Adaptation and Therapeutic Exploitation of the Plasma Membrane of African Trypanosomes



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
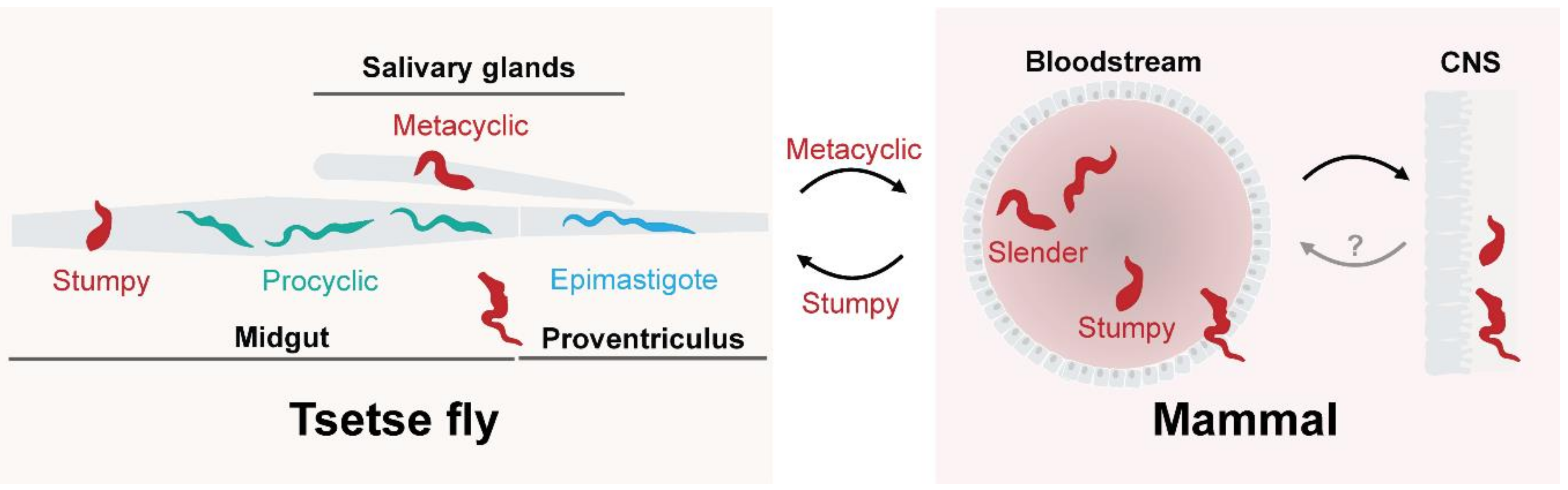
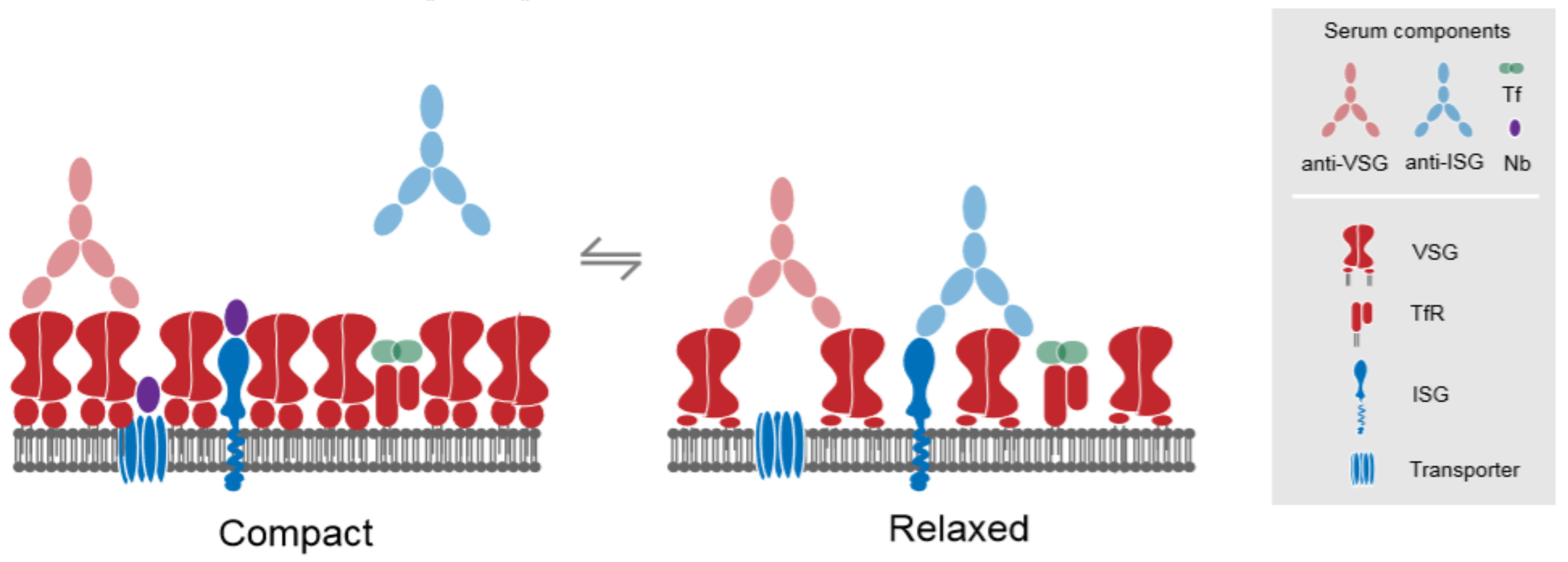
2. A Dynamic Surface for Host and Environmental Interactions
3. The Major Surface Proteins and Their Trafficking Itinerates
4. Invariant Surface Glycoproteins
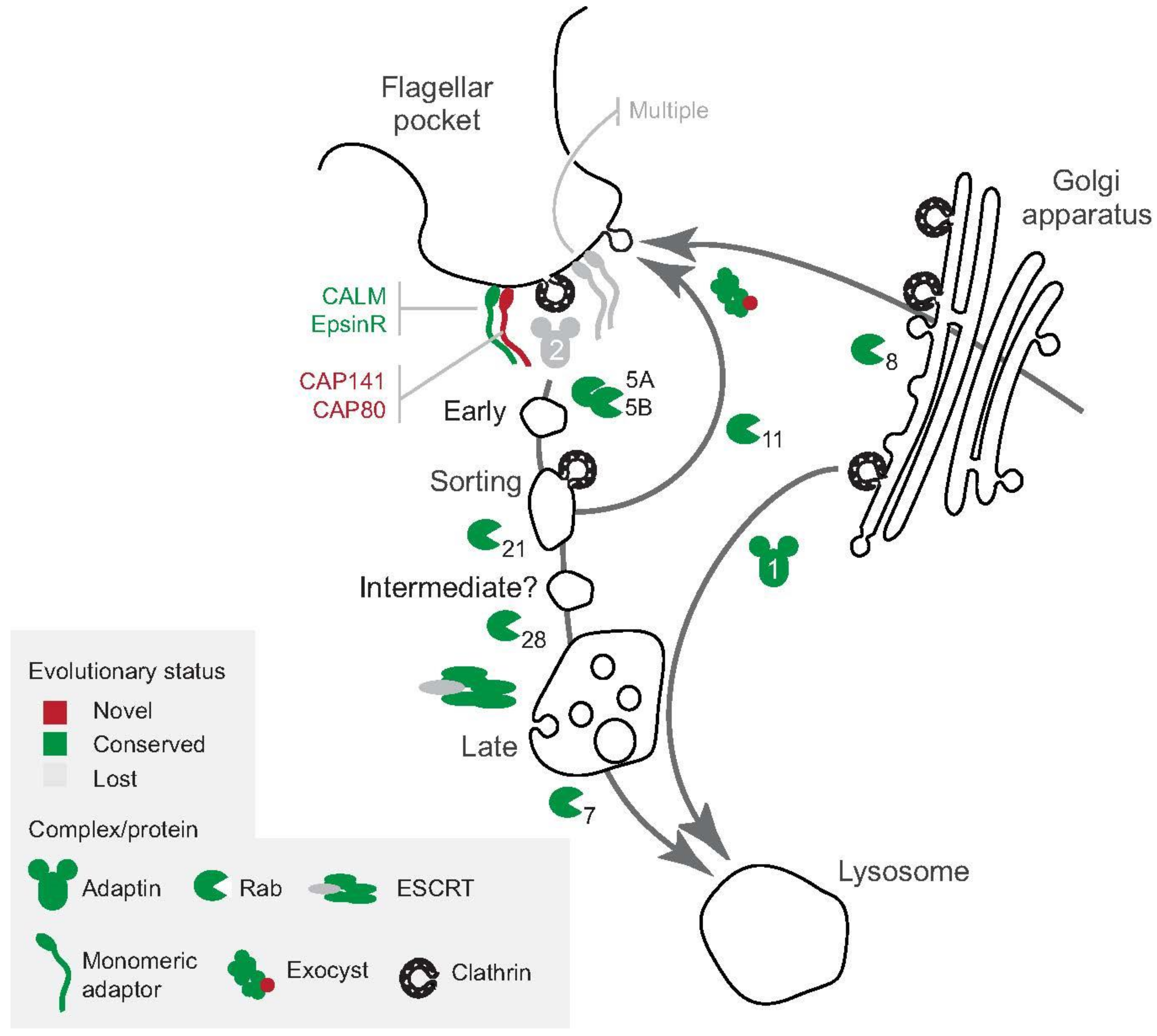
5. The Flagellar Pocket and Contact with the Environment
6. Complex Interactions between Drugs and Trafficking Revealed by Genetics
7. Can We Harness Endocytic Machinery for Therapy?
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Simpson, A.G.B.; Stevens, J.R.; Lukeš, J. The evolution and diversity of kinetoplastid flagellates. Trends Parasitol. 2006, 22, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Dacks, J.B.; Field, M.C.; Buick, R.; Eme, L.; Gribaldo, S.; Roger, A.J.; Brochier-Armanet, C.; Devos, D.P. The changing view of eukaryogenesis—fossils, cells, lineages and how they all come together. J. Cell Sci. 2016, 129, 3695–3703. [Google Scholar] [CrossRef] [PubMed]
- Schlacht, A.; Herman, E.K.; Klute, M.J.; Field, M.C.; Dacks, J.B. Missing pieces of an ancient puzzle: Evolution of the eukaryotic membrane-trafficking system. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Horn, D. Antigenic variation in African trypanosomes. Mol. Biochem. Parasitol. 2014, 195, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Carrington, M. The trypanosome flagellar pocket. Nat. Rev. Microbiol. 2009, 7, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Pays, E.; Vanhollebeke, B.; Uzureau, P.; Lecordier, L.; Pérez-Morga, D. The molecular arms race between African trypanosomes and humans. Nat. Rev. Microbiol. 2014, 12, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Carrington, M. Intracellular membrane transport systems in Trypanosoma brucei. Traffic 2004, 5, 905–913. [Google Scholar] [CrossRef] [PubMed]
- Overath, P.; Engstler, M. Endocytosis, membrane recycling and sorting of GPI-anchored proteins: Trypanosoma brucei as a model system. Mol. Microbiol. 2004, 53, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Lumb, J.H.; Adung’a, V.O.; Jones, N.G.; Engstler, M. Chapter 1: Macromolecular Trafficking and Immune Evasion in African Trypanosomes. In International Review of Cell and Molecular Biology, 1st ed.; Jeon, K.W., Ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2009; Volume 278, pp. 1–67. [Google Scholar]
- Bonhivers, M.; Nowacki, S.; Landrein, N.; Robinson, D.R. Biogenesis of the trypanosome endo-exocytotic organelle is cytoskeleton mediated. PLoS Biol. 2008, 6, 1033–1046. [Google Scholar] [CrossRef] [PubMed]
- Boehm, C.M.; Obado, S.; Gadelha, C.; Kaupisch, A.; Manna, P.T.; Gould, G.W.; Munson, M.; Chait, B.T.; Rout, M.P.; Field, M.C. The Trypanosome Exocyst: A Conserved Structure Revealing a New Role in Endocytosis. PLoS Pathog. 2017, 13, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Salcedo, J.A.; Unciti-Broceta, J.D.; Soriano, M. Could specific cell targeting overcome resistance associated with current treatments for African trypanosomiasis? Nanomedicine 2015, 10, 3515–3517. [Google Scholar] [CrossRef] [PubMed]
