Iron-Rich Nanoparticles in Natural Aquatic Environments



1
Department of Earth Sciences, Stellenbosch University, Stellenbosch 7600, South Africa
2
Department of Geosciences, Princeton University, Princeton, NJ 08544, USA
*
Author to whom correspondence should be addressed.
Minerals 2019, 9(5), 287; https://doi.org/10.3390/min9050287
Submission received: 8 April 2019
/
Revised: 26 April 2019
/
Accepted: 27 April 2019
/
Published: 11 May 2019
(This article belongs to the Special Issue Nanomineralogy)
Abstract
:Naturally-occurring iron nanoparticles constitute a quantitatively-important and biogeochemically-active component of the broader Earth ecosystem. Yet detailed insights into their chemical speciation is sparse compared to the body of work conducted on engineered Fe nanoparticles. The present contribution briefly reviews the analytical approaches that can be used to characterize natural Fe nanoparticles, before detailing a dedicated synchrotron-based X-ray spectro-microscopic investigation into the speciation of suspended Fe nanoparticles collected from fluvial, marine, and lacustrine surface waters. Ferrous, ferric and magnetite classes of Fe nanoparticles (10–100 nm) were identified, and all three classes exhibited a high degree of heterogeneity in the local bonding environment around the Fe center. The heterogeneity is attributed to the possible presence of nanoparticle aggregates, and to the low degrees of crystallinity and ubiquitous presence of impurities (Al and organic moieties) in natural samples. This heterogeneity further precludes a spectroscopic distinction between the Fe nanoparticles and the larger sized Fe-rich particles that were evaluated. The presented results provide an important baseline for natural nanoparticle speciation in pristine aquatic systems, highlight the degree of inter-particle variability, which should be parameterized in future accurate biogeochemical models, and may inform predictions of the fate of released engineered Fe nanoparticles as they evolve and transform in natural systems.
1. Introduction
Iron (Fe) is the most abundant transition metal in the Earth’s crust, with important and biogeochemically-active repositories existing in all four of the Earth’s major subsystems (viz. lithosphere, hydrosphere, atmosphere and biosphere). In these natural environments iron particles exist over a broad range of sizes, however with the recent advent of the so-called “nanotechnology revolution”, it is the smallest size classes (i.e., nanoparticles defined as having one dimension <100 nm; e.g., [1]) that are receiving special scientific and industrial attention. Two key reasons for this gain in prominence include: (1) On account of their high surface area to size ratio and because of quantum effects, nanoparticles commonly exhibit differing chemical behavior to their bulk analogues; and (2) technological advancement and human activity is rampantly increasing the concentrations of incidental and engineered nanoparticles released into the natural environment (e.g., References [2,3,4]). Iron nanoparticles are of particular interest since their surface chemistry renders them as important transporting agents for nutrients and pollutants in natural systems [5,6], their reactivity has been exploited for environmental and hazardous waste site remediation (notably engineered Fe0 nanoparticles [7]), and they may be actively cycled in biogeochemical systems where Fe is an essential nutrient for driving primary productivity (which in turn influences atmospheric CO2 levels) [8,9].
Despite their ubiquity and importance, natural Fe nanoparticles have received relatively limited scientific attention relative to the large body of work that focusses on the synthesis, behavior, stability, toxicity, and fate of engineered Fe nanoparticles (e.g., References [7,10,11]). A recent review presented by Sharma and coworkers [12] highlights that, relative to engineered nanoparticles, environmental inorganic nanoparticles may differ in both their biogeochemical behavior and their respective ecotoxicities. For this to be validated however, a greater body of work focused specifically on identifying the speciation of natural Fe nanoparticles is required. Examples of previous work include Neubauer et al. [13] who use Transmission Electron Microscopy (TEM) to investigate soil Fe nanomineralogy and its association with ambient heavy metals; and Carbone et al. [14] who use Electron Paramagnetic Resonance (EPR) spectroscopy to investigate incidental Fe nanoparticles formed in an Acid Mine Drainage environment. The present contribution seeks to augment the existing body of work by (1) briefly reviewing the prominent analytical techniques capable of evaluating natural Fe nanoparticles; and (2) applying a newly-developed synchrotron-based soft X-ray spectromicroscopy approach towards evaluating natural Fe nanoparticles and nanoparticle aggregates collected from a selection of fluvial, marine, and lacustrine aquatic environments. The approach exploits the chemical information contained within the Fe L2,3-edge X-ray Absorption Near-Edge Structure (XANES) spectra (pertaining to the local bonding environment around the Fe metal center [15]) and is augmented with additional information collected at the Aluminum K-edge and at the Carbon K-edge. It is foreseen that the new insights derived from this novel study approach will add meaningfully to our understanding of Fe nanoparticle speciation and biogeochemical behavior in natural systems. This, in turn, may better inform the development of future generations of biogeochemical models (e.g., Reference [16]) and may assist in predictions related to the fate of engineered Fe0 nanoparticles, which readily experience surface oxidation in the natural environment.
