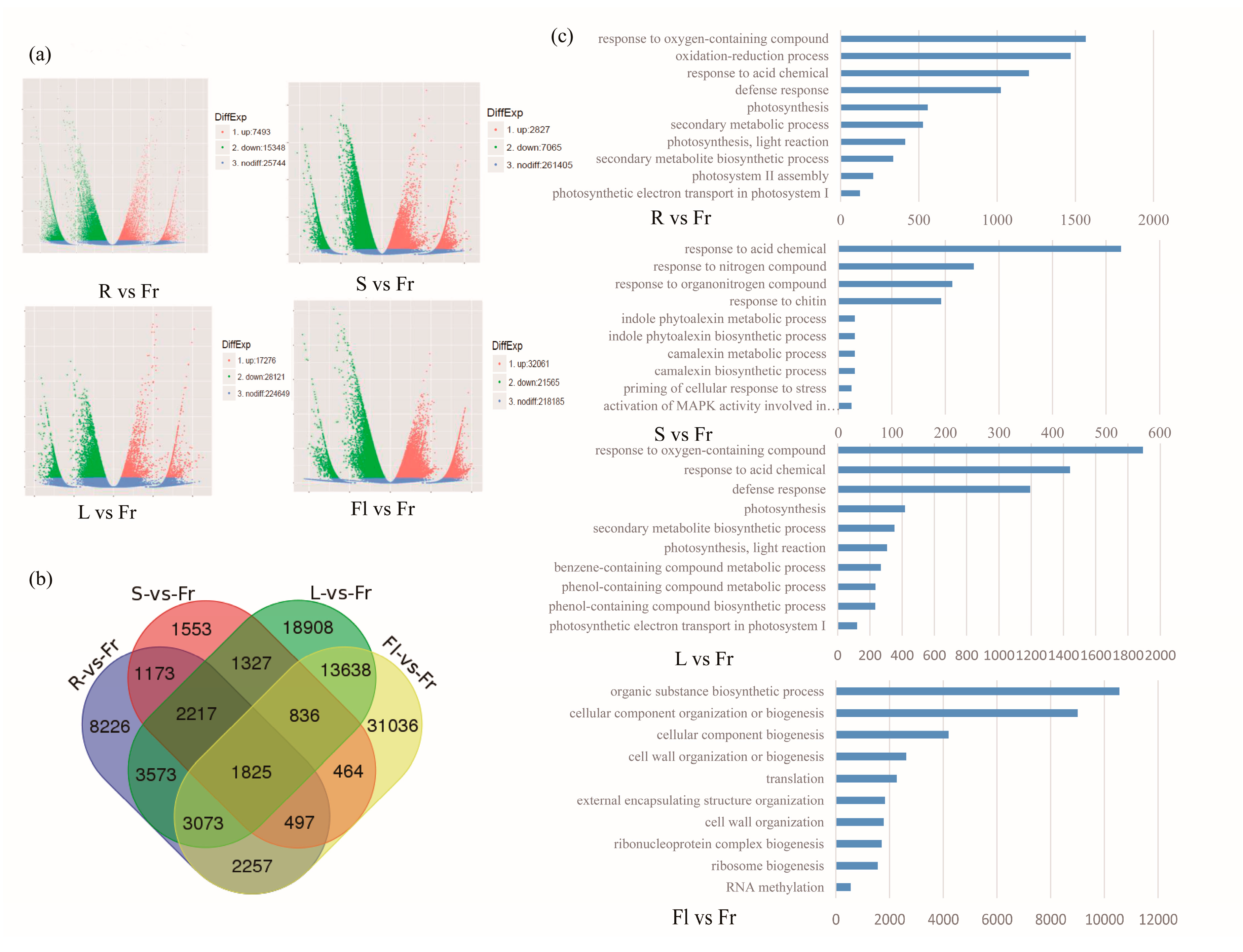
3.3. Differential Gene Expression Analysis and KEGG Pathway Classification among Different Organs
3.4. Differentially Expressed Genes Comparison of Total Flavonoids and Polysaccharides in Five Different Organs of Okra
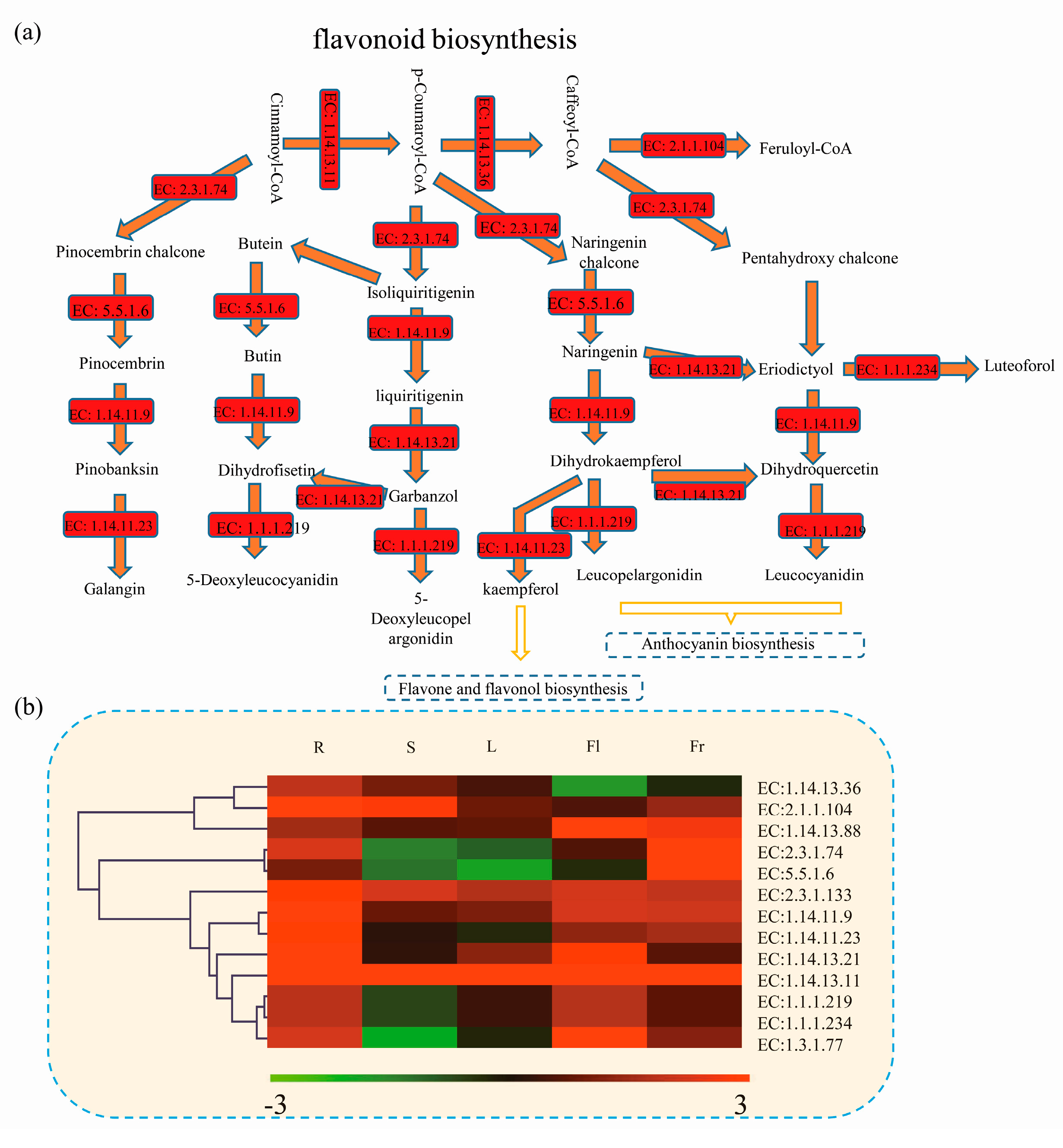
3.5. Differentially Expressed Genes Related to Flavonoid Biosynthetic Pathway in Okra
3.6. Differentially Expressed Genes Related to Fructose and Mannose Metabolism and Glycosyltransferase Genes in Okra
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sami, R.; Jiang, L.Z.; Yang, L.; Ma, Y.; Jing, J. Evaluation of fatty acid and amino acid compositions in okra (Abelmoschus esculentus) grown in different geographical locations. Biomed. Res. Int. 2013, 2013, 574283. [Google Scholar] [CrossRef] [PubMed]
- Savello, P.A.; Martin, F.W.; Hill, J.M. Nutritional composition of okra seed meal. J. Agric. Food. Chem. 1980, 28, 1163–1166. [Google Scholar] [CrossRef] [PubMed]
- Adelakun, O.E.; Oyelade, O.J.; Ade-Omowaye, B.I.; Adeyemi, I.A.; Van de Venter, M. Chemical composition and the antioxidative properties of nigerian okra seed (Abelmoschus esculentus moench) flour. Food. Chem. Toxic. 2009, 47, 1123–1126. [Google Scholar] [CrossRef]
- Liao, H.; Dong, W.; Shi, X.; Liu, H.; Yuan, K. Analysis and comparison of the active components and antioxidant activities of extracts from Abelmoschus esculentus l. Pharmacog. Mag. 2012, 8, 156–161. [Google Scholar]
- Mishra, N.; Kumar, D.; Rizvi, S.I. Protective effect of Abelmoschus esculentus against alloxan-induced diabetes in wistar strain rats. J. Diet. Suppl. 2016, 13, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.H.; Miao, F.T.; Zhang, X.; Wang, Q.H.; Lei, N.; Guo, L.C. Therapeutic effect of okra extract on gestational diabetes mellitus rats induced by streptozotocin. Asian Pac. J. Trop. Med. 2015, 8, 1038–1042. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, C.C.C.R.; Cruz, P.A.; Da Fonseca, M.M.R.; Xavier-Filho, L. Antibacterial properties of the extract of Abelmoschus esculentus. Biotechnol. Bioprocess. Eng. 2011, 16, 971–977. [Google Scholar] [CrossRef]
- Andrews, B. Industrial Gums. Polysaccharides and Their Derivatives, 3rd ed.; Whistler, R.L., Bemiller, J.N., Eds.; Academic Press: New York, NY, USA, 1993; 642 p, ISBN 0-12-746253-8. [Google Scholar]
