Abstract
The demographic history of anatomically modern humans (AMH) involves multiple migration events, population extinctions and genetic adaptations. As genome-wide data from complete genome sequencing becomes increasingly abundant and available even from extinct hominins, new insights of the evolutionary history of our species are discovered. It is currently known that AMH interbred with archaic hominins once they left the African continent. Modern non-African human genomes carry fragments of archaic origin. This review focuses on the fitness consequences of archaic interbreeding in current human populations. We discuss new insights and challenges that researchers face when interpreting the potential impact of introgression on fitness and testing hypotheses about the role of selection within the context of health and disease.
1. Widespread Interbreeding between Hominins
The demographic history of anatomically modern humans (AMH) is complex, and involves a large number of migrations, genetic admixtures and introgressions, population extinctions and genetic adaptations, which overlap both in time and in space (see Figure 1). Due to this complexity, the evolutionary history of humankind is still far from being fully understood [1]. During the last 30 years, the most accepted demographic scenario for explaining recent evolution of AMH has been the Out of Africa model (OOA). According to this model, AMH evolved in Africa around 100–200 thousand years ago (kya) in East Africa [2,3] and migrated to the rest of the world around 50–60 kya [4,5,6,7]. This widely accepted dating of Homo sapiens emergence was recently challenged by Hublin et al. [8], who found AMH fossils of 300 kya at Jebel Irhoud in Morocco [9]. Similarly, Herschkovitz et al. [10] described a H. sapiens maxilla of 177 to 194 kya in Misliya Cave, Israel. All these studies suggest that the members of the H. sapiens clade left Africa earlier than previously thought, probably in several waves of OOA migration at different stages of evolution. Classical “pure” OOA assumes that admixture with other archaic populations such as Neanderthals or Denisovans, present at the time of the rise of AMH, either did not occur or was negligible [11].
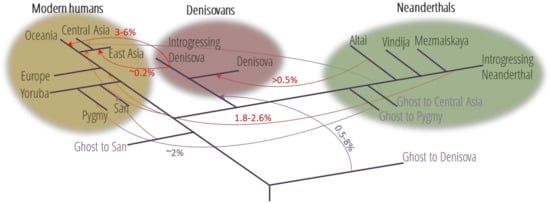
Figure 1.
Family tree of the four groups of early humans living in Eurasia 50,000 years ago and the inferred gene flow between the groups due to interbreeding (based on [12,13,14,15,16]). The direction and estimated magnitude of inferred gene flow events are shown. Branch lengths and timing of gene flows are not scaled. Light violet color indicates introgression events from unknown archaic populations (Ghost).
However, genomic studies of ancient DNA have revealed that AMH interbred with other hominid lineages, such as Neanderthals and Denisovans, present in Eurasia since 300 kya up to 30–50 kya. The admixture with Neanderthals occurred around 37–86 kya, and most likely between 47 and 65 kya [17,18,19,20]. The event of admixture with Denisovan took place within similar time span, ~44–54 kya [21]. Whole-genome sequences from ancient specimens [22] have revealed that Non-African populations outside Oceania carry between 1.8% and 2.6% of Neanderthal DNA (Figure 1) [13]. As described in [18], East Asians carry somewhat more Neanderthal DNA (2.3% to 2.6%) than people in Western Eurasia (1.8% to 2.4%). In contrast, DNA introgressed into modern humans from Denisovans is found mostly in Australo-Melanesians, which may account for up to 6% of Denisovan DNA in their genomes and, to a lesser extent, in South Asians [23] and East Asians [24]. These estimates are averages across the modern human genome. However, specific regions of the genome may have degrees of Neanderthal ancestry as high as 64% in Europeans and 62% in Asians.
New studies based on current genetic diversity are suggesting that the events of archaic introgression in AMH did occur after out of Africa migration with other hidden ”ghost” archaic populations [14]. The main difficulty for the inference of archaic introgression in African modern human genomes from African archaic populations is mainly due to the current absence of genetic material from the remains of archaic hominins that could be used as a proxy for studying the source of introgression, as the climate of African continent is not favorable for DNA preservation. Nevertheless, there is growing evidence that archaic introgression occurred also within this continent [15,25,26,27,28,29], raising the exciting possibility that other unknown archaic groups may have contributed to human genetic diversity. Therefore, recent work suggests that apparently distinct species can exchange the genetic material along their evolutionary history [30]. The biological implications of such introgression, including their consequences on modern human health, is reviewed in the following sections.
2. Selection against Introgressed Regions at the Level of Genomes and Individual Loci
Introgressed alleles in a foreign genetic background frequently have negative fitness effects regardless of the amount of adaptive introgression. Martin and Jiggins [31] made two important considerations when dealing with models of selection against introgressed genomic tracts. First, as many of the factors that influence selection—such as recombination rate and gene density—are interdependent, the models that account for combined effects of both factors are more feasible, especially if they incorporate specific predictions such as the decline in selective sweep strength with increasing distance from selected loci. Juric et al. [32] modelled the level of Neanderthal ancestry in human populations as a function of the recombination distance from nearby selected alleles and estimated both the density of selected loci and the strength of selection. Second, interpretations of the landscape of ancient introgression into human species may vary depending on underlining assumptions. For example, the majority of models assume that introgressed blocks are selected independently of each other in the genetic background of the recipient population. However, Harris and Nielsen [33] showed that much of the selection against introgression may occur in early generations, since early generation hybrids should have complex ancestries in which epistasis can lead to non-additive fitness effects. Another assumption pointed out in this study is that weakly deleterious mutations segregating in the donor population would be the main driver of selection against Neanderthal introgression in humans. Under such a model, the lower effective population size in Neanderthals would have led to the accumulation of weakly deleterious alleles that, once introgressed into humans, would reduce the relative fitness of the hybrid. However, in such context, even if both species bear recessive deleterious alleles but at differing sites, hybrids might have enhanced overdominant fitness variation regardless these deleterious recessives, which leads to the conclusion that Neanderthal introgression may have initially been favored by selection in humans [33].