- Matthews, K.R. The developmental cell biology of Trypanosoma brucei. Cell 2009, 118, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Goulding, D.; Field, M.C. Clathrin-mediated endocytosis is essential in Trypanosoma brucei. EMBO J. 2003, 22, 4991–5002. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.T.; Kelly, S.; Field, M.C. Adaptin evolution in kinetoplastids and emergence of the variant surface glycoprotein coat in African trypanosomatids. Mol. Phylogenet. Evol. 2013, 67, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.T.; Obado, S.O.; Boehm, C.; Gadelha, C.; Sali, A.; Chait, B.T.; Rout, M.P.; Field, M.C. Lineage-specific proteins essential for endocytosis in trypanosomes. J. Cell Sci. 2017, 130, 1379–1392. [Google Scholar] [CrossRef] [PubMed]
- Adung’a, V.O.; Gadelha, C.; Field, M.C. Proteomic Analysis of Clathrin Interactions in Trypanosomes Reveals Dynamic Evolution of Endocytosis. Traffic 2013, 14, 440–457. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Natesan, S.K.A.; Gabernet-Castello, C.; Koumandou, V.L. Intracellular trafficking in the trypanosomatids. Traffic 2007, 8, 629–639. [Google Scholar] [CrossRef] [PubMed]
- Morgan, G.W.; Hall, B.S.; Denny, P.W.; Field, M.C.; Carrington, M. The endocytic apparatus of the kinetoplastida. Part II: Machinery and components of the system. Trends Parasitol. 2002, 18, 540–546. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.; Brighouse, A.; Gabernet-Castello, C.; Field, M.C.; Dacks, J.B. Sculpting the endomembrane system in deep time: High resolution phylogenetics of Rab GTPases. J. Cell Sci. 2012, 125, 2500–2508. [Google Scholar] [CrossRef] [PubMed]
- Ackers, J.P.; Dhir, V.; Field, M.C. A bioinformatic analysis of the RAB genes of Trypanosoma brucei. Mol. Biochem. Parasitol. 2005, 141, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Hall, B.S.; Nesbeth, D.N.; Field, H.I.; Field, M.C. Differential endocytic functions of Trypanosoma brucei Rab5 isoforms reveal a glycosylphosphatidylinositol-specific endosomal pathway. J. Biol. Chem. 2002, 277, 9529–9539. [Google Scholar] [CrossRef] [PubMed]
- Engstler, M.; Pfohl, T.; Herminghaus, S.; Boshart, M.; Wiegertjes, G.; Heddergott, N.; Overath, P. Hydrodynamic Flow-Mediated Protein Sorting on the Cell Surface of Trypanosomes. Cell 2007, 131, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Koltzscher, M.; Neumann, C.; Kö, S.; Gerke, V. Ca 2+-dependent Binding and Activation of Dormant Ezrin by Dimeric S100P. Mol. Biol. Cell 2003, 14, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Tazeh, N.N.; Silverman, J.S.; Schwartz, K.J.; Sevova, E.S.; Sutterwala, S.S.; Bangs, J.D. Role of AP-1 in developmentally regulated lysosomal trafficking in Trypanosoma brucei. Eukaryot. Cell 2009, 8, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Silverman, J.S.; Schwartz, K.J.; Hajduk, S.L.; Bangs, J.D. Late endosomal Rab7 regulates lysosomal trafficking of endocytic but not biosynthetic cargo in Trypanosoma brucei. Mol. Microbiol. 2011, 82, 664–678. [Google Scholar] [CrossRef] [PubMed]
- Lumb, J.H.; Leung, K.F.; DuBois, K.N.; Field, M.C. Rab28 function in trypanosomes: Interactions with retromer and ESCRT pathways. J. Cell Sci. 2011, 124, 3771–3783. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.; Leung, K.F.; Field, M.C. The ancient small GTPase Rab21 functions in intermediate endocytic steps in trypanosomes. Eukaryot. Cell 2014, 13, 304–319. [Google Scholar] [CrossRef] [PubMed]
