Insights into Avian Incomplete Dosage Compensation: Sex-Biased Gene Expression Coevolves with Sex Chromosome Degeneration in the Common Whitethroat

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Species and Sequencing
2.2. DNA Analyses
2.2.1. Extracting Gametolog Sequences
2.2.2. Gametolog Sequence Alignment and Processing
2.3. Gene Expression Analyses
2.4. Statistics
3. Results
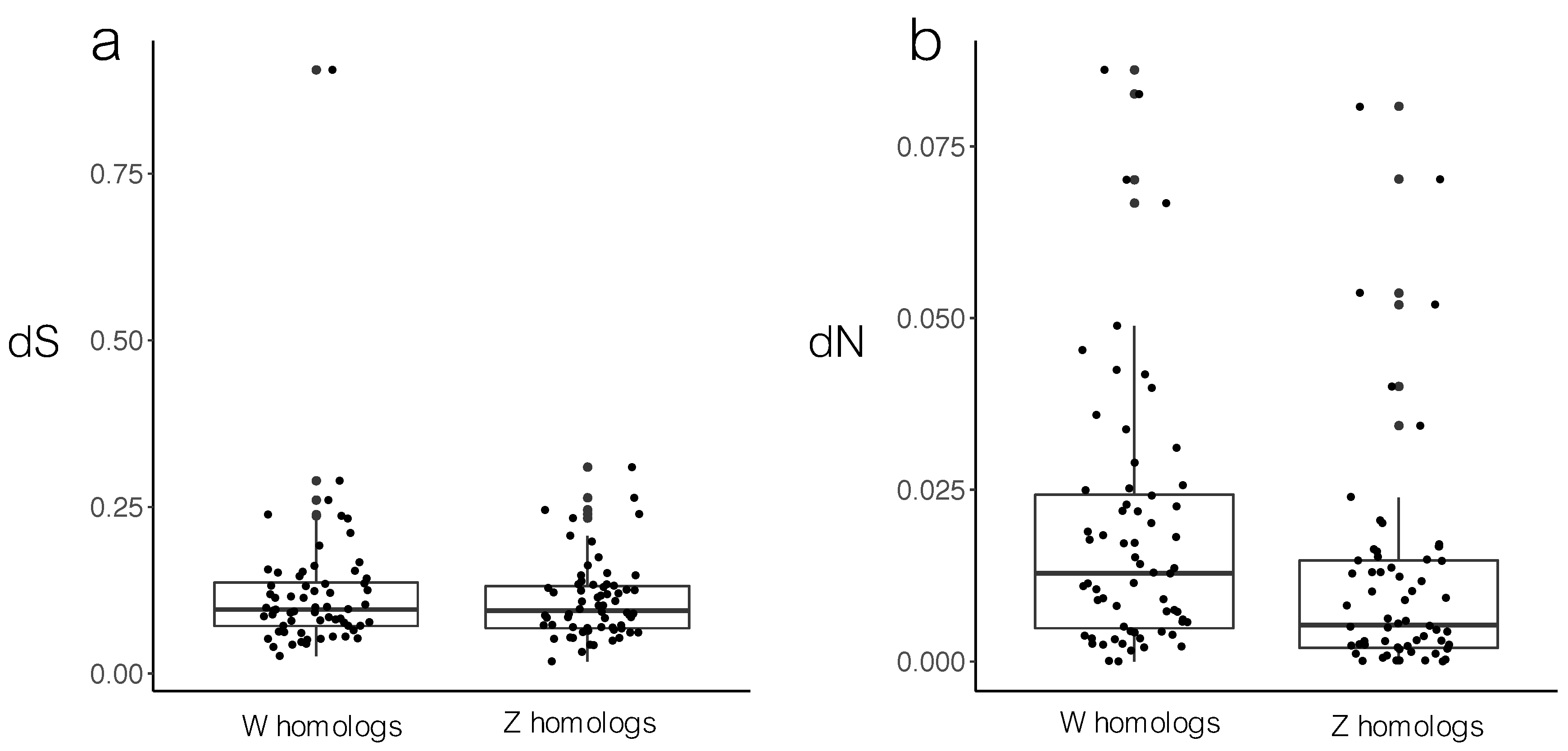
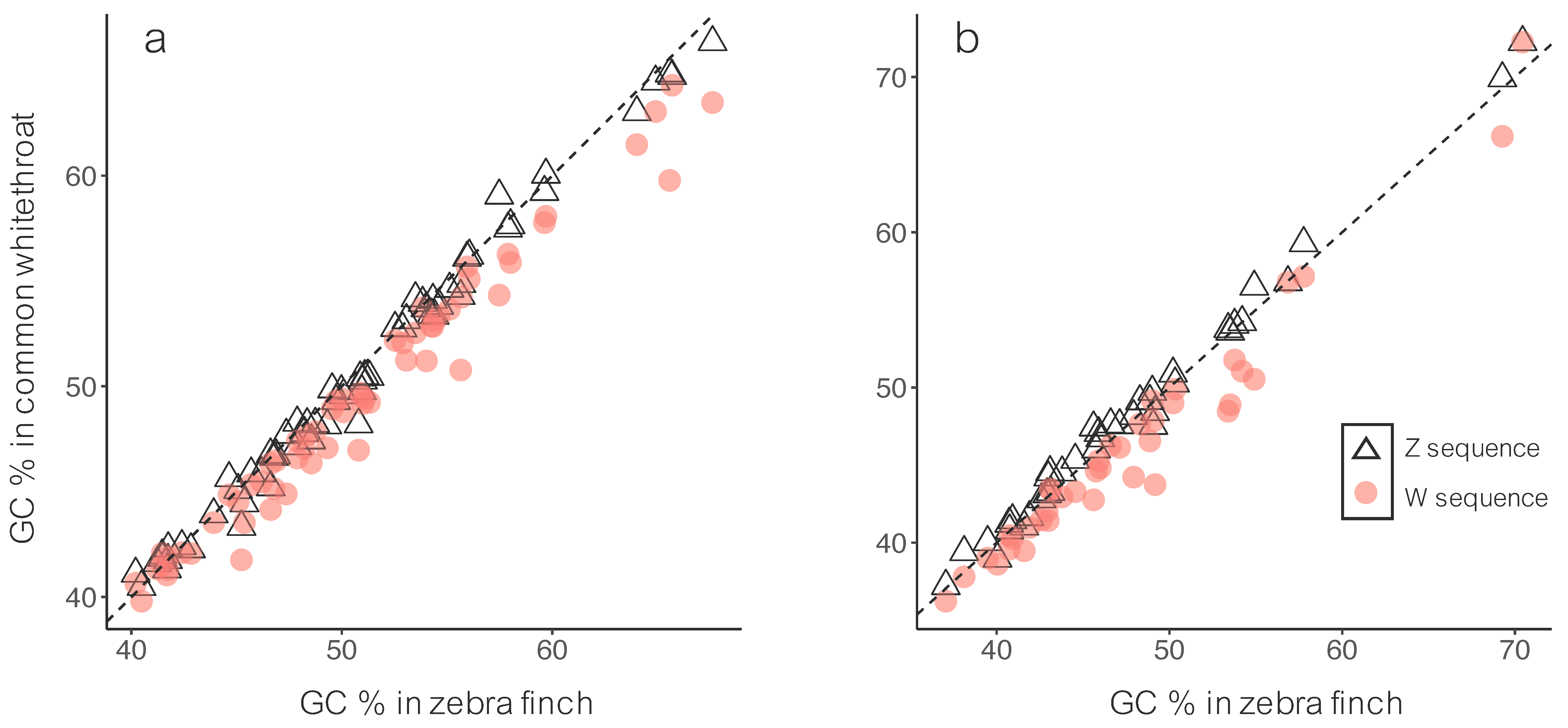
3.1. Sequence Divergence
3.2. Gene Expression
4. Discussion
4.1. Sequence Divergence
4.2. Gene Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ohno, S. Sex Chromosomes and Sex-Linked Genes; Springer: Berlin, Germany, 1967. [Google Scholar]
- Livernois, A.M.; Graves, J.A.M.; Waters, P.D. The origin and evolution of vertebrate sex chromosomes and dosage compensation. Heredity 2012, 108, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Cortez, D.; Marin, R.; Toledo-Flores, D.; Froidevaux, L.; Liechti, A.; Waters, P.D.; Grutzner, F.; Kaessmann, H. Origins and functional evolution of Y chromosomes across mammals. Nature 2014, 508, 488–493. [Google Scholar] [CrossRef] [PubMed]
- Beukeboom, L.W.; Perrin, N. The Evolution of Sex Determination; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Charlesworth, B. The evolution of sex chromosomes. Science 1991, 251, 1030–1033. [Google Scholar] [CrossRef] [PubMed]
- Bachtrog, D. Y-chromosome evolution: Emerging insights into processes of Y-chromosome degeneration. Nat. Rev. Genet. 2013, 14, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Abbott, J.K.; Norden, A.K.; Hansson, B. Sex chromosome evolution: Historical insights and future perspectives. Proc. R. Soc. Lond. B 2017, 284, 20162806. [Google Scholar] [CrossRef] [PubMed]
- Rice, W.R. Sex chromosomes and the evolution of sexual dimorphism. Evolution 1984, 38, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Rice, W.R. Genetic hitchhiking and the evolution of reduced genetic activity of the Y sex chromosome. Genetics 1987, 116, 161–167. [Google Scholar] [PubMed]
- Ponnikas, S.; Sigeman, H.; Abbott, J.; Hansson, B. Why do sex chromosomes stop recombining? Trends Genet. 2018, 34, 492–503. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Charlesworth, D. The degeneration of Y chromosomes. Phil. Trans. R. Soc. Lond. B 2000, 355, 1563–1572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlesworth, D.; Charlesworth, B.; Marais, G. Steps in the evolution of heteromorphic sex chromosomes. Heredity 2005, 95, 118–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergero, R.; Charlesworth, D. The evolution of restricted recombination in sex chromosomes. Trends Ecol. Evol. 2009, 24, 94–102. [Google Scholar] [CrossRef] [PubMed]