Disproportionate roles for sex chromosomes in species differences and hybrid incompatibility constitutes a consistent pattern in speciation [34,35]. The compelling evidence of these processes has been reported in the genomes of non-African humans, which have sequences devoid of introgressed variation (“deserts”) from Neanderthals and Denisovans, possibly driven by selection against introgression described by Sankararaman et al. [21,36]. Furthermore, the authors indicated that the introgression deserts of Neanderthal and Denisovan DNA in modern humans are largely overlapping. Of particular interest is a significant reduction in admixture associated with genes showing testes-specific expression, suggesting that admixture may have led to reduced male fertility and supporting evidence of reduced introgression on sex chromosomes [21,36,37]. However, this genomic evidence must be interpreted with caution [38]. When selection against introgression occurs at a large number of loci throughout the genome, its combined effects on many loci can leave detectable patterns, even though selection on any individual locus may be weak [31]. Moreover, weaker signals of introgression have been observed in parts of the genome with high gene density and/or low recombination [21], agreeing with theoretical work, which predicted that the strength of selection against introgression depends on the density of selected sites and the recombination rate [39].
Evidence for the role of purifying selection in shaping the introgression landscape comes from particular categories of genes experiencing different amounts of introgression as previously demonstrated for non-human species [40,41,42,43]. This is also true for the autosomal regions deficient in both Neanderthal and Denisovan ancestries, which contain a significant enrichment of genes transcribed in meiotic germ cells [44,45]. The phenotypic traits affected by archaic introgression are summarized in Figure 2, and their corresponding genomic regions with the type of selective regime acting on them are listed in the Table S1. Taking into account that there has been strong selection against archaic introgression among protein-coding genes [21,46,47], functional regions contributing to the uniqueness of some modern human traits could be identified if they are strongly depleted of archaic ancestry [48]. For example, no Neanderthal ancestry has been detected around the forkhead box protein P2 (FOXP2) gene [21], mutations of which are associated with language disorders [49]. Similarly, Neanderthals and Denisovans carry a single copy of AMY1 gene, encoding an amylase enzyme responsible for starch digestion [21]. In contrast, AMH carry multiple copies of the gene [50,51] and there is no evidence of Neanderthal introgression [21]. This has been interpreted as an evidence that the production of larger amounts of salivary amylase for starch digestion has been under positive selection in modern humans compared to archaic species [52]. Moreover, regions depleted of both Neanderthal and Denisova ancestry are enriched for genes expressed in specific brain regions (e.g., the ventral frontal cortex-ventrolateral prefrontal cortex in infants and the striatum in adulthood; [48]). Another genomic study of Chintalapati et al. [53] on small indels introgressed from Neanderthal demonstrated that negative selection affected these variants more than other variants segregating in modern humans and confirmed that deletions evolved under more constraint than insertions, the vast majority of them laying in the intronic regions. Besides, introgressed variants that may influence on the phenotype of their carriers were identified (Table S1). Among them, an introgressed deletion associated with a decrease in the time to menarche may constitute an example of a former Neanderthal-specific trait contributing to modern human phenotypic diversity [53] (Table S1).
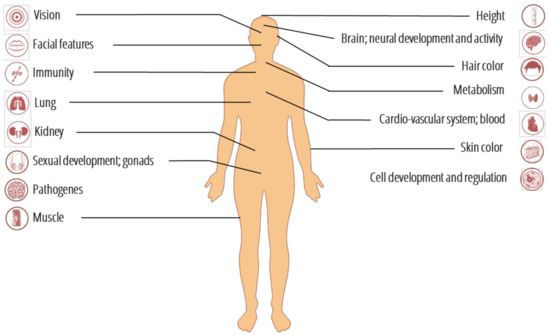
Figure 2.
The modern human organs and systems affected by introgressed variants from ancient genomes (see Table S1 for details).
Further evidence of the deleterious effect of Neanderthal introgression can be identified at the expression level. Analysis of gene expression of Neanderthal alleles in current individuals shows a significant downregulation in the testes and brain compared to other tissues [54,55].
3. Genomic Signatures of Adaptive Introgression from Archaic to Modern Humans
The footprint of purifying selection against archaic alleles is widespread in the human genome. Nevertheless, given that archaic species evolved for long times in environments for which early AMH were not biologically adapted, interbreeding between anatomically modern humans with archaic species could have facilitated adaptation to specific environments [56,57] (see Table S1). This evolutionary process could bring variants at a higher frequency than de novo mutations, providing linked blocks of sequence with multiple functional mutations, potentially including co-adapted alleles [58]. This process, known as adaptive introgression, has risen to prominence based on a series of high profile examples in human genomes [56,59,60] (see Figure 2). For example, genes involved in functions related to keratin filaments, sugar metabolism, muscle contraction, body fat distribution, enamel thickness, oocyte meiosis, brain size and functioning have been targeted by adaptive introgression from Neanderthals in different non-African genomes [29,36,56,61,62,63,64,65]. Genes involved in the variation of skin pigmentation and hair morphology (BNC2, MC1R) also show the signature of positive selection as the result of adaptation to diverse habitats with different degree of insolation (Table S1) [54,66,67]. Advantageous immune variants introduced into the modern human population from archaic genomes have substantially contributed in the present-day diversity of immune genes [56,68,69,70,71,72]. Since innate immunity genes have evolved under stronger purifying selection than the rest of the genome [73], this enrichment of introgressed alleles suggests the presence of strong positive selection at the immune system. A broader overview on ancient pathogens transmitted into modern human genomes through sexual interactions with archaic hominins and their impact on AMH immunity evolution can be found in the review of Pimenoff et al. [74] published in this Special Issue on Evolutionary Medicine. Similarly to innate immunity genes, EGLN1 and EPAS1 genes, associated with hemoglobin concentration and response to hypoxia, display a high degree of Denisovan ancestry in Tibetans, suggesting that this population acquired advantageous alleles for high altitude life through ancient admixture [75,76,77]. In contrast to these evidences of positive selection, evidence for balancing selection in humans is largely circumstantial [78]. However, host defense genes such as those encoding several membrane glycoproteins, the KIR regions that coevolve with HLA ligands, and other genes encoding proteins involved in cell migration or innate immunity, apparently are subject to this otherwise rare selective regime [79,80,81,82,83]. The HLA region, a paradigm of balancing selection in humans, harbors functional variants that were probably introgressed from Neanderthals and Denisovans [84]. An alternative explanation by Yasukochi and Ohashi [85] based on phylogenetic analysis does not support the introgression hypothesis and concludes that it is highly likely that the supposedly introgressed allelic lineage HLA-B*73 has been maintained in the direct ancestors of modern humans [85].