- Herman, E.K.; Ali, M.; Field, M.C.; Dacks, J.B. Regulation of early endosomes across eukaryotes: Evolution and functional homology of Vps9 proteins. Traffic 2018, 19, 546–563. [Google Scholar] [CrossRef] [PubMed]
- Umaer, K.; Bush, P.J.; Bangs, J.D. Rab11 mediates selective recycling and endocytic trafficking in Trypanosoma brucei. Traffic 2018, 19, 406–420. [Google Scholar] [CrossRef] [PubMed]
- Gabernet-Castello, C.; DuBois, K.N.; Nimmo, C.; Field, M.C. Rab11 function in Trypanosoma brucei: Identification of conserved and novel interaction partners. Eukaryot. Cell 2011, 10, 1082–1094. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, T.R.; Morgan, G.W.; Field, M.C. A developmentally regulated Rab11 homologue in Trypanosoma brucei is involved in recycling processes. J. Cell Sci. 2001, 114, 2617–2626. [Google Scholar] [PubMed]
- Mugnier, M.R.; Stebbins, C.E.; Papavasiliou, F.N. Masters of Disguise: Antigenic Variation and the VSG Coat in Trypanosoma brucei. PLoS Pathog. 2016, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bartossek, T.; Jones, N.G.; Schäfer, C.; Cvitković, M.; Glogger, M.; Mott, H.R.; Kuper, J.; Brennich, M.; Carrington, M.; Smith, A.S.; et al. Structural basis for the shielding function of the dynamic trypanosome variant surface glycoprotein coat. Nat. Microbiol. 2017, 2, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.E.; Demick, K.P.; Mansfield, J.M. Trypanosomes Expressing a Mosaic Variant Surface Glycoprotein Coat Escape Early Detection by the Immune System Trypanosomes Expressing a Mosaic Variant Surface Glycoprotein Coat Escape Early Detection by the Immune System. Infect. Immun. 2005, 73, 2690–2697. [Google Scholar] [CrossRef] [PubMed]
- Pinger, J.; Chowdhury, S.; Papavasiliou, F.N. Variant surface glycoprotein density defines an immune evasion threshold for African trypanosomes undergoing antigenic variation. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Manna, P.T.; Boehm, C.; Leung, K.F.; Natesan, S.K.; Field, M.C. Life and times: Synthesis, trafficking, and evolution of VSG. Trends Parasitol. 2014, 30, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.K.; Vasileva, N.; Gluenz, E.; Terry, S.; Portman, N.; Kramer, S.; Carrington, M.; Michaeli, S.; Gull, K.; Rudenko, G. Blocking variant surface glycoprotein synthesis in Trypanosoma brucei triggers a general arrest in translation initiation. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Sheader, K.; Vaughan, S.; Minchin, J.; Hughes, K.; Gull, K.; Rudenko, G. Variant surface glycoprotein RNA interference triggers a precytokinesis cell cycle arrest in African trypanosomes. Proc. Natl. Acad. Sci. USA 2005, 102, 8716–8721. [Google Scholar] [CrossRef] [PubMed]
- Boothroyd, J.C.; Paynter, C.A.; Cross, G.A.M.; Bernards, A.; Borst, P. Variant surface glycoproteins of Trypanosoma brucei are synthesised with cleavable hydrophobic sequences at the carboxy and amino termini. Nucleic Acids Res. 1981, 9, 4735–4744. [Google Scholar] [CrossRef] [PubMed]
- Lustig, Y.; Vagima, Y.; Goldshmidt, H.; Erlanger, A.; Ozeri, V.; Vince, J.; McConville, M.J.; Dwyer, D.M.; Landfear, S.M.; Michaeli, S. Down-regulation of the trypanosomatid signal recognition particle affects the biogenesis of polytopic membrane proteins but not of signal peptide-containing proteins. Eukaryot. Cell 2007, 6, 1865–1875. [Google Scholar] [CrossRef] [PubMed]