2. Methods for Evaluating Fe Nanomineralogy
The current section diverges from a traditional “Methods” section in that it includes a brief but pertinent review of a range of analytical methodologies that can be and have been used to study natural Fe nanominerals and nanoparticles. This review is augmented with a detailed description of the Fe L-edge XANES research protocol that we have developed specifically for evaluating the chemistry, valence speciation and local coordination in natural Fe nanoparticles. For a more thorough assessment of the science underpinning this analytical technique (i.e., X-ray interactions with Fe metal centers), the reader is referred to Reference [15].
2.1. Review of Current Methods of Studying Fe Nanominerals
Iron nanoparticles have a complex composition and structure in nature, thus a diverse suit of methods is required to analyze them. Due to their effects on nanoparticle reactivity, the main analytical parameters of interest include size, shape, size distribution, aggregation, surface area, crystal structure, surface chemistry, chemical composition, speciation, and coordination. The methods that are commonly employed therefore are primarily for microscopy and imaging, for physico-chemical characterization or for a combination of both. A number of instrumental techniques (XRD, BET, NMR, SEM-EDS, AFM, SEM, TEM, FTIR, and 3-D Tomography; Table 1) are now readily available in common laboratories that together can characterize a large set of properties of interest rather quickly and economically.
While hard to access, in recent years there is an increased use of synchrotron-based XRD and spectroscopy techniques to study Fe-nanoparticles. Some advantages of synchrotron techniques include a need for only small amounts of sample material, no need for sample homogenization or excessive preparation, the option to selectively target specific and sub-micrometer scale areas of interest, and in some cases, analyses can be carried out on wet samples at room temperature and pressure. These X-ray based techniques (e.g., EXAFS, XANES, STXM, and XPS; Table 1) encompass a wide range of approaches that measure the X-rays or particles emitted during electronic transition caused by Fe-nanoparticles exposed to high intensity energy beams. The techniques provide element specific data and offer information on chemical state of iron species, interatomic distances, electronic structure, oxidation states, and ligand bonding. For example, micro-focused synchrotron XRD has been used to understand the formation condition or role of surface reactions among iron minerals found in biofilms or Fe-rich nodules [18,19]. More recently and using TEM coupled with L-edge X-ray Absorption Spectroscopy (XAS), Hirst et al. [32] evaluated nanoparticles from a boreal river system and found that the Fe(III)-rich nanoparticles comprised predominantly ferrihydrite aggregates, with minor goethite and hematite also observed. A detailed discussion and application of analytical techniques to study Fe-nanoparticles is beyond the scope of this article and readers are referred to recent published reviews [36,37].
2.2. Fe L-Edge X-ray Absorption Spectroscopy as a Powerful Tool for Investigating Fe Nanomineralogy in Natural Systems
Synchrotron X-ray spectromicroscopy at the Fe L2,3-absorption edge is well suited towards imaging and evaluating the chemical and mineralogical speciation of natural Fe particles. The technique is capable of providing spatial resolutions as small as 10 nm, and spectral resolutions on the order of 0.1 eV and smaller. Importantly, the Fe L-edge (~700 eV) falls within the soft X-ray range and thus provides complementary information to the Fe K-edge (~7100 eV) but without the significant beam damage artifacts that may be caused to natural samples by the more energetic X-rays. Soft X-ray spectroscopy is also favored over hard X-rays because of better resolution of fine structure, higher absorption cross section, and lower intrinsic lifetime broadening (e.g., Reference [38]). As the Fe L-edge probes chemical information related to the local coordination around the metal center, its application is not limited to crystalline mineral phases and it is thus our preferred analytical approach when evaluating natural nanoparticles, which are commonly amorphous or poorly crystalline (e.g., Reference [34]).