Increasing evidence suggests that regulatory variants play a central role in adaptive processes [86,87,88]. A compelling example of local adaptation detected on the expression level is at the apelin receptor gene APLNR. Apelin is a signal peptide that influences several aspects of cardiac, digestive, brain, and vascular function, including regulation of oxygen levels. This gene exhibits strong allele-specific expression favoring the Neanderthal allele in brain tissues, but allele-specific expression favoring the modern human allele in non-brain tissues [55]. There are also a number of examples of local adaptation driven by regulatory variants resulting in population differences in immune responses [71,72,89]. Despite the evidence that functional archaic alleles (non-synonymous and associated with expression) have decreased in frequency over the past 8500 years, four loci were identified where the archaic alleles associated with differential expression show large increases in frequency over time. Among these are introgressed alleles modifying expression of the OAS1/OAS2/OAS3 genes involved in innate immunity and whose tissue-specific effects suggest that they may be functionally relevant [71,89]. Archaic alleles in OAS1 are associated with higher expression in subcutaneous adipose tissue and sun-exposed skin, while higher expression of OAS2 in the thyroid and OAS3 in the pancreas and vagina is associated with archaic alleles. In contrast, individuals carrying archaic alleles show down-regulation of OAS1 in esophagus mucosa and spleen, OAS2 in fibroblasts, and OAS3 in fibroblasts as well as in esophagus mucosa, spleen and three brain regions (hippocampus, putamen, and caudate nucleus) [84]. Other examples of local adaptation influencing the levels of expression include expression of gene ERAP2, involved in susceptibility to Crohn’s disease; CCR1, limiting leukocyte recruitment and preventing inflammatory responses; HLA-DQA1, associated with susceptibility to celiac disease; and TLR1, associated with markedly lower levels of inflammatory response gene expression [71,72]. Apparently, introgression from Neanderthals also contributed to the diversification of transcriptional responses to infection in human populations. The introgressed genetic segments in European genomes contain regulatory variants with effects on steady-state expression and responses to TLR7/8 stimulation and influenza virus [72,78]. Furthermore, the archaic variants of several expression quantitative trait loci (eQTLs) have been reported as potential candidates for adaptive introgression conferring better adaptation through the regulation of gene expression. Examples are the gene DARS associated with neuroinflammatory and white matter disorders [71]; the archaic variants of OAS locus apparently associated with diverse flavivirus resistance phenotypes [90]; and PNMA1 harboring a response eQTL for influenza virus and stimulating interferon production [72]. Another regulatory archaic variant that modifies the expression of TNPO3 in the brain is associated with multiple autoimmune phenotypes [83]. All these studies clearly show that selection and archaic admixture affect substantially present-day inter-population differences in immune responses, at least in terms of transcriptional variability, supporting the notion that variation in gene expression has been an important vehicle for human adaptation [86]. Furthermore, it has been shown that the higher frequency archaic variants contribute significantly more to gene expression changes than lower frequency archaic variants. This suggests that at least some of the archaic alleles that modify gene expression may have been driven to higher frequencies in many human populations by positive selection, supporting the idea that changes in gene expression are likely to have important adaptive effects in humans [89].
However, whatever the potential benefits of archaic introgression in the past, alleles of Neanderthal origin have been also associated with several neurological, dermatological, and immunological phenotypes, indicating an influence of ancient admixture on current disease risk in humans [91,92,93]. For example, introgressed alleles associated with the immune system response can increase the risk of inflammation or autoimmunity under environmental factors changing overtime [94,95,96,97,98]. The case of celiac disease neatly illustrates the tradeoff between past selection and current maladaptation. Taskent et al. [93] detected evidence of Neanderthal introgression in the chemokine receptor (CCR) gene family constituting the risk alleles for celiac disease, which was possibly maintained by selective forces in early European population. Furthermore, population genetic analyses have shown that the high frequency of several risk alleles of genes associated with celiac disease such as IL12A, IL18RAP and SH2B3 [99] in Europeans results from past positive selection events [94,100]. Another example comes from a nonsynonymous variant of the ZNF365D gene present in ~32% of Europeans and absent from Africans, which was inherited from Neanderthals and is associated with a higher risk of Crohn’s disease [36]. Likewise, variants of gene cluster TLR6-1-10 inherited from Neanderthals and Denisovans and present in Europeans and Asians has been associated with greater susceptibility to allergies [101].
Further investigations are required, but the studies published to date have provided invaluable resources and increased our understanding of the molecular and cellular processes underlined by introgressed genetic variants and different selective regimes acting on them.
4. Conclusions and Perspectives
The ongoing deluge of sequencing data from thousands of individuals and different populations worldwide, including some archaic hominins and ancient AMH genomes, has provided new insights into the evolutionary history of our species. Genomic studies of introgression between early Eurasians and archaic human species, such as Neanderthals and Denisovans, are beginning to offer novel insights into the evolutionary and phenotypical consequences of hybridization. There is quite common evidence for widespread selection against introgression across the genome, but adaptive introgression may also be considered an important force driving adaptation of modern humans to new environments. However, additional human dataset advances, integration of different sources of information and development of new statistical and analytical methods are critical for understanding the biological and medical implications of such signals of selection.
Table S1 illustrates that our knowledge of the functional consequences of the introgressed variation is essentially based on populations with European ancestry. Informative data from other ethnic groups and sequencing additional samples from ancient hominins will further deepen our knowledge of the contribution of archaic hominins to the diversity of human traits and complex diseases. Furthermore, it will help to identify the functional changes that have contributed to human adaptation and survival over time. Moreover, multigenerational prospective cohort studies from multiple human populations will allow direct measurements of genetic variation and selection intensity for common traits in contemporary populations, performed in a range of nutritional, cultural and geographic conditions, constituting the best way of characterization of the magnitude and importance of complex ecological, epidemiological, demographic and evolutionary shifts. In addition to the genome databases of European origins as the Framingham cohort in the USA, the Uppsala Birth Cohort in Sweden and the Lifelines Cohort in the Netherlands such cohorts now include the H3Africa Initiative on Human Heredity and Health in Africa and the Tohoku Medical Megabank Project in Japan. Data desideratum supplemented with genomic and medical information will further increase our understanding of the antagonistic pleiotropic effects that contribute to the burden of non-infectious diseases and provide new clues to disease causes, potential therapies and possible adverse effects of novel therapies [96].