- Smeds, L.; Kawakami, T.; Burri, R.; Bolivar, P.; Husby, A.; Qvarnstrom, A.; Uebbing, S.; Ellegren, H. Genomic identification and characterization of the pseudoautosomal region in highly differentiated avian sex chromosomes. Nat. Commun. 2014, 5, 5448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.E.; Moghadam, H.K.; Mank, J.E. Trade-off between selection for dosage compensation and masculinization on the avian Z chromosome. Genetics 2012, 192, 1433–1445. [Google Scholar] [CrossRef] [PubMed]
- Bellott, D.W.; Hughes, J.F.; Skaletsky, H.; Brown, L.G.; Pyntikova, T.; Cho, T.J.; Koutseva, N.; Zaghlul, S.; Graves, T.; Rock, S.; et al. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 2014, 508, 494–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeds, L.; Warmuth, V.; Bolivar, P.; Uebbing, S.; Burri, R.; Suh, A.; Nater, A.; Bures, S.; Garamszegi, L.Z.; Hogner, S.; et al. Evolutionary analysis of the female-specific avian W chromosome. Nat. Commun. 2015, 6, 7330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Julien, P.; Brawand, D.; Soumillon, M.; Necsulea, A.; Liechti, A.; Schutz, F.; Daish, T.; Grutzner, F.; Kaessmann, H. Mechanisms and evolutionary patterns of mammalian and avian dosage compensation. PLoS Biol. 2012, 10, e1001328. [Google Scholar] [CrossRef] [PubMed]
- Graves, J.A.M. Evolution of vertebrate sex chromosomes and dosage compensation. Nat. Rev. Genet. 2016, 17, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Lindsley, D.L.; Sandler, L.; Jacobs, P.A.; Nozawa, H.; Parry, D.M.; Carpente, A.T.; Baker, B.S.; Denell, R.E.; Hessler, A.; Davis, B.K.; et al. Segmental aneuploidy and genetic gross structure of Drosophila genome. Genetics 1972, 71, 157–184. [Google Scholar] [PubMed]
- Rosenbusch, B. The incidence of aneuploidy in human oocytes assessed by conventional cytogenetic analysis. Hereditas 2004, 141, 97–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birchler, J.A.; Fernandez, H.R.; Kavi, H.H. Commonalities in compensation. BioEssays 2006, 28, 565–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, V.; Parisi, M.; Sturgill, D.; Nuttall, R.; Doctolero, M.; Dudko, O.K.; Malley, J.D.; Eastman, P.S.; Oliver, B. Global analysis of X-chromosome dosage compensation. J. Biol. 2006, 5, 3. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M. The evolution of genetic networks by non-adaptive processes. Nat. Rev. Genet. 2007, 8, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, J.C.; Kelly, W.G.; Parming, B. Chromatin remodeling in dosage compensation. Annu. Rev. Genet. 2005, 39, 615–651. [Google Scholar] [CrossRef] [PubMed]