- Wickner, W.; Schekman, R. Protein Translocation Across Biological Membranes. Science 2005, 310, 1452–1456. [Google Scholar] [CrossRef] [PubMed]
- Pinger, J.; Nešić, D.; Ali, L.; Aresta-Branco, F.; Lilic, M.; Chowdhury, S.; Kim, H.-S.; Verdi, J.; Raper, J.; Ferguson, M.A.J.; et al. African trypanosomes evade immune clearance by O-glycosylation of the VSG surface coat. Nat. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Böhme, U.; Cross, G.A.M. Structural features affecting variant surface glycoprotein expression in Trypanosoma brucei. Mol. Biochem. Parasitol. 2003, 128, 135–145. [Google Scholar] [CrossRef]
- Field, M.C.; Sergeenko, T.; Wang, Y.N.; Böhm, S.; Carrington, M. Chaperone requirements for biosynthesis of the trypanosome variant surface glycoprotein. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Tiengwe, C.; Muratore, K.A.; Bangs, J.D. Variant Surface Glycoprotein, Transferrin Receptor, and ERAD in Trypanosoma brucei. Cell Microbiol. 2016, 18, 1673–1688. [Google Scholar] [CrossRef] [PubMed]
- Manthri, S.; Güther, M.L.S.; Izquierdo, L.; Acosta-serrano, A.; Ferguson, M.A.J. Deletion of the TbALG3 gene demonstrates site-specific N-glycosylation and N-glycan processing in Trypanosoma brucei. Glycobiology 2008, 18, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Bangs, J.D.; Doerings, T.L.; Englund, P.T.; Hartll, G.W. Biosynthesis of a Variant Surface Glycoprotein of Trypanosoma brucei. J. Biol. Chem. 1988, 263, 17697–17705. [Google Scholar] [PubMed]
- Mowatt, M.R.; Clayton, C.E. Developmental regulation of a novel repetitive protein of Trypanosoma brucei. Mol. Cell. Biol. 1987, 7, 2838–2844. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Serrano, A.; Vassella, E.; Liniger, M.; Renggli, C.K.; Brun, R.; Roditi, I.; Englund, P.T. The surface coat of procyclic Trypanosoma brucei: Programmed expression and proteolytic cleavage of procyclin in the tsetse fly. Proc. Natl. Acad. Sci. USA 2001, 98, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Filigheddu, N.; Gnocchi, V.F.; Coscia, M.; Cappelli, M.; Porporato, P.E.; Taulli, R. Ghrelin and Des-Acyl Ghrelin Promote Differentiation and Fusion of C2C12 Skeletal Muscle Cells. Mol. Biol. Cell 2007, 18, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Schlaeppi, A.C.; Malherbe, T.; Bütikofer, P. Coordinate Expression of GPEET Procyclin and Its Membrane-associated Kinase in Trypanosoma brucei Procyclic Forms. J. Biol. Chem. 2003, 278, 49980–49987. [Google Scholar] [CrossRef] [PubMed]
- Ziegelbauer, K.; Overath, P. Identification of invariant surface glycoproteins in the bloodstream stage of Trypanosoma brucei. J. Biol. Chem. 1992, 267, 10791–10796. [Google Scholar] [PubMed]
- Zoltner, M.; Leung, K.F.; Alsford, S.; Horn, D.; Field, M.C. Modulation of the Surface Proteome through Multiple Ubiquitylation Pathways in African Trypanosomes. PLoS Pathog. 2015, 11, 1–26. [Google Scholar] [CrossRef]