In the case of integrating different sources of information, studies of genetic variants with regulatory effects on gene expression (eQTL) have already provided insight into the genetic and evolutionary determinants of population phenotypic diversity [102]. One of the first global approaches is the Genotype-Tissue Expression (GTEx) project. GTEx explores the landscape of gene expression across 54 different tissues, providing the richest catalog of tissue-specific and shared eQTL [103,104]. It was already used in the analyses of expression patterns of introgressed haplotypes in the recent studies conducted by McCoy et al. [55] and Dannemann et al. [89]. The future population genetic analyses should extend this across-tissue rationale to multiple populations from different ethnic backgrounds to provide a comprehensive picture of the physiological mechanisms underlying adaptation to environmental pressure and the maintenance of homeostasis.
In the case of new methods, the challenge for the future will be to develop robust statistical models and computational methods for detecting selection, quantifying the frequency of adaptive introgression more widely and understanding the circumstances where it is likely to play a predominant role in adaptation. An exciting future prospect is that our interpretations of observations in nature will be aided by simulation studies [33] and empirical studies of the consequences of introgression for phenotype and fitness [55]. In this context, there are no accurate estimates of the timing of most of the signatures of selection now being detected in the human genome (lactase persistence as an exception) [105,106], or good methods for estimating the ages and natures of the environments in which past selection occurred [97]. Future studies should be able to discriminate with confidence between different time-scales of selection, for example, as a result of the agricultural revolution 8000–10,000 years ago or the industrial revolution 100–300 years ago.
In conclusion, the integration of all of these datasets into a clinical, epidemiological, and population genetics framework will provide new insights on the history of adaptations in the genus Homo, and the ways our genetic and non-genetic makeup, together with changes in our environment and cultural behaviors, influence phenotypic variation in both health and disease.
Supplementary Materials
The following are available online at http://www.mdpi.com/2073-4425/9/7/358/s1, Table S1: Phenotypic implications of putatively introgressed genomic regions.
Author Contributions
O.D. selected relevant literature for the review; O.D. and O.L. reviewed the literature and wrote the manuscript.
Funding
This work was supported by a BFU2015-68759-P (MINECO/FEDER) grant and the support of Secretaria d’Universitats i Recerca del Departament d’Economia i Coneixement de la Generalitat de Catalunya (GRC 2017 SGR 937). O.L was also supported by a Ramón y Cajal grant from the Spanish Ministerio de Economia y Competitividad (MINECO) with reference RYC-2013-14797.
Acknowledgments
We thank Antonio Barbadilla and two anonymous reviewers for helpful comments on the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nielsen, R.; Akey, J.M.; Jakobsson, M.; Pritchard, J.K.; Tishkoff, S.; Willerslev, E. Tracing the peopling of the world through genomics. Nature 2017, 541, 302–310. [Google Scholar] [CrossRef] [PubMed]
- White, T.D.; Asfaw, B.; DeGusta, D.; Gilbert, H.; Richards, G.D.; Suwa, G.; Howell, F.C. Pleistocene Homo sapiens from Middle Awash, Ethiopia. Nature 2003, 423, 742–747. [Google Scholar] [CrossRef] [PubMed]
- McDougall, I.; Brown, F.H.; Fleagle, J.G. Stratigraphic placement and age of modern humans from Kibish, Ethiopia. Nature 2005, 433, 733–736. [Google Scholar] [CrossRef]
- Mercier, N.; Valladas, H.; Bar-Yosef, O.; Vandermeersch, B.; Stringer, C.; Joron, J.-L. Thermoluminescence Date for the Mousterian Burial site of Es-Skhul, Mt. Carmel. J. Archaeol. Sci. 1993, 20, 169–174. [Google Scholar] [CrossRef]
- Stringer, C.B.; Grün, R.; Schwarcz, H.P.; Goldberg, P. ESR dates for the hominid burial site of Es Skhul in Israel. Nature 1989, 338, 756–758. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, H.P.; Grün, R.; Vandermeersch, B.; Bar-Yosef, O.; Valladas, H.; Tchernov, E. ESR dates for the hominid burial site of Qafzeh in Israel. Hum. Evol. 1988, 17, 733–737. [Google Scholar] [CrossRef]
- Liu, W.; Martinón-Torres, M.; Cai, Y.J.; Xing, S.; Tong, H.W.; Pei, S.W.; Sier, M.J.; Wu, X.H.; Edwards, R.L.; Cheng, H.; et al. The earliest unequivocally modern humans in southern China. Nature 2015, 526, 696–699. [Google Scholar] [CrossRef] [PubMed]
- Hublin, J.J.; Ben-Ncer, A.; Bailey, S.E.; Freidline, S.E.; Neubauer, S.; Skinner, M.M.; Bergmann, I.; Le Cabec, A.; Benazzi, S.; Harvati, K.; et al. New fossils from Jebel Irhoud, Morocco and the pan-African origin of Homo sapiens. Nature 2017, 546, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Richter, D.; Grün, R.; Joannes-Boyau, R.; Steele, T.E.; Amani, F.; Rué, M.; Fernandes, P.; Raynal, J.P.; Geraads, D.; Ben-Ncer, A.; et al. The age of the hominin fossils from Jebel Irhoud, Morocco, and the origins of the Middle Stone Age. Nature 2017, 546, 293–296. [Google Scholar] [CrossRef] [PubMed]