- Costantino, A.J.; Romanchik-Cerpovicz, J.E. Physical and sensory measures indicate moderate fat replacement in frozen dairy dessert is feasible using okra gum as a milk-fat ingredient substitute. J. Am. Diet. Assoc. 2004, 104, 44. [Google Scholar] [CrossRef]
- Romanchik Cerpovicz, J.E.; Costantino, A.C.; Gunn, L.H. Sensory evaluation ratings and melting characteristics show that okra gum is an acceptable milk-fat ingredient substitute in chocolate frozen dairy dessert. J. Am. Diet. Assoc. 2006, 106, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.M.; Jiang, Y.J.; Hwang, P.Y.; Shen, F.S. Characterization of the okra mucilage by interaction with gal, galnac and glcnac specific lectins. Biochim. Biophys. Acta 1995, 1243, 157–160. [Google Scholar] [CrossRef]
- Lengsfeld, C.; Titgemeyer, F.; Faller, G.; Hensel, A. Glycosylated compounds from okra inhibit adhesion of Helicobacter pylori to human gastric mucosa. J. Agric. Food Chem. 2004, 52, 1495. [Google Scholar] [CrossRef] [PubMed]
- Deters, A.M.; Lengsfeld, C.; Hensel, A. Oligo- and polysaccharides exhibit a structure-dependent bioactivity on human keratinocytes in vitro. J. Ethnopharmacol. 2005, 102, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Zhu, L.Y.; Yang, B.S.; Shi, L.J.; Liu, Y.; Jiang, A.M.; Zhao, L.L.; Song, G.; Liu, T.F. Antitumor and immunomodulating activity of a polysaccharide from Sophora flavescens ait. Int. J. Biol. Macromol. 2012, 51, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Bonin, C.P.; Potter, I.; Vanzin, G.F.; Reiter, W.D. The mur1 gene of Arabidopsis thaliana encodes an isoform of gdp-d-mannose-4,6-dehydratase, catalyzing the first step in the de novo synthesis of GDP-l-fucose. Proc. Natl. Acad. Sci. USA 1997, 94, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Cai, Z.B.; Li, W.; Wang, H.T.; Yan, W.Q.; Zhou, Y.L.; Wang, G.J.; Cui, J.W.; Wang, F. Anti-tumor and immunomodulating activities of a polysaccharide from the root of Sanguisorba officinalis l. Int. J. Biol. Macromol. 2012, 51, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Zhao, T.; Feng, W.; Wang, W.; Zou, Y.; Zheng, D.; Takase, M.; Li, Q.; Wu, H.; Yang, L.; et al. Purification, characterization and immunomodulating activity of a polysaccharide from flowers of Abelmoschus esculentus. Carbohyd. Polym. 2014, 106, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Jiao, H.; Cheng, Y.; Xu, K.; Jia, X.; Shi, Q.; Guo, S.; Wang, M.; Du, L.; Wang, F. In vitro and in vivo immunomodulatory activity of okra (Abelmoschus esculentus l.) polysaccharides. J. Med. Food. 2016, 27, 457–465. [Google Scholar]