- Alsford, S.; Eckert, S.; Baker, N.; Glover, L.; Sanchez-Flores, A.; Leung, K.F.; Turner, D.J.; Field, M.C.; Berriman, M.; Horn, D. High-throughput decoding of antitrypanosomal drug efficacy and resistance. Nature 2012, 482, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Alsford, S.; Field, M.C.; Horn, D. Receptor-mediated endocytosis for drug delivery in African trypanosomes: Fulfilling Paul Ehrlich’s vision of chemotherapy. Trends Parasitol. 2013, 29, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Zoltner, M.; Horn, D.; de Koning, H.P.; Field, M.C. Exploiting the Achilles’ heel of membrane trafficking in trypanosomes. Curr. Opin. Microbiol. 2016, 34, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Chung, W.L.; Leung, K.F.; Carrington, M.; Field, M.C. Ubiquitylation is required for degradation of transmembrane surface proteins in Trypanosomes. Traffic 2008, 9, 1681–1697. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.F.; Riley, F.S.; Carrington, M.; Field, M.C. Ubiquitylation and developmental regulation of invariant surface protein expression in trypanosomes. Eukaryot. Cell 2011, 10, 916–931. [Google Scholar] [CrossRef] [PubMed]
- Barquilla, A.; Crespo, J.L.; Navarro, M. Rapamycin inhibits trypanosome cell growth by preventing TOR complex 2 formation. Proc. Natl. Acad. Sci. USA 2008, 105, 14579–14584. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.S.; Gabernet-Castello, C.; Voak, A.; Goulding, D.; Natesan, S.K.; Field, M.C. TbVps34, the trypanosome orthologue of Vps34, is required for Golgi complex segregation. J. Biol. Chem. 2006, 281, 27600–27612. [Google Scholar] [CrossRef] [PubMed]
- Pays, E.; Lips, S.; Nolan, D.; Vanhamme, L.; Pérez-Morga, D. The VSG expression sites of Trypanosoma brucei: Multipurpose tools for the adaptation of the parasite to mammalian hosts. Mol. Biochem. Parasitol. 2001, 114, 1–16. [Google Scholar] [CrossRef]
- Mussmann, R.; Engstler, M.; Gerrits, H.; Kieft, R.; Toaldo, C.B.; Onderwater, J.; Koerten, H.; Van Luenen, H.G.A.M.; Borst, P. Factors affecting the level and localization of the transferrin receptor in Trypanosoma brucei. J. Biol. Chem. 2004, 279, 40690–40698. [Google Scholar] [CrossRef] [PubMed]
- Tiengwe, C.; Bush, P.J.; Bangs, J.D. Controlling transferrin receptor trafficking with GPI-valence in bloodstream stage African trypanosomes. PLOS Pathog. 2017, 13, e1006366. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.C.; Mclatchie, A.P.; Kelly, J.M. Evidence that transport of iron from the lysosome to the cytosol in African trypanosomes is mediated by a mucolipin orthologue. Mol. Microbiol. 2013, 89, 420–432. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, K.J.; Peck, R.F.; Tazeh, N.N.; Bangs, J.D. GPI valence and the fate of secretory membrane proteins in African trypanosomes. J. Cell Sci. 2005, 118, 5499–5511. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.S.; Smith, E.; Langer, W.; Jacobs, L.A.; Goulding, D.; Field, M.C. Developmental variation in Rab11-dependent trafficking in Trypanosoma brucei. Eukaryot. Cell 2005, 4, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Raper, J.; Fung, R.; Ghiso, J.; Nussenzweig, V.; Tomlinson, S. Characterization of a Novel Trypanosome Lytic Factor from Human Serum Characterization of a Novel Trypanosome Lytic Factor from Human Serum. Infect. Immun. 1999, 67, 1910–1916. [Google Scholar] [PubMed]
- Tomlinson, S.; Jansen, A.; Koudinov, A.; Ghiso, J.A.; Rifkin, M.R.; Ohtaki, S.; Nussenzweig, V. High-density-lipoprotein-independent killing of Trypanosoma brucei by human serum. Mol. Biochem. Parasitol. 1995, 70, 131–138. [Google Scholar] [CrossRef]