- Hershkovitz, I.; Weber, G.W.; Quam, R.; Duval, M.; Grün, R.; Kinsley, L.; Ayalon, A.; Bar-Matthews, M.; Valladas, H.; Mercier, N.; et al. The earliest modern humans outside Africa. Science 2018, 359, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Lalueza-Fox, C.; Gilbert, M.T.P. Paleogenomics of Archaic Hominins Review. Curr. Biol. 2011, 21, R1002–R1009. [Google Scholar] [CrossRef] [PubMed]
- Prüfer, K.; Racimo, F.; Patterson, N.; Jay, F.; Sankararaman, S.; Sawyer, S.; Heinze, A.; Renaud, G.; Sudmant, P.H.; De Filippo, C.; et al. The complete genome sequence of a Neanderthal from the Altai Mountains. Nature 2014, 505, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Prüfer, K.; de Filippo, C.; Grote, S.; Mafessoni, F.; Korlević, P.; Hajdinjak, M.; Vernot, B.; Skov, L.; Hsieh, P.; Peyrégne, S.; et al. A high-coverage Neandertal genome from Vindija Cave in Croatia. Science 2017, 505, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Mondal, M.; Casals, F.; Xu, T.; Dall’Olio, G.M.; Pybus, M.; Netea, M.G.; Comas, D.; Laayouni, H.; Li, Q.; Majumder, P.P.; et al. Genomic analysis of Andamanese provides insights into ancient human migration into Asia and adaptation. Nat. Genet. 2016, 48, 1066–1070. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, P.; Woerner, A.E.; Wall, J.D.; Lachance, J.; Tishkoff, S.A.; Gutenkunst, R.N.; Hammer, M.F. Model-based analyses of whole-genome data reveal a complex evolutionary history involving archaic introgression in Central African Pygmies. Genome Res. 2016, 26, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Medina-Gomez, C.; Lao, O.; Rivadeneira, F. Evolution of complex traits in human populations. In Evolutionary Biology: Self/Nonself Evolution, Species and Complex Traits Evolution, Methods and Concepts; Pontarotti, P., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 165–186. ISBN 978-3-319-61568-4. [Google Scholar]
- Sankararaman, S.; Patterson, N.; Li, H.; Pääbo, S.; Reich, D. The date of interbreeding between Neandertals and modern humans. PLoS Genet. 2012, 8, e1002947. [Google Scholar] [CrossRef] [PubMed]
- Wall, J.D.; Yang, M.A.; Jay, F.; Kim, S.K.; Durand, E.Y.; Stevison, L.S.; Gignoux, C.; Woerner, A.; Hammer, M.F.; Slatkin, M. Higher levels of Neanderthal ancestry in east Asians than in Europeans. Genetics 2013. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Li, H.; Moorjani, P.; Jay, F.; Slepchenko, S.M.; Bondarev, A.-P.; Pääbo, S. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature 2014, 514, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Seguin-Orlando, A.; Korneliussen, T.S.; Sikora, M.; Malaspinas, A.S.; Manica, A.; Moltke, I.; Albrechtsen, A.; Ko, A.; Margaryan, A.; Moiseyev, V.; et al. Genomic structure in Europeans dating back at least 36,200 years. Science 2014, 346, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Sankararaman, S.; Mallick, S.; Patterson, N.; Reich, D. The Combined landscape of Denisovan and Neanderthal ancestry in present-day humans. Curr. Boil. 2016, 26, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Green, R.E.; Krause, J.; Briggs, A.W.; Maricic, T.; Stenzel, U.; Kircher, M.; Patterson, N.; Li, H.; Zhai, W.; Fritz, M.H.; et al. A draft sequence of the Neandertal genome. Science 2010, 328, 710–722. [Google Scholar] [CrossRef] [PubMed]
- Mallick, S.; Li, H.; Lipson, M.; Mathieson, I.; Gymrek, M.; Racimo, F.; Zhao, M.; Chennagiri, N.; Nordenfelt, S.; Tandon, A.; et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature 2016, 538, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Browning, S.R.; Zhou, Y.; Tucci, S.; Akey, J.M. Analysis of Human Sequence Data Reveals Two Pulses of Archaic Denisovan Admixture. Cell 2018, 178, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Labuda, D.; Zietkiewicz, E.; Yotova, V. Archaic lineages in the history of modern humans. Genetics 2000, 156, 799–808. [Google Scholar] [PubMed]
- Hammer, M.F.; Woerner, A.E.; Mendez, F.L.; Watkins, J.C.; Wall, J.D. Genetic evidence for archaic admixture in Africa. Proc. Natl. Acad. Sci. USA 2011, 108, 15123–15128. [Google Scholar] [CrossRef] [PubMed]
- Lachance, J.; Vernot, B.; Elbers, C.C.; Ferwerda, B.; Froment, A.; Bodo, J.M.; Lema, G.; Fu, W.; Nyambo, T.B.; Rebbeck, T.R.; et al. Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell 2012, 150, 457–469. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Pavlidis, P.; Taskent, R.O.; Alachiotis, N.; Flanagan, C.; Degiorgio, M.; Blekhman, R.; Ruhl, S.; Gokcumen, O. Archaic Hominin Introgression in Africa Contributes to Functional Salivary MUC7 Genetic Variation. Mol. Biol. Evol. 2017, 34, 2704–2715. [Google Scholar] [CrossRef] [PubMed]
- Zanolli, C.; Hourset, M.; Esclassan, R.; Mollereau, C. Neanderthal and Denisova tooth protein variants in present-day humans. PLoS ONE 2017, 12. [Google Scholar] [CrossRef] [PubMed]
- Mallet, J.; Besansky, N.; Hahn, M.W. How reticulated are species? BioEssays 2016, 38, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.H.; Jiggins, C.D. Interpreting the genomic landscape of introgression. Curr. Opin. Genet. Dev. 2017, 47, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Juric, I.; Aeschbacher, S.; Coop, G. The strength of selection against Neanderthal introgression. PLoS Genet. 2016, 12, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Harris, K.; Nielsen, R. The genetic cost of neanderthal introgression. Genetics 2016, 203, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Presgraves, D.C. Sex chromosomes and speciation in Drosophila. Trends Genet. 2008, 24, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Qvarnström, A.; Bailey, R.I. Speciation through evolution of sex-linked genes. Heredity 2009, 102, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Sankararaman, S.; Mallick, S.; Dannemann, M.; Prüfer, K.; Kelso, J.; Pääbo, S.; Patterson, N.; Reich, D. The landscape of Neandertal ancestry in present-day humans. Nature 2014, 20, 354–357. [Google Scholar] [CrossRef] [PubMed]