Data presented in this submission were collected using Scanning Transmission X-ray Microscopy (STXM) at the Molecular Environmental Sciences (MES) beamline 11.0.2 at the Advanced Light Source, Lawrence Berkeley National Laboratory, CA, USA [39]. This soft X-ray beamline is especially designed to ensure that natural particles can be investigated in their pristine geochemical state under ambient physicochemical conditions (i.e., without need for excessive sample drying which induces shrinkage and loss of water from chemically-active hydration shells). X-ray absorption near-edge structure (XANES) spectra were collected in transmission mode in a 1 atm. He environment at room temperature. XANES spectra were collected primarily at the Fe L3-edge (703–715 eV), with supporting data related to elemental associations and substitutions collected at the C K-edge (283–300 eV) and the Al K-edge (1563–1572 eV). The spectral and spatial resolutions were 0.2 eV and 12 nm, respectively, and were achieved using a 17 nm zone plate, a 1200 L/mm grating and 25 mm exit slitsas key parameters in the experimental end-station. The Fe nanoparticles evaluated in this study were sampled from various Southern African marine and fluvial water masses, and the details of sample collection and preparation are described elsewhere [33,34,40].
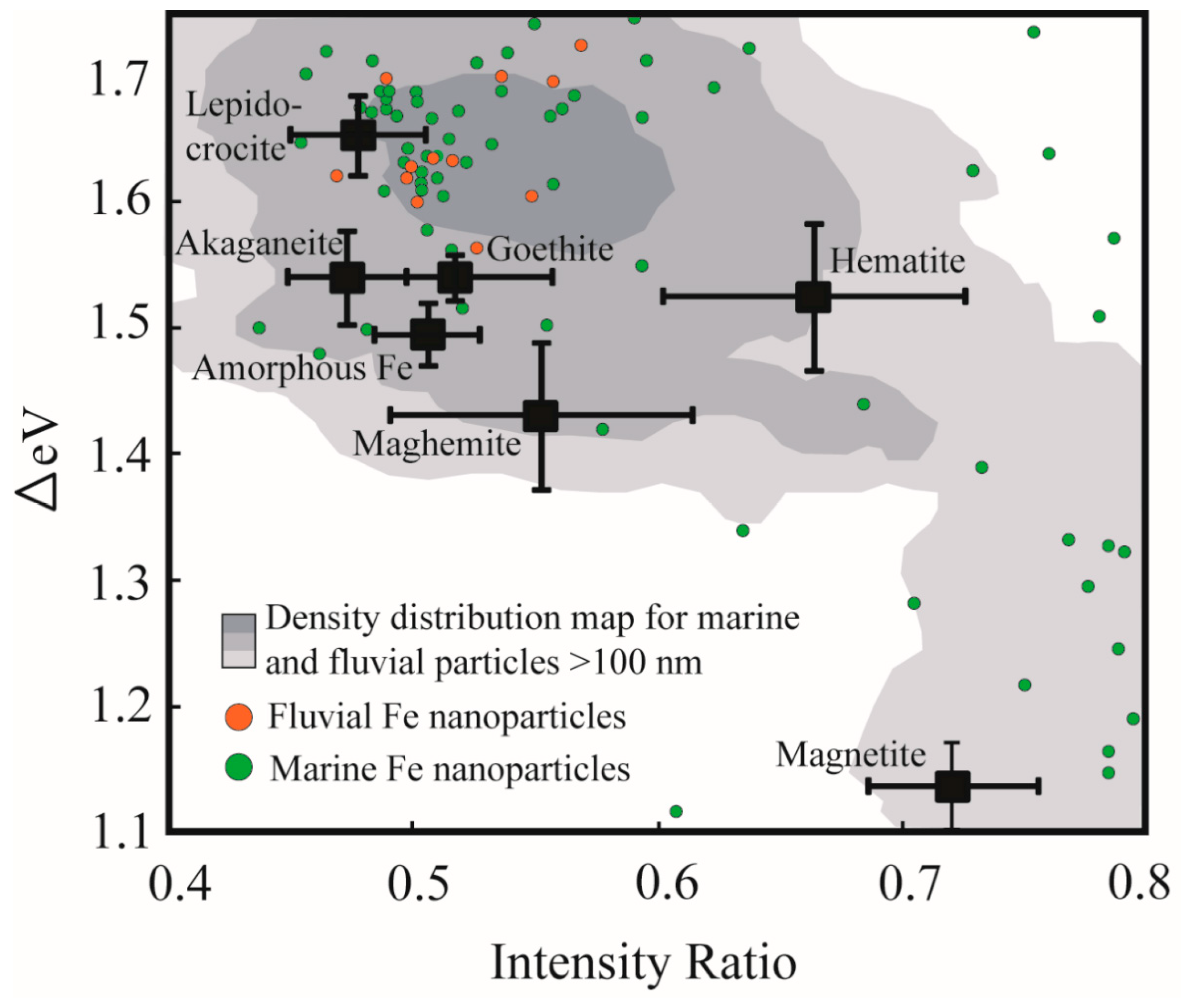
The Fe L2,3 X-ray absorption edge contains a wealth of information related to the mineral structure and chemistry of Fe containing minerals. A host of earlier workers have used Fe L-edge spectroscopy to identify the valence state of iron in mineral structures [41,42,43], to identify the structural coordination of Fe in nano-particulate-ferrihydrite [44], and to probe Fe speciation in samples collected from natural biogeochemical systems [45,46,47,48]. A recent systematic study into the Fe L-edge spectral response to Fe mineralogy has highlighted how the split peaks of the Fe L3-edge (Figure 1) can be used to identify Fe mineral speciation [15]. Briefly, this peak splitting arises largely because of the effects of ligand field splitting which results in the Fe 3d orbitals separating into t2g and eg subsets occurring at different energy positions [15,38,49]. The peak splitting can be parameterized according to the energy difference between the two peaks (ΔeV) and according to the ratio of the respective peaks’ intensities (Figure 1B). These two parameters vary in response to Fe mineralogy (i.e., factors inherent to mineral structure such as Fe valence state, coordination number, strength of the ligand field, levels of distortion to the local Fe polyhedron, nature of ligand bonding and the composition of the resultant molecular orbitals [15]), and a two parameter ΔeV versus intensity ratio plot can thus be used to identify Fe mineral speciation (Figure 2: black squares indicating standard Fe-oxide and Fe-oxyhydroxide spectral signatures). The two parameter plot has previously been used to investigate sub-micrometer sized environmental particles from glacial [50], lacustrine [40], marine [33], and fluvial systems [34], and the present study seeks to extend on this body of research by utilizing the spectroscopic tool specifically to evaluate environmental particles existing in the nano size-domain (i.e., having at least one size dimension <100 nm).