- Liao, H.; Liu, H.; Yuan, K. A new flavonol glycoside from the Abelmoschus esculentus linn. Pharmacog. Mag. 2012, 8, 12–15. [Google Scholar]
- Lin, Y.; Lu, M.F.; Liao, H.B.; Li, Y.X.; Han, W.; Yuan, K. Content determination of the flavonoids in the different parts and different species of Abelmoschus esculentus l. By reversed phase-high performance liquid chromatograph and colorimetric method. Pharmacog. Mag. 2014, 10, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Tang, H.T.; Liu, H.Q.; Wang, J.; Zhang, P.; Tang, Y. Simultaneous determination of five flavonoids and specific chromatograms analysis of huangkui capsule. J. Chin. Med. Mater. 2013, 36, 132–136. [Google Scholar]
- Wammanda, D.T.; Kadams, A.M.; Jonah, P.M. Combining ability analysis and heterosis in a diallel cross of okra (Abelmoschus esculentus l. Moench). Afr. J. Agric. Res. 2010, 5, 2108–2115. [Google Scholar]
- Olayiwola, M.O.; Ariyo, O.J. Relative discriminatory ability of gge biplot and ysi in the analysis of genotype×environment interaction in okra (Abelmoschus esculentus). Int. J. Plant. Breed. Genet. 2013, 7, 146–158. [Google Scholar] [CrossRef]
- Islam, M.; Hossain, N.; Alamgir, M.; Kibria, M.G. Comparative effects of biogas plant residues, poultry manure and inorganic fertilizer on growth and yield of ladies finger. Iosrjournals Org. 2014, 7, 2319–2372. [Google Scholar] [CrossRef]
- Mairuae, N.; Connor, J.R.; Lee, S.Y.; Cheepsunthorn, P.; Tongjaroenbuangam, W. The effects of okra (Abelmoschus esculentus linn.) on the cellular events associated with Alzheimer's disease in a stably expressed HFE neuroblastoma SH-SY5Y cell line. Neurosci. Lett. 2015, 603, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Monte, L.G.; Santi-Gadelha, T.; Reis, L.B.; Braganhol, E.; Prietsch, R.F.; Dellagostin, O.A.; RR, E.L.; Gadelha, C.A.; Conceicao, F.R.; Pinto, L.S. Lectin of Abelmoschus esculentus (okra) promotes selective antitumor effects in human breast cancer cells. Biotechnol. Lett. 2014, 36, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Gulsen, O.; Karagul, S.; Abak, K. Diversity and relationships among turkish okra germplasm by srap and phenotypic marker polymorphism. Biologia 2007, 62, 41–45. [Google Scholar] [CrossRef]