- Garrigan, D.; Mobasher, Z.; Severson, T.; Wilder, J.A.; Hammer, M.F. Evidence for archaic Asian ancestry on the human X chromosome. Mol. Biol. Evol. 2005, 22, 189–192. [Google Scholar] [CrossRef] [PubMed]
- Garrigan, D.; Kingan, S.B.; Geneva, A.J.; Andolfatto, P.; Clark, A.G.; Thornton, K.R.; Presgraves, D.C. Genome sequencing reveals complex speciation in the Drosophila simulans clade. Genome Res. 2012, 22, 1499–1511. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.; Bengtsson, B.O. The barrier to genetic exchange between hydridizing populations. Heredity 1986, 57, 357–376. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, M.C.; Pease, J.B.; Steele, A.; Waterhouse, R.M.; Neafsey, D.E.; Sharakhov, I.V.; Jiang, X.; Hall, A.B.; Catteruccia, F.; Kakani, E.; et al. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 2015, 347, 1258524. [Google Scholar] [CrossRef] [PubMed]
- Janoušek, V.; Munclinger, P.; Wang, L.; Teeter, K.C.; Tucker, P.K. Functional organization of the genome may shape the species boundary in the house mouse. Mol. Boil. Evol. 2015, 32, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Runemark, A.; Trier, C.N.; Eroukhmanoff, F.; Hermansen, J.S.; Matschiner, M.; Ravinet, M.; Elgvin, T.O.; Sætre, G.P. Variation and constraints in hybrid genome formation. Nat. Ecol. Evol. 2018, 2, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Schumer, M.; Cui, R.; Powell, D.L.; Rosenthal, G.G.; Andolfatto, P. Ancient hybridization and genomic stabilization in a swordtail fish. Mol. Ecol. 2016, 25, 2661–2679. [Google Scholar] [CrossRef] [PubMed]
- Jégou, B.; Sankararaman, S.; Rolland, A.D.; Reich, D.; Chalmel, F. Meiotic genes are enriched in regions of reduced archaic ancestry. Mol. Boil. Evol. 2017, 34, 1974–1980. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ho, W.C.S.; Boon, S.S.; Law, P.T.Y.; Chan, M.C.W.; DeSalle, R.; Burk, R.D.; Chan, P.K.S. Ancient Evolution and Dispersion of Human Papillomavirus Type 58 Variants. J. Virol. 2017, JVI.01285-17. [Google Scholar] [CrossRef] [PubMed]
- Fu, Q.; Posth, C.; Hajdinjak, M.; Petr, M.; Mallick, S.; Fernandes, D.; Furtwängler, A.; Haak, W.; Meyer, M.; Mittnik, A.; et al. The genetic history of Ice Age Europe. Nature 2016, 534, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Vernot, B.; Akey, J.M. Human evolution: Genomic gifts from Archaic hominins. Curr. Biol. 2014, 24, R845–R848. [Google Scholar] [CrossRef] [PubMed]
- Vernot, B.; Tucci, S.; Kelso, J.; Schraiber, J.G.; Wolf, A.B.; Gittelman, R.M.; Dannemann, M.; Grote, S.; McCoy, R.C.; Norton, H.; et al. Excavating Neandertal and Denisovan DNA from the genomes of Melanesian individuals. Science 2016. [Google Scholar] [CrossRef] [PubMed]
- Konopka, G.; Roberts, T.F. Insights into the neural and genetic basis of vocal communication. Cell 2016, 164, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.H.; Dominy, N.J.; Claw, K.G.; Lee, A.S.; Fiegler, H.; Redon, R.; Werner, J.; Villanea, F.A.; Mountain, J.L.; Misra, R.; et al. Diet and the evolution of human amylase gene copy number variation. Nat. Genet. 2007, 39, 1256–1260. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.L.; Saus, E.; Smalley, S.V.; Cataldo, L.R.; Alberti, G.; Parada, J.; Gratacòs, M.; Estivill, X. Copy number polymorphism of the salivary amylase gene: Implications in human nutrition research. Lifestyle Genom. 2012, 5, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.H.; Kistler, L.; Kelaita, M.A.; Sams, A.J. Insights into hominin phenotypic and dietary evolution from ancient DNA sequence data. J. Hum. Evol. 2015, 79, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Chintalapati, M.; Dannemann, M.; Prüfer, K. Using the Neandertal genome to study the evolution of small insertions and deletions in modern humans. BMC Evol. Biol. 2017, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- Dannemann, M.; Kelso, J. The Contribution of Neanderthals to Phenotypic Variation in Modern Humans. Am. J. Hum. Genet. 2017, 101, 578–589. [Google Scholar] [CrossRef] [PubMed]
- McCoy, R.C.; Wakefield, J.; Akey, J.M. Impacts of Neanderthal-introgressed sequences on the landscape of human gene expression. Cell 2017, 168, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Racimo, F.; Sankararaman, S.; Nielsen, R.; Huerta-Sánchez, E. Evidence for archaic adaptive introgression in humans. Nat. Rev. Genet. 2015, 16, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Gittelman, R.M.; Schraiber, J.G.; Vernot, B.; Mikacenic, C.; Wurfel, M.M.; Akey, J.M. Archaic Hominin Admixture Facilitated Adaptation to Out-of-Africa Environments. Curr. Biol. 2016, 26, 3375–3382. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, P.W. Adaptive introgression in animals: Examples and comparison to new mutation and standing variation as sources of adaptive variation. Mol. Ecol. 2013, 22, 4606–4618. [Google Scholar] [CrossRef] [PubMed]
- Kelso, J.; Prüfer, K. Ancient humans and the origin of modern humans. Curr. Opin. Genet. Dev. 2014, 29, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Vattathil, S.; Akey, J.M. Small Amounts of Archaic Admixture Provide Big Insights into Human History. Cell 2015, 163, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Pittman, A.; Myers, A.; Gwinn-Hardy, K.; Fung, H. C.; de Silva, R.; Hutton, M.; Duckworth, J. Evidence suggesting that Homo neanderthalensis contributed the H2 MAPT haplotype to Homo sapiens. Biochem. Soc. Trans. 2005, 33, 582–585. [Google Scholar] [CrossRef] [PubMed]