3. Results
The complete dataset includes four fluvial nanoparticles, ten coastal marine nanoparticles, and 122 open ocean nanoparticles. Of these particles, the majority comprises Fe in its ferric form although three ferrous nanoparticles were also identified. Carbon K-edge XANES confirmed an association between these ferrous nanoparticles and organic carbon [40], suggesting that organic functional groupssuch as carboxamidemay play a role in stabilizing reduced forms of Fe in oxic water masses. Additionally, at one of the open ocean marine sampling sites (located in sub-Antarctica frontal zone of the Southern Ocean) the nanoparticle mineralogy predominantly matched that of magnetite (Figure 3A). Throughout the samples evaluated, the nanoparticle morphology was generally rounded, although several examples (e.g., Figure 3B,D—particle corresponding to spectrum F) were elongate and classified as nanoparticles because of a single dimension being in the <100 nm size range.
Figure 2 provides an overview of the spectral signatures of the nanoparticles identified in this study (green- and orange point data), and compares these data to larger µm-scale natural Fe particles (grey shading) and standard Fe oxides and Fe oxy-hydroxides [15]. Most of the data points fall outside of the fields for the standard Fe mineral phases, alluding to the fact that most of these evaluated natural nanoparticles are aggregates, are amorphous in nature or contain ionic substituents that render their chemistry impure relative to the standards. The data points are relatively scattered on the ΔeV versus intensity ratio diagram, although there exists some clustering near to the position of the lepidocrocite standard. This clustering agrees well with the density distribution of larger (>100 nm) natural Fe-rich particles indicating a degree of similarity in mineral chemistry between the two datasets. This clustering occurs at high ΔeV values (between 1.6 and 1.7) which suggests that the natural Fe nanoparticles predominantly comprise Fe(III) in octahedral coordination, experience a relatively high degree of ligand field splitting, and may be characterized by distorted (larger volume) local Fe coordination polyhedral [15]. Similarly, the clustering of data favours lower intensity ratio values (between 0.45–0.55) indicative that a significant concentration of hydroxyl ligands coordinate to the Fe metal center (i.e., compare FeOOH standards at low intensity ratio values to Fe2O3 standards at intensity ratios> 0.55). A secondary clustering (i.e., Δ1.1–1.3; intensity ratio >0.7; Figure 3E) occurs near to the standard magnetite data point. Again, the agreement between the standard data point and the natural particles is not perfect, likely reflecting structural and chemical impurities. The level of disagreement may also reflect a possible particle size effect in the nanoparticle size regime, since it is noted that a proportion of the >100 nm natural Fe particles (grey shading) do show good overlap with the standard magnetite data points.