- Stephens, N.A.; Hajduk, S.L. Endosomal localization of the serum resistance-associated protein in African trypanosomes confers human infectivity. Eukaryot. Cell 2011, 10, 1023–1033. [Google Scholar] [CrossRef] [PubMed]
- Xong, H.; Vanhamme, L.; Chamekh, M.; Chimfwembe, C.; Van Den Abbeele, J.; Pays, A.; Van Meirvenne, N.; Hamers, R.; De Baetselier, P.; Pays, E. A VSG expression site-associated gene confers resistance to human serum in Trypanosoma rhodesiense. Cell 1998, 95, 839–846. [Google Scholar] [CrossRef]
- Vanhollebeke, B.; De Muylder, G.; Nielsen, M.J.; Pays, A.; Tebabi, P.; Dieu, M.; Raes, M.; Moestrup, S.K.; Pays, E. A Haptoglobin-Hemoglobin Receptor Conveys Innate Immunity to Trypanosoma brucei in Humans. Science 2008, 320, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Currier, R.B.; Cooper, A.; Burrell-Saward, H.; MacLeod, A.; Alsford, S. Decoding the network of Trypanosoma brucei proteins that determines sensitivity to apolipoprotein-L1. PLoS Pathog. 2018, 14, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Peck, R.F.; Shiflett, A.M.; Schwartz, K.J.; McCann, A.; Hajduk, S.L.; Bangs, J.D. The LAMP-like protein p67 plays an essential role in the lysosome of African trypanosomes. Mol. Microbiol. 2008, 68, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.L.; Schwartz, K.J.; Balber, A.E.; Bangs, J.D. Developmentally regulated trafficking of the lysosomal membrane protein p67 in Trypanosoma brucei. J. Cell Sci. 2002, 115, 3253–3263. [Google Scholar] [PubMed]
- Hicke, L.; Dunn, R. Regulation of Membrane Protein Transport by Ubiquitin and Ubiquitin-Binding Proteins. Annu. Rev. Cell Dev. Biol. 2003, 19, 141–172. [Google Scholar] [CrossRef] [PubMed]
- Haglund, K.; Dikic, I. The role of ubiquitylation in receptor endocytosis and endosomal sorting. J. Cell Sci. 2012, 125, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; Alsford, S.; Horn, D. Genome-wide RNAi screens in African trypanosomes identify the nifurtimox activator NTR and the eflornithine transporter AAT6. Mol. Biochem. Parasitol. 2011, 176, 55–57. [Google Scholar] [CrossRef] [PubMed]
- Field, M.C.; Horn, D.; Fairlamb, A.H.; Ferguson, M.A.J.; Gray, D.W.; Read, K.D.; De Rycker, M.; Torrie, L.S.; Wyatt, P.G.; Wyllie, S.; et al. Anti-trypanosomatid drug discovery: An ongoing challenge and a continuing need. Nat. Rev. Microbiol. 2017, 15, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; Glover, L.; Munday, J.C.; Aguinaga Andrés, D.; Barrett, M.P.; de Koning, H.P.; Horn, D. Aquaglyceroporin 2 controls susceptibility to melarsoprol and pentamidine in African trypanosomes. Proc. Natl. Acad. Sci. USA 2012, 109, 10996–11001. [Google Scholar] [CrossRef] [PubMed]
- Munday, J.C.; Settimo, L.; de Koning, H.P. Transport proteins determine drug sensitivity and resistance in a protozoan parasite, Trypanosoma brucei. Front. Pharmacol. 2015, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- De Koning, H.P. Uptake of pentamidine in Trypanosoma brucei brucei is mediated by three distinct transporters: Implications for cross-resistance with arsenicals. Mol. Pharmacol. 2001, 59, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.S.; Macêdo, J.P.; Steinmann, M.E.; Salgado, A.G.; Bütikofer, P.; Sigel, E.; Rentsch, D.; Mäser, P. Transporters of Trypanosoma brucei—phylogeny, physiology, pharmacology. FEBS J. 2018, 285, 1012–1023. [Google Scholar] [CrossRef] [PubMed]