- Straub, T.; Becker, P.B. Dosage compensation: The beginning and end of generalization. Nat. Rev. Genet. 2007, 8, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Mank, J.E.; Hosken, D.J.; Wedell, N. Some inconvenient truths about sex chromosome dosage compensation and the potential role of sexual conflict. Evolution 2011, 65, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Melamed, E.; Arnold, A.P. Regional differences in dosage compensation on the chicken Z chromosome. Genome Biol. 2007, 8, R202. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H.; Hultin-Rosenberg, L.; Brunstrom, B.; Dencker, L.; Kultima, K.; Scholz, B. Faced with inequality: Chicken do not have a general dosage compensation of sex-linked genes. BMC Biol. 2007, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Melamed, E.; Yang, X.; Kampf, K.; Wang, S.; Yehya, N.; Van Nas, A.; Replogle, K.; Band, M.; Clayton, D.; et al. Dosage compensation is less effective in birds than in mammals. J. Biol. 2007, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Deakin, J.E.; Hore, T.A.; Koina, E.; Marshall Graves, J.A. The status of dosage compensation in the multiple X chromosomes of the platypus. PLoS Genet. 2008, 4, e1000140. [Google Scholar] [CrossRef] [PubMed]
- Itoh, Y.; Replogle, K.; Kim, Y.H.; Wade, J.; Clayton, D.F.; Arnold, A.P. Sex bias and dosage compensation in the zebra finch versus chicken genomes: General and specialized patterns among birds. Genome Res. 2010, 20, 512–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, J.B.; Bryk, J. General lack of global dosage compensation in ZZ/ZW systems? Broadening the perspective with RNA-seq. BMC Genom. 2011, 12, 91. [Google Scholar] [CrossRef] [PubMed]
- Leder, E.H.; Cano, J.M.; Leinonen, T.; O’Hara, R.B.; Nikinmaa, M.; Primmer, C.R.; Merila, J. Female-biased expression on the X chromosome as a key step in sex chromosome evolution in threespine sticklebacks. Mol. Biol. Evol. 2010, 27, 1495–1503. [Google Scholar] [CrossRef] [PubMed]
- Naurin, S.; Hasselquist, D.; Bensch, S.; Hansson, B. Sex-biased gene expression on the avian Z chromosome: Highly expressed genes show higher male-biased expression. PLoS ONE 2012, 7, e46854. [Google Scholar] [CrossRef] [PubMed]
- Adolfsson, S.; Ellegren, H. Lack of dosage compensation accompanies the arrested stage of sex chromosome evolution in ostriches. Mol. Biol. Evol. 2013, 30, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Vicoso, B.; Emerson, J.J.; Zektser, Y.; Mahajan, S.; Bachtrog, D. Comparative sex chromosome genomics in snakes: Differentiation, evolutionary strata, and lack of global dosage compensation. PLoS Biol. 2013, 11, e1001643. [Google Scholar] [CrossRef] [PubMed]
- Vicoso, B.; Kaiser, V.B.; Bachtrog, D. Sex-biased gene expression at homomorphic sex chromosomes in emus and its implication for sex chromosome evolution. Proc. Natl. Acad. Sci. USA 2013, 110, 6453–6458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naurin, S.; Hansson, B.; Hasselquist, D.; Kim, Y.H.; Bensch, S. The sex-biased brain: Sexual dimorphism in gene-expression in two species of songbirds. BMC Genom. 2011, 12, 37. [Google Scholar] [CrossRef] [PubMed]