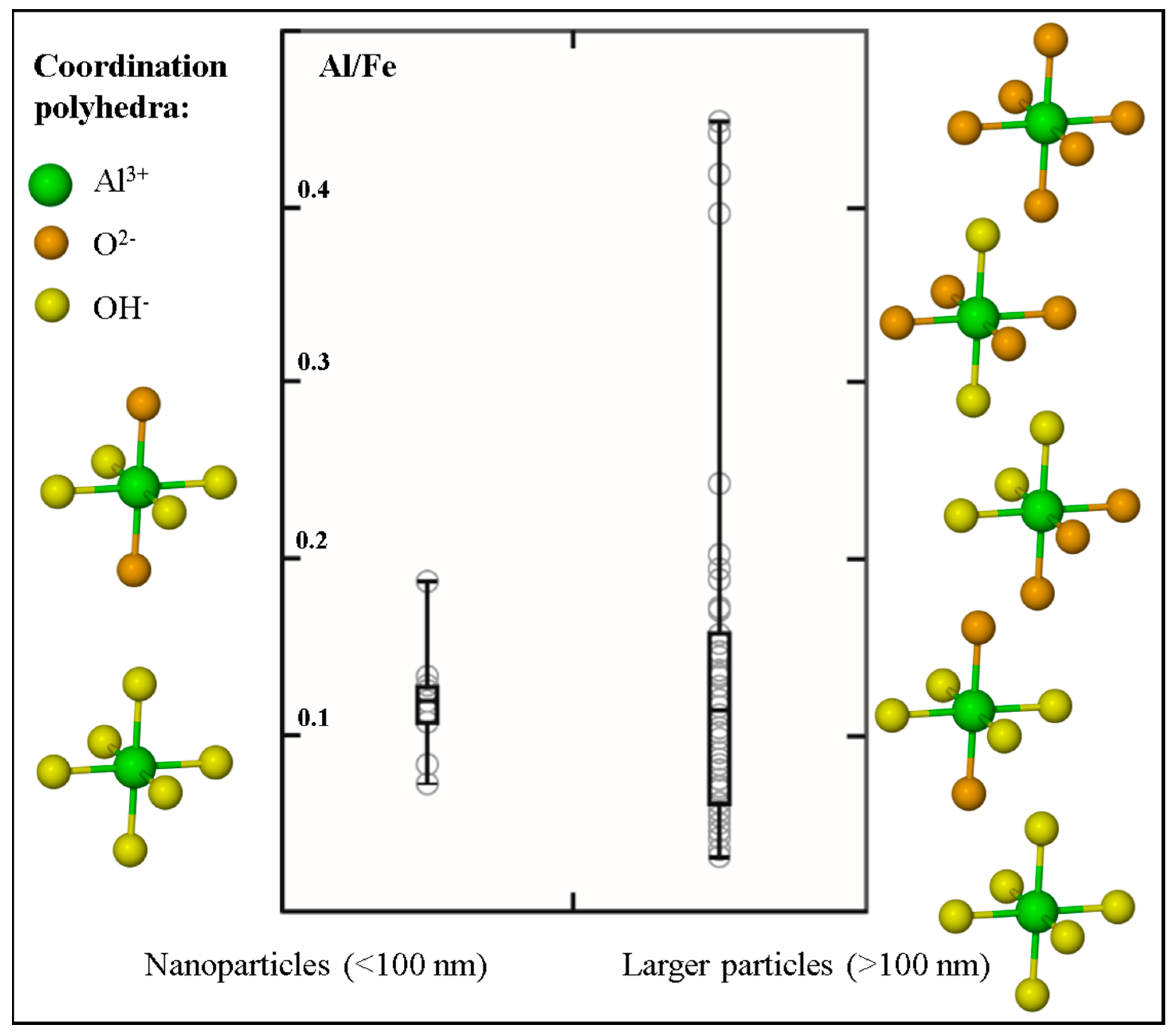
Similarities in cation size and charge result in Al-for-Fe substitution reactions being common phenomena in Fe oxides and Fe oxy-hydroxides. For example, natural soil Fe oxide minerals have previously been investigated and were found to contain levels of Al substitution reaching up to 36% [51]. Using Al subtraction maps generated by subtracting an X-ray absorption map collected below the Al K-edge (e.g., 1565 eV) from the X-ray absorption map collected at the Al K-edge (1572 eV), we were able to measure and compare the Al/Fe ratio in a subset of natural aquatic Fe nanoparticles to the Al/Fe ratio measured in natural aquatic particles >100 nm in size (Figure 4). The Al/Fe ratio in the nanoparticles ranges between 0.07 and 0.18; whereas the larger particles show a more extensive range spanning between 0.03 and 0.45. A student t-test conducted on the two datasets however, revealed that there is no significant difference (α = 0.05) between the Al/Fe ratios measured in the two size fractions. The Al K-edge XANES spectra were also used to evaluate the ligand coordination around the Al metal center in the natural nanoparticles and larger particles [52]. The side panels in Figure 4 summarize these findings (further details in Reference [34]) and show that Al associated with Fe-rich nanoparticles is high hydroxylated (i.e., coordinated to between four (coordination environment similar to Al in 1:1 clays) and six (Al(OH)3-like coordination) hydroxyl ligands). In comparison, the larger sized Fe-rich particles show a much more diverse array in the local coordination environment of the contained Al, with the number of hydroxyl ligands ranging between none (i.e., Al is coordinated only to oxygen ligands in a local bonding environment characteristic of Al in primary silicates) and six.
4. Discussion
The nanoparticles evaluated in this study were collected from relatively pristine aquatic systems and are thus expected to reflect the natural class of nanoparticles, although the presence of engineered and incidental nanoparticles cannot be completely ruled out. In contrast to the volume of work that has recently been conducted on engineered nanoparticles, natural inorganic nanoparticles have received a surprisingly lesser amount of scientific attention, despite some studies showing that the fate and toxicity of these natural nanoparticles differ from those of engineered ones (Reference [12], and references therein). Using a specialized synchrotron-based X-ray spectro-microscopic tool, this study thus seeks to provide new and additional insights into the characterization of biogeochemically-active natural Fe-rich nanoparticles.