- Setó-Salvia, N.; Sánchez-Quinto, F.; Carbonell, E.; Lorenzo, C.; Comas, D.; Clarimón, J. Using the Neanderthal and Denisova Genetic Data to Understand the Common MAPT 17q21 Inversion in Modern Humans. Hum. Biol. 2012, 84, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.D.; Mekel-Bobrov, N.; Vallender, E.J.; Hudson, R.R.; Lahn, B.T. Evidence that the adaptive allele of the brain size gene microcephalin introgressed into Homo sapiens from an archaic Homo lineage. PNAS 2006, 103, 18178–18183. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.D.; Kippenhan, J.Sh.; Eisenberg, D.P.; Kohn, Ph.D.; Dickinson, D.; Mattay, V.S.; Chen, Q.; Weinberger, D.R.; Saad, Z.S.; Berman, K.F. Neanderthal-Derived Genetic Variation Shapes Modern Human Cranium and Brain. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Racimo, F.; Gokhman, D.; Fumagalli, M.; Ko, A.; Hansen, T.; Moltke, I.; Albrechtsen, A.; Carmel, L.; Huerta-Sanchez, E.; Nielsen, R. Archaic adaptive introgression in TBX15/WARS2. Mol. Biol. Evol. 2017, 34, 509–524. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Hu, Y.; Xu, S.; Wang, J.; Jin, L. Neanderthal introgression at chromosome 3p21.31 was under positive natural selection in east asians. Mol. Boil. Evol. 2014, 31, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Frost, P.; Kleisner, K.; Flegr, J. Health status by gender, hair color, and eye color: Red-haired women are the most divergent. PLoS ONE 2017, 12, e0190238. [Google Scholar] [CrossRef] [PubMed]
- Mendez, F.L.; Watkins, J.C.; Hammer, M.F. Global genetic variation at OAS1 provides evidence of archaic admixture in Melanesian populations. Mol. Biol. Evol. 2012, 29, 1513–1520. [Google Scholar] [CrossRef] [PubMed]
- Mendez, F.L.; Watkins, J.C.; Hammer, M.F. Neandertal origin of genetic variation at the cluster of OAS immunity genes. Mol. Biol. Evol. 2013, 30, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, M.; Quintana-Murci, L. Immunité innée et maladies chez l’homme. Med. Sci. 2016, 32, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Nédélec, Y.; Sanz, J.; Baharian, G.; Szpiech, Z.A.; Pacis, A.; Dumaine, A.; Grenier, J.C.; Freiman, A.; Sams, A.J.; Hebert, S.; et al. Genetic ancestry and natural selection drive population differences in immune responses to pathogens. Cell 2016, 167, 657–669.e21. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; Rotival, M.; Pothlichet, J.; Loh, Y.H.E.; Dannemann, M.; Zidane, N.; Laval, G.; Patin, E.; Harmant, C.; Lopez, M.; et al. Genetic adaptation and Neandertal admixture shaped the immune system of human populations. Cell 2016, 167, 643–656.e17. [Google Scholar] [CrossRef] [PubMed]
- Deschamps, M.; Laval, G.; Fagny, M.; Itan, Y.; Abel, L.; Casanova, J.L.; Patin, E.; Quintana-Murci, L. Genomic Signatures of Selective Pressures and Introgression from Archaic Hominins at Human Innate Immunity Genes. Am. J. Hum. Genet. 2016, 1, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Pimenoff, V.N.; Houldcroft, C.J.; Rifkin, R.F.; Underdown, S. The role of aDNA in understanding the coevolutionary patterns of human sexually transmitted infections. Genes 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Petousi, N.; Glusman, G.; Yu, Y.; Bohlender, R.; Tashi, T.; Downie, J.M.; Roach, J.C.; Cole, A.M.; Lorenzo, F.R.; et al. Evolutionary history of Tibetans inferred from whole-genome sequencing. PLoS Genet. 2017, 13, e1006675. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Sánchez, E.; Jin, X.; Bianba, Z.; Peter, B.M.; Vinckenbosch, N.; Liang, Y.; Yi, X.; He, M.; Somel, M.; Ni, P.; et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 2014, 512, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Tashi, T.; Scott Reading, N.; Wuren, T.; Zhang, X.; Moore, L.G.; Hu, H.; Tang, F.; Shestakova, A.; Lorenzo, F.; Burjanivova, T.; et al. Gain-of-function EGLN1 prolyl hydroxylase (PHD2 D4E:C127S) in combination with EPAS1 (HIF-2α) polymorphism lowers hemoglobin concentration in Tibetan highlanders. J. Mol. Med. 2017, 95, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Quach, H.; Quintana-Murci, L. Living in an adaptive world: Genomic dissection of the genus Homo and its immune response. J. Exp. Med. 2017, 214, 877–894. [Google Scholar] [CrossRef] [PubMed]
- Andrés, A.M.; Hubisz, M.J.; Indap, A.; Torgerson, D.G.; Degenhardt, J.D.; Boyko, A.R.; Gutenkunst, R.N.; White, T.J.; Green, E.D.; Bustamante, C.D.; et al. Targets of balancing selection in the human genome. Mol. Biol. Evol. 2009, 26, 2755–2764. [Google Scholar] [CrossRef] [PubMed]
- DeGiorgio, M.; Lohmueller, K.E.; Nielsen, R. A Model-Based Approach for Identifying Signatures of Ancient Balancing Selection in Genetic Data. PLoS Genet. 2014, 10, e1004561. [Google Scholar] [CrossRef] [PubMed]
- Leffler, E.M.; Gao, Z.; Pfeifer, S.; Ségurel, L.; Auton, A.; Venn, O.; Bowden, R.; Bontrop, R.; Wall, J.D.; Sella, G.; et al. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 2013, 340, 1578–1582. [Google Scholar] [CrossRef] [PubMed]