- Yuan, C.Y.; Zhang, C.; Wang, P.; Hu, S.; Chang, H.P.; Xiao, W.J.; Lu, X.T.; Jiang, S.B.; Ye, J.Z.; Guo, X.H. Genetic diversity analysis of okra (Abelmoschus esculentus l.) by inter-simple sequence repeat (ISSR) markers. Genet. Mol. Res. 2014, 13, 3165–3175. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef] [PubMed]
- Pertea, G.; Huang, X.; Liang, F.; Antonescu, V.; Sultana, R.; Karamycheva, S.; Lee, Y.; White, J.; Cheung, F.; Parvizi, B. TIGR gene indices clustering tools (TGICL): A software system for fast clustering of large EST datasets. Bioinformatics 2003, 19, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2go: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Fang, L.; Zheng, H.K.; Zhang, Y.; Chen, J.; Zhang, Z.J.; Wang, J.; Li, S.T.; Li, R.Q.; Bolund, L.; et al. Wego: A web tool for plotting go annotations. Nucleic Acids Res. 2006, 34, W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-Seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Saed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: a free, open-source system for microarray data management and analysis. Biotechniques 2003, 34, 374–378. [Google Scholar]
- Shen, C.; Guo, H.; Chen, H.; Shi, Y.; Meng, Y.; Lu, J.; Feng, S.; Wang, H. Identification and analysis of genes associated with the synthesis of bioactive constituents in Dendrobium officinale using RNA-Seq. Sci. Rep. 2017, 7, 187. [Google Scholar] [CrossRef] [PubMed]
- Lao, J.; Oikawa, A.; Bromley, J.R.; Mcinerney, P.; Suttangkakul, A.; Smith-Moritz, A.M.; Plahar, H.; Chiu, T.Y.; Ebert, B. The plant glycosyltransferase clone collection for functional genomics. Plant J. 2014, 79, 517. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.H.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.H.; Chyau, C.C.; Wang, C.J.; Lin, H.T.; Huang, C.N.; Ker, Y.B. Abelmoschus esculentus fractions potently inhibited the pathogenic targets associated with diabetic renal epithelial to mesenchymal transition. Food Funct. 2016, 7, 728–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; He, C.M.; Wu, K.L.; da Silva, J.A.T.; Zeng, S.J.; Zhang, X.H.; Yu, Z.M.; Xia, H.Q.; Duan, J. Transcriptome analysis of Dendrobium officinale and its application to the identification of genes associated with polysaccharide synthesis. Front. Plant Sci. 2016, 7, 5. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Q.; Wang, B.; Hua, W.P.; Niu, J.F.; Dang, K.K.; Qiang, Y.; Wang, Z.Z. De novo assembly and analysis of Polygonatum sibiricum transcriptome and identification of genes involved in polysaccharide biosynthesis. Int. J. Mol. Sci. 2017, 18, 1950. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Jiang, B.; Fang, J.; Shen, Y.; Fang, Z.; Rm, S.K.; Yi, K.; Shen, C.; Yan, D.; Zheng, B. Analysis of transcriptome in hickory (Carya cathayensis), and uncover the dynamics in the hormonal signaling pathway during graft process. BMC Genomics 2016, 17, 935. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.; Xuan, L.; Yiu, S.M.; Lim, B.L. De novo assembly and characterization of Camelina sativa transcriptome by paired-end sequencing. BMC Genomics 2013, 14, 146. [Google Scholar]
- Kumar, D.S.; Tony, D.E.; Kumar, P.; Kumar, K.A.; Rao, D.B.S.; Nadendla, R. A review on: Abelmoschus esculentus (okra). International research journal of pharmaceutical and applied sciences. IRJPAS 2013, 3, 129–132. [Google Scholar] [CrossRef]
- Mathiba, M.T.; Reddy, G.V.S.; Ntushelo, K.; Kanu, S.A. The potential of omics technologies as tools to understand the environmental factors influencing okra (Abelmoschus esculentus) growth and adaptation. S. Afr. J. Plant Soil 2017, 35, 1–8. [Google Scholar] [CrossRef]
- Schafleitner, R.; Kumar, S.; Lin, C.-y.; Hegde, S.G.; Ebert, A. The okra (Abelmoschus esculentus) transcriptome as a source for gene sequence information and molecular markers for diversity analysis. Gene 2013, 517, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Dagnoko, S.; Haougui, A.; Ratnadass, A.; Pasternak, D.; Kouame, C. Okra (Abelmoschus spp.) in west and central africa: Potential and progress on its improvement. Afr. J. Agric. Res. 2010, 5, 3590–3598. [Google Scholar]
- van Eldik, G.J.; Reijnen, W.H.; Ruiter, R.K.; van Herpen, M.M.A.; Schrauwen, J.A.M.; Wullems, G.J. Regulation of flavonol biosynthesis during anther and pistil development, and during pollen tube growth in Solanum tuberosum. Plant J. 1997, 11, 105–113. [Google Scholar]
- Hemleben, V.; Dressel, A.; Epping, B.; Lukacin, R.; Martens, S.; Austin, M.B. Characterization and structural features of a chalcone synthase mutation in a white-flowering line of Matthiola incana r. Br. (brassicaceae). Plant Mol. Biol. 2004, 55, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Petroni, K.; Tonelli, C. Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 2011, 181, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Aharoni, A.; Vos, C.H.R.D.; Wein, M.; Sun, Z.; Greco, R.; Kroon, A.; Mol, J.N.M.; O’Connell, A.P. The strawberry famyb1 transcription factor suppresses anthocyanin and flavonol accumulation in transgenic tobacco. Plant J. 2001, 28, 319. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.T.B.; Tanner, M.E. Mechanism and active site residues of GDP-fucose synthase. J. Am. Chem. Soc. 2008, 130, 17593–17602. [Google Scholar] [CrossRef] [PubMed]
- Murata, T. Purification and some properties of phosphomannomutase from corms of Amorphophallus-konjac c-koch. Plant Cell Physiol. 1976, 17, 1099–1109. [Google Scholar]
- Badejo, A.A.; Eltelib, H.A.; Fukunaga, K.; Fujikawa, Y.; Esaka, M. Increase in ascorbate content of transgenic tobacco plants overexpressing the acerola (Malpighia glabra) phosphomannomutase gene. Plant Cell Physiol. 2009, 50, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.Q.; Yu, C.M.; Qin, H.J.; Liu, X.; Zhang, A.M.; Johansen, I.E.; Wang, D.W. Molecular and functional analysis of phosphomannomutase (PMM) from higher plants and genetic evidence for the involvement of pmm in ascorbic acid biosynthesis in Arabidopsis and Nicotiana benthamiana. Plant J. 2007, 49, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Hoeberichts, F.A.; Vaeck, E.; Kiddle, G.; Coppens, E.; van de Cotte, B.; Adamantidis, A.; Ormenese, S.; Foyer, C.H.; Zabeau, M.; Inze, D.; et al. A temperature-sensitive mutation in the Arabidopsis thaliana phosphomannomutase gene disrupts protein glycosylation and triggers cell death. J. Biol. Chem. 2008, 283, 5708–5718. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.C.; Zhang, C.; Chen, H.Q.; Yang, Q.; Zhou, X.; Gu, Z.N.; Zhang, H.; Chen, W.; Chen, Y.Q. Biochemical characterization of an isoform of gdp-d-mannose-4,6-dehydratase from Mortierella alpina. Biotechnol Lett. 2016, 38, 1761–1768. [Google Scholar] [CrossRef] [PubMed]
- Rayon, C.; Cabanes-Macheteau, M.; Loutelier-Bourhis, C.; Salliot-Maire, I.; Lemoine, J.; Reiter, W.D.; Lerouge, P.; Faye, L. Characterization of n-glycans from Arabidopsis. Application to a fucose-deficient mutant. Plant Physiol. 1999, 119, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Li, Y.; Lim, E.K.; Bowles, D.J. Higher plant glycosyltransferases. Genome Biol. 2001, 2, REVIEWS3004. [Google Scholar] [CrossRef] [PubMed]