Because of its combined imaging and local coordination environment probe capabilities, soft X-ray Fe L-edge XANES and STXM techniques have gained prominence as tools for studying natural sub-micrometer particles and nanoparticle in their pristine state [34,50]. Furthermore, and as correlative study approaches continue to evolve, it is foreseen that the detailed chemical information contained in the Fe L-edge spectrum will be used to complement other study methodologies (Table 1) to more fully explore the emerging field of natural nanoparticle research. In contrast to engineered nanoparticles, and in contrast to experimental results produced in controlled laboratory environments; natural nanoparticles are produced by a diverse array of formation mechanisms (including bottom-up (aggregation and growth of nanomaterial precursors); top-down (e.g., weathering reactions), and biologically-mediated reactions) and are influenced by variable environmental conditions (e.g., pH, Eh, temperature, salinity, solar radiation and the presence of organic matter) [1,2,12]. Given this vast range in influencing parameters, it is thus not surprising that our dataset shows a high degree of scatter on the ΔeV versus intensity ratio diagram (Figure 2). This scattering reflects divergence in the local Fe coordination environment away from what is expected for pure crystalline Fe mineral standards (Figure 2, black squares), and is caused for example, by variations in the ligand type and geometry and by structural distortions to the local Fe coordination polyhedra [15]. Given the measured presence of Al in the Fe-rich nanoparticles (Figure 4), we suggest that one of the reasons for the scatter is the ubiquitous presence of cation substitutions within the nanoparticle structure, including but not limited to Al and Si. Two other possible reasons for the scatter include: (1) The likelihood that natural particles are poorly-crystalline or amorphous, and (2) the possibility that what we have measured as discrete 10–100 nm sized particles actually represent aggregates of smaller (<10 nm) particles with variable chemistry and bound together by organic coatings (e.g., humic acids [53]).