- Mank, J.E.; Ellegren, H. All dosage compensation is local: Gene-by-gene regulation of sex-biased expression on the chicken Z chromosome. Heredity 2009, 102, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Mank, J.E. The W, X, Y and Z of sex-chromosome dosage compensation. Trends Genet. 2009, 25, 226–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pala, I.; Naurin, S.; Stervander, M.; Hasselquist, D.; Bensch, S.; Hansson, B. Evidence of a neo-sex chromosome in birds. Heredity 2012, 108, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Pala, I.; Hasselquist, D.; Bensch, S.; Hansson, B. Patterns of molecular evolution of an avian neo-sex chromosome. Mol. Biol. Evol. 2012, 29, 3741–3754. [Google Scholar] [CrossRef] [PubMed]
- Moyle, R.G.; Oliveros, C.H.; Andersen, M.J.; Hosner, P.A.; Benz, B.W.; Manthey, J.D.; Travers, S.L.; Brown, R.M.; Faircloth, B.C. Tectonic collision and uplift of Wallacea triggered the global songbird radiation. Nat. Commun. 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Meunier, J.; Duret, L. Recombination drives the evolution of GC-content in the human genome. Mol. Biol. Evol. 2004, 21, 984–990. [Google Scholar] [CrossRef] [PubMed]
- del Hoyo, J.; Elliott, A.; Chren, D.A. Handbook of the Birds of the World, Volume 15: Weavers to New World Warblers; Lynx Edicions: Barcelona, Spain, 2010. [Google Scholar]
- Cramp, S. Handbook of the Birds of Europe, the Middle East and north Africa; Oxford University Press: Oxford, UK, 1992. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Naurin, S.; Bensch, S.; Hansson, B.; Johansson, T.; Clayton, D.F.; Albrekt, A.S.; von Schantz, T.; Hasselquist, D. A microarray for large-scale genomic and transcriptional analyses of the zebra finch (Taeniopygia guttata) and other passerines. Mol. Ecol. Res. 2008, 8, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, F.; Amode, M.R.; Barrell, D.; Beal, K.; Billis, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fitzgerald, S.; et al. Ensembl 2015. Nucleic Acids Res. 2015, 43, D662–D669. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Proc, G.P.D. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Garrison, E.; Marth, G. Haplotype-based variant detection from short-read sequencing. arXiv, 2012; arXiv:1207.3907. [Google Scholar]
- Quinlan, A.R.; Hall, I.M. Bedtools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI reference sequences (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007, 35, D61–D65. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Punta, M.; Coggill, P.C.; Eberhardt, R.Y.; Mistry, J.; Tate, J.; Boursnell, C.; Pang, N.; Forslund, K.; Ceric, G.; Clements, J.; et al. The Pfam protein families database. Nucleic Acids Res. 2012, 40, D290–D301. [Google Scholar] [CrossRef] [PubMed]