- Keegstra, K.; Raikhel, N. Plant glycosyltransferases. Curr. Opin. Plant. Biol. 2001, 4, 219–224. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Total Raw Reads | Total Clean Reads | Total Clean Nucleotides (nt) | Q20 Percentage | Q30 Percentage | N Percentage | GC Percentage |
|---|---|---|---|---|---|---|---|
| Root1 | 48,257,634 | 46,916,458 | 7,037,468,700 | 97.80% | 94.49% | 0.01% | 44.72% |
| Root2 | 48,419,454 | 47,097,672 | 7,064,650,800 | 97.92% | 94.74% | 0.01% | 44.86% |
| Root3 | 47,422,632 | 46,112,238 | 6,916,835,700 | 97.79% | 94.47% | 0.00% | 44.82% |
| Stem1 | 48,647,618 | 47,316,422 | 7,097,463,300 | 97.88% | 94.62% | 0.01% | 44.91% |
| Stem2 | 48,693,636 | 47,323,464 | 7,098,519,600 | 97.94% | 94.75% | 0.01% | 44.96% |
| Stem3 | 44,275,350 | 43,132,910 | 6,469,936,500 | 97.21% | 93.29% | 0.00% | 44.61% |
| Leaf1 | 48,625,474 | 47,341,802 | 7,101,270,300 | 98.00% | 94.84% | 0.01% | 45.61% |
| Leaf2 | 47,424,186 | 46,142,720 | 6,921,408,000 | 97.85% | 94.52% | 0.00% | 45.95% |
| Leaf3 | 50,618,708 | 49,272,238 | 7,390,835,700 | 97.84% | 94.49% | 0.00% | 45.78% |
| Flower1 | 44,078,602 | 42,777,662 | 6,416,649,300 | 97.89% | 94.64% | 0.00% | 45.22% |
| Flower2 | 47,743,650 | 45,883,906 | 6,882,585,900 | 97.85% | 94.51% | 0.00% | 45.90% |
| Flower3 | 43,314,640 | 41,960,012 | 6,294,001,800 | 97.85% | 94.52% | 0.00% | 45.59% |
| Fruit1 | 51,485,706 | 49,905,802 | 7,485,870,300 | 97.45% | 93.84% | 0.00% | 45.26% |
| Fruit2 | 49,436,520 | 48,083,880 | 7,212,582,000 | 98.01% | 94.90% | 0.01% | 44.67% |
| Fruit3 | 47,886,442 | 46,568,924 | 6,985,338,600 | 97.92% | 94.70% | 0.00% | 44.83% |
| All | 716,330,252 | 695,836,110 | 104,375 Gb |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, C.; Dong, W.; Gen, W.; Xu, B.; Shen, C.; Yu, C. De Novo Transcriptome Assembly and Characterization of the Synthesis Genes of Bioactive Constituents in Abelmoschus esculentus (L.) Moench. Genes 2018, 9, 130. https://doi.org/10.3390/genes9030130
Zhang C, Dong W, Gen W, Xu B, Shen C, Yu C. De Novo Transcriptome Assembly and Characterization of the Synthesis Genes of Bioactive Constituents in Abelmoschus esculentus (L.) Moench. Genes. 2018; 9(3):130. https://doi.org/10.3390/genes9030130
Chicago/Turabian StyleZhang, Chenghao, Wenqi Dong, Wei Gen, Baoyu Xu, Chenjia Shen, and Chenliang Yu. 2018. "De Novo Transcriptome Assembly and Characterization of the Synthesis Genes of Bioactive Constituents in Abelmoschus esculentus (L.) Moench" Genes 9, no. 3: 130. https://doi.org/10.3390/genes9030130