- Norman, P.J.; Abi-Rached, L.; Gendzekhadze, K.; Korbel, D.; Gleimer, M.; Rowley, D.; Bruno, D.; Carrington, C.V.F.; Chandanayingyong, D.; Chang, Y.H.; et al. Unusual selection on the KIR3DL1/S1 natural killer cell receptor in Africans. Nat. Genet. 2007, 39, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Single, R.M.; Martin, M.P.; Gao, X.; Meyer, D.; Yeager, M.; Kidd, J.R.; Kidd, K.K.; Carrington, M. Global diversity and evidence for coevolution of KIR and HLA. Nat. Genet. 2007, 39, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Abi-Rached, L.; Jobin, M.J.; Kulkarni, S.; McWhinnie, A.; Dalva, K.; Gragert, L.; Babrzadeh, F.; Gharizadeh, B.; Luo, M.; Plummer, F.A.; et al. The shaping of modern human immune systems by multiregional admixture with archaic humans. Science 2011, 334, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Yasukochi, Y.; Ohashi, J. Elucidating the origin of HLA-B*73 allelic lineage: Did modern humans benefit by archaic introgression? Immunogenetics 2017, 69, 63–67. [Google Scholar] [CrossRef] [PubMed]
- Fraser, H.B. Gene expression drives local adaptation in humans. Genome Res. 2013, 23, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, J.K. Joint analysis of functional genomic data and genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 2014, 94, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Schaub, M.A.; Boyle, A.P.; Kundaje, A.; Batzoglou, S.; Snyder, M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012, 22, 1748–1759. [Google Scholar] [CrossRef] [PubMed]
- Dannemann, M.; Prüfer, K.; Kelso, J. Functional implications of Neandertal introgression in modern humans. Genome Biol. 2017, 18, 61. [Google Scholar] [CrossRef] [PubMed]
- Sams, A.J.; Dumaine, A.; Nédélec, Y.; Yotova, V.; Alfieri, C.; Tanner, J.E.; Messer, P.W.; Barreiro, L.B. Adaptively introgressed Neandertal haplotype at the OAS locus functionally impacts innate immune responses in humans. Genome Biol. 2016, 17, 246. [Google Scholar] [CrossRef] [PubMed]
- Mozzi, A.; Forni, D.; Cagliani, R.; Pozzoli, U.; Clerici, M.; Sironi, M. Distinct selective forces and Neanderthal introgression shaped genetic diversity at genes involved in neurodevelopmental disorders. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Simonti, C.N.; Vernot, B.; Bastarache, L.; Bottinger, E.; Carrell, D.S.; Chisholm, R.L.; Crosslin, D.R.; Hebbring, S.J.; Jarvik, G.P.; Kullo, I.J.; et al. The phenotypic legacy of admixture between modern humans and Neandertals. Science 2016, 351, 737–741. [Google Scholar] [CrossRef] [PubMed]
- Taskent, R.O.; Alioglu, N.D.; Fer, E.; Melike Donertas, H.; Somel, M.; Gokcumen, O. Variation and functional impact of Neanderthal ancestry in Western Asia. Genome Boil. Evol. 2017, 9, 3516–3524. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, L.B.; Quintana-Murci, L. From evolutionary genetics to human immunology: How selection shapes host defence genes. Nat. Rev. Genet. 2010, 11, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Brinkworth, J.F.; Barreiro, L.B. The contribution of natural selection to present-day susceptibility to chronic inflammatory and autoimmune disease. Curr. Opin. Immunol. 2014, 31, 66–78. [Google Scholar] [CrossRef] [PubMed]
- Corbett, S.; Courtiol, A.; Lummaa, V.; Moorad, J.; Stearns, S. The transition to modernity and chronic disease: Mismatch and natural selection. Nat. Rev. Genet. 2018, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sironi, M.; Clerici, M. The hygiene hypothesis: An evolutionary perspective. Microbes Infect. 2010, 12, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Stearns, S.C. Evolutionary medicine: Its scope, interest and potential. Proc. R. Soc. B Biol. Sci. 2012, 279, 4305–4321. [Google Scholar] [CrossRef] [PubMed]
- Hunt, K.A.; Zhernakova, A.; Turner, G.; Heap, G.A.R.; Franke, L.; Bruinenberg, M.; Romanos, J.; Dinesen, L.C.; Ryan, A.W.; Panesar, D.; et al. Newly identified genetic risk variants for celiac disease related to the immune response. Nat. Genet. 2008, 40, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Zhernakova, A.; Elbers, C.C.; Ferwerda, B.; Romanos, J.; Trynka, G.; Dubois, P.C.; de Kovel, C.G.F.; Franke, L.; Oosting, M.; Barisani, D.; et al. Evolutionary and Functional Analysis of Celiac Risk Loci Reveals SH2B3 as a Protective Factor against Bacterial Infection. Am. J. Hum. Genet. 2010, 86, 970–977. [Google Scholar] [CrossRef] [PubMed]
- Dannemann, M.; Andrés, A.M.; Kelso, J. Introgression of Neandertal-and Denisovan-like haplotypes contributes to adaptive variation in human Toll-like receptors. Am. J. Hum. Genet. 2016, 98, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.P.; Knight, J.C. Genetics of gene expression in immunity to infection. Curr. Opin. Immunol. 2014, 30, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Aguet, F.; Brown, A.A.; Castel, S.E.; Davis, J.R.; He, Y.; Jo, B.; Mohammadi, P.; Park, Y.S.; Parsana, P.; Segrè, A.V.; et al. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Ardlie, K.G.; DeLuca, D.S.; Segrè, A.V.; Sullivan, T.J.; Young, T.R.; Gelfand, E.T.; Trowbridge, C.A.; Maller, J.B.; Tukiainen, T.; Lek, M.; et al. The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Field, Y.; Boyle, E.A.; Telis, N.; Gao, Z.; Gaulton, K.J.; Golan, D.; Yengo, L.; Rocheleau, G.; Froguel, P.; McCarthy, M.I.; et al. Detection of human adaptation during the past 2000 years. Science 2016, 354, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Tishkoff, S.A.; Reed, F.A.; Ranciaro, A.; Voight, B.F.; Babbitt, C.C.; Silverman, J.S.; Powell, K.; Mortensen, H.M.; Hirbo, J.B.; Osman, M.; et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 2007. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).