- Vincent, I.M.; Creek, D.; Watson, D.G.; Kamleh, M.A.; Woods, D.J.; Wong, P.E.; Burchmore, R.J.S.; Barrett, M.P. A molecular mechanism for eflornithine resistance in African trypanosomes. PLoS Pathog. 2010, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Macedo, J.P.; Currier, R.B.; Wirdnam, C.; Horn, D.; Alsford, S.; Rentsch, D. Ornithine uptake and the modulation of drug sensitivity in Trypanosoma brucei. FASEB J. 2017, 31, 4649–4660. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; de Koning, H.P.; Mäser, P.; Horn, D. Drug resistance in African trypanosomiasis: The melarsoprol and pentamidine story. Trends Parasitol. 2013, 29, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Graf, F.E.; Baker, N.; Munday, J.C.; de Koning, H.P.; Horn, D.; Mäser, P. Chimerization at the AQP2-AQP3 locus is the genetic basis of melarsoprol-pentamidine cross-resistance in clinical Trypanosoma brucei gambiense isolates. Int. J. Parasitol. Drugs Drug Resist. 2015, 5, 65–68. [Google Scholar] [CrossRef] [PubMed]
- Beitz, E. Aquaporins from pathogenic protozoan parasites: Structure, function and potential for chemotherapy. Biol. Cell 2005, 97, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S.; Anderson, M.O.; Papadopoulos, M.C. Aquaporins: Important but elusive drug targets. Nat. Rev. Drug Discov. 2014, 13, 259–277. [Google Scholar] [CrossRef] [PubMed]
- Montalvetti, A.; Rohloff, P.; Docampo, R.A. functional aquaporin co-localizes with the vacuolar proton pyrophosphatase to acidocalcisomes and the contractile vacuole complex of Trypanosoma cruzi. J. Biol. Chem. 2004, 279, 38673–38682. [Google Scholar] [CrossRef] [PubMed]
- Bassarak, B.; Uzcátegui, N.L.; Schönfeld, C.; Duszenko, M. Functional Characterization of Three Aquagly- ceroporins from Trypanosoma brucei in Osmoregulation and Glycerol Transport. Cell. Physiol. Biochem. 2011, 27, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Nejsum, L.N.; Zelenina, M.; Aperia, A.; Frøkiaer, J.; Nielsen, S. Bidirectional regulation of AQP2 trafficking and recycling: Involvement of AQP2-S256 phosphorylation. Am. J. Physiol. Renal Physiol. 2005, 288, F930–F938. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.J.; Matsuzaki, T.; Bouley, R.; Hasler, U.; Qin, Q.-H.; Brown, D. The phosphorylation state of serine 256 is dominant over that of serine 261 in the regulation of AQP2 trafficking in renal epithelial cells. Am. J. Physiol. Renal Physiol. 2008, 295, F290–F294. [Google Scholar] [CrossRef] [PubMed]
- Tamma, G.; Robben, J.H.; Trimpert, C.; Boone, M.; Deen, P.M.T. Regulation of AQP2 localization by S256 and S261 phosphorylation and ubiquitination. AJP Cell Physiol. 2011, 300, C636–C646. [Google Scholar] [CrossRef] [PubMed]
- Kamsteeg, E.J.; Hendriks, G.; Boone, M.; Konings, I.B.M.; Oorschot, V.; van der Sluijs, P.; Klumperman, J.; Deen, P.M.T. Short-chain ubiquitination mediates the regulated endocytosis of the aquaporin-2 water channel. Proc. Natl. Acad. Sci. USA 2006, 103. [Google Scholar] [CrossRef] [PubMed]
- Mollapour, M.; Piper, P.W. Hog1 Mitogen-Activated Protein Kinase Phosphorylation Targets the Yeast Fps1 Aquaglyceroporin for Endocytosis, Thereby Rendering Cells Resistant to Acetic Acid. Mol. Cell. Biol. 2007, 27, 6446–6456. [Google Scholar] [CrossRef] [PubMed]