- Loytynoja, A. Phylogeny-aware alignment with PRANK. Methods Mol. Biol. 2014, 1079, 155–170. [Google Scholar] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. Paml 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. Emboss: The european molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2014; Available online: http://www.R-project.org/ (accessed on 9 April 2018).
- Kawakami, T.; Smeds, L.; Backstrom, N.; Husby, A.; Qvarnstrom, A.; Mugal, C.F.; Olason, P.; Ellegren, H. A high-density linkage map enables a second-generation collared flycatcher genome assembly and reveals the patterns of avian recombination rate variation and chromosomal evolution. Mol. Ecol. 2014, 23, 4035–4058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smeds, L.; Mugal, C.F.; Qvarnstrom, A.; Ellegren, H. High-resolution mapping of crossover and non-crossover recombination events by whole-genome re-sequencing of an avian pedigree. PLoS Genet. 2016, 12, e1006044. [Google Scholar] [CrossRef] [PubMed]
- Naurin, S.; Hansson, B.; Bensch, S.; Hasselquist, D. Why does dosage compensation differ between XY and ZW taxa? Trends Genet. 2010, 26, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Rice, W.R. The accumulation of sexually antagonistic genes as a selective agent promoting the evolution of reduced recombination between primitive sex chromosomes. Evolution 1987, 41, 911–914. [Google Scholar] [CrossRef] [PubMed]
- Ellegren, H.; Fridolfsson, A.K. Male-driven evolution of DNA sequences in birds. Nat. Genet. 1997, 17, 182–184. [Google Scholar] [CrossRef] [PubMed]
- Makova, K.D.; Li, W.H. Strong male-driven evolution of DNA sequences in humans and apes. Nature 2002, 416, 624–626. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A.J. Intra-sexual selection in Drosophila. Heredity 1948, 2, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Trivers, R.L. Parental investment and sexual selection. In Sexual Selection and the Descent of Man; Campbell, B., Ed.; Aldine: Chicago, IL, USA, 1972; pp. 136–179. [Google Scholar]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sigeman, H.; Ponnikas, S.; Videvall, E.; Zhang, H.; Chauhan, P.; Naurin, S.; Hansson, B. Insights into Avian Incomplete Dosage Compensation: Sex-Biased Gene Expression Coevolves with Sex Chromosome Degeneration in the Common Whitethroat. Genes 2018, 9, 373. https://doi.org/10.3390/genes9080373
Sigeman H, Ponnikas S, Videvall E, Zhang H, Chauhan P, Naurin S, Hansson B. Insights into Avian Incomplete Dosage Compensation: Sex-Biased Gene Expression Coevolves with Sex Chromosome Degeneration in the Common Whitethroat. Genes. 2018; 9(8):373. https://doi.org/10.3390/genes9080373
Chicago/Turabian StyleSigeman, Hanna, Suvi Ponnikas, Elin Videvall, Hongkai Zhang, Pallavi Chauhan, Sara Naurin, and Bengt Hansson. 2018. "Insights into Avian Incomplete Dosage Compensation: Sex-Biased Gene Expression Coevolves with Sex Chromosome Degeneration in the Common Whitethroat" Genes 9, no. 8: 373. https://doi.org/10.3390/genes9080373