- Mandal, G.; Sharma, M.; Kruse, M.; Sander-Juelch, C.; Munro, L.A.; Wang, Y.; Vilg, J.V.; Tamás, M.J.; Bhattacharjee, H.; Wiese, M.; Mukhopadhyay, R. Modulation of Leishmania major aquaglyceroporin activity by a mitogen-activated protein kinase. Mol. Microbiol. 2012, 85, 1204–1218. [Google Scholar] [CrossRef] [PubMed]
- Baker, N.; Hamilton, G.; Wilkes, J.M.; Hutchinson, S.; Barrett, M.P.; Horn, D. Vacuolar ATPase depletion affects mitochondrial ATPase function, kinetoplast dependency, and drug sensitivity in trypanosomes. Proc. Natl. Acad. Sci. USA 2015, 112, 9112–9117. [Google Scholar] [CrossRef] [PubMed]
- Lanteri, C.A.; Tidwell, R.R.; Meshnick, S.R. The mitochondrion is a site of trypanocidal action of the aromatic diamidine DB75 in bloodstream forms of Trypanosoma brucei. Antimicrob. Agents Chemother. 2008, 52, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Moreno, S.N.; Gadelha, F.R.; Docampo, R. Crystal violet as an uncoupler of oxidative phosphorylation in rat liver mitochondria. J. Biol. Chem. 1988, 263, 12493–12499. [Google Scholar] [PubMed]
- Bray, P.G.; Barrett, M.P.; Ward, S.A.; De Koning, H.P. Pentamidine uptake and resistance in pathogenic protozoa: Past, present and future. Trends Parasitol. 2003, 19, 232–239. [Google Scholar] [CrossRef]
- Stijlemans, B.; De Baetselier, P.; Caljon, G.; Van Den Abbeele, J.; Van Ginderachter, J.A.; Magez, S. Nanobodies as tools to understand, diagnose, and treat African trypanosomiasis. Front. Immunol. 2017, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Salcedo, J.A.; Unciti-Broceta, J.D.; Valverde-Pozo, J.; Soriano, M. New approaches to overcome transport related drug resistance in trypanosomatid parasites. Front. Pharmacol. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Unciti-Broceta, J.D.; Arias, J.L.; Maceira, J.; Soriano, M.; Ortiz-González, M.; Hernández-Quero, J.; Muñóz-Torres, M.; de Koning, H.P.; Magez, S.; Garcia-Salcedo, J.A. Specific Cell Targeting Therapy Bypasses Drug Resistance Mechanisms in African Trypanosomiasis. PLoS Pathog. 2015, 11, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.L.; Unciti-Broceta, J.D.; Maceira, J.; Del Castillo, T.; Hernández-Quero, J.; Magez, S.; Soriano, M.; García-Salcedo, J.A. Nanobody conjugated PLGA nanoparticles for active targeting of African Trypanosomiasis. J. Control. Release 2015, 197, 190–198. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quintana, J.F.; Del Pino, R.C.; Yamada, K.; Zhang, N.; Field, M.C. Adaptation and Therapeutic Exploitation of the Plasma Membrane of African Trypanosomes. Genes 2018, 9, 368. https://doi.org/10.3390/genes9070368
Quintana JF, Del Pino RC, Yamada K, Zhang N, Field MC. Adaptation and Therapeutic Exploitation of the Plasma Membrane of African Trypanosomes. Genes. 2018; 9(7):368. https://doi.org/10.3390/genes9070368
Chicago/Turabian StyleQuintana, Juan F., Ricardo Canavate Del Pino, Kayo Yamada, Ning Zhang, and Mark C. Field. 2018. "Adaptation and Therapeutic Exploitation of the Plasma Membrane of African Trypanosomes" Genes 9, no. 7: 368. https://doi.org/10.3390/genes9070368
APA StyleQuintana, J. F., Del Pino, R. C., Yamada, K., Zhang, N., & Field, M. C. (2018). Adaptation and Therapeutic Exploitation of the Plasma Membrane of African Trypanosomes. Genes, 9(7), 368. https://doi.org/10.3390/genes9070368