Our research broadly identifies three groups of Fe nanoparticle mineralogy in the natural aquatic systems evaluated. A small number of Fe(II)-rich nanoparticles were identified despite all samples being collected from oxygen replete waters where Fe(II) is the thermodynamically less-favored form of Fe. Based on carbon K-edge XANES analyses, we have previously suggested that the Fe(II) presence may be a result of active reduction of Fe by microbial communities [40], or may be due to the stabilizing effect of Fe(II)-specific organic functional groups [48]. Recent work focused on Fe nanoparticulate speciation in glacial sediments invokes the role of amorphous silica in Fe(II) preservation [50], highlighting the need for further work to more firmly elucidate the mechanisms that stabilize ferrous Fe in natural nanoparticles and to constrain the interactions between these phases and the biosphere.
The second broad group identified in this study-in which Fe is present in its oxidized (Fe(III)) valence state-represents the dominant Fe nanoparticle speciation identified in natural aquatic environments. No distinctive difference was observed between the chemical speciation of the nanoparticles and that of the larger sized particles (Figure 2), likely reflecting that in heterogenous natural systems nanosize effects (e.g., quantum effects) have a secondary influence on XANES spectral signatures relative to the more obvious effects of inter-particle variability caused by differences in environmental parameters. Because of their strong surface complexing capacity, their high surface area to charge ratio and their resistance to sedimentation, ferric oxide nanoparticles have been highlighted as important contaminant and nutrient transporting agents in fluvial systems [1,5,6,54]. Furthermore, within the marine domain where important biologically-mediated air-sea CO2 exchange takes place, Fe particles (including these Fe(III) nanoparticles) collected in the nominally-defined “dissolved” size fraction (<0.2 or <0.45 µm filter pore sizes) are suggested as being important repositories of potentially bioavailable Fe [8,9].The Fe L-edge spectral signatures (i.e., intensity ratio < 0.55) suggest that these phases are predominantly hydroxyl-bearing moieties, which is corroborated by the prevalence of highly-hydroxylated Al metal centers associated with these Fe nanoparticles (Figure 4). This Al is further noteworthy in that its presence (especially as a cation substituent) is expected to modify the particles’ surface chemistry by decreasing the net concentration of reactive Fe sites through replacement by less-reactive Al [55,56].This impact on surface reactivity in turn affects the nutrient- and pollutant transporting capacity of the natural Fe nanoparticles, especially when compared to model experimental systems in which observations are based on the behavior of pure mineral standards. Aluminum substitution is further known to modify the solubility and dissolution kinetics of model Fe oxides (e.g., Reference [57], and references therein). The importance and accessibility of the marine Fe nanoparticulate pool as a repository of bioavailable Fe for driving primary productivity (and thus for influencing global climate feedback loops) must therefore be interpreted in a context of the observed and associated Al presence.
Finally, the presence of magnetite nanoparticles in marine surface waters of the Southern Ocean (Figure 2 and Figure 3) has previously been related to Patagonian atmospheric Fe inputs [33], but may also derive from magnetotactic bacteria [58]. The possible presence of biogenic Fe nanoparticles may be extended to the Fe(III) phases discussed above, since certain genera of Fe-oxidising bacteria are known to produce ferrihydrite and Fe(III) oxyhydroxides [12].Magnetic Fe nanoparticles have recently received increased attention since they are known to cause complex and potentially harmful interactions with living matter (e.g., through the production of excess reactive oxygen species [59]). However, it is important to highlight that the ecotoxicology of natural magnetite nanoparticles and the other forms of natural Fe nanoparticles identified in this study is not well constrained. Indeed, by virtue of their small size, they have a much higher likelihood of crossing cellular barriers (e.g., Reference [60]). However, ecotoxicological effects of the natural nanoparticles are expected to be restricted (relative to engineered nanoparticles) due to the potential presence of organic surface coatings [12]—such as those identified from our carbon K-edge XANES spectro-microscopy.
5. Conclusions
The discussion above highlights the diversity in nanoparticle mineral chemistry and speciation in natural aquatic environments. Given that the production of engineered Fe nanoparticles is expected to increase significantly in forthcoming years (2010 estimates at up to 42,000 t per year [61]), such a study focused on relatively pristine aquatic systems forms an important baseline from which to interpret future environmental change. The most common type of engineered Fe nanoparticle is the Fe0 class of nanoparticles, which are commonly used in environmental remediation efforts [7]. These engineered nanoparticles are however expected to experience significant levels of surface oxidation and surface reactions when released into natural aquatic environments [62]. The insights provided here related to the associations of natural Fe nanoparticles with Al and with organic functional groups, and may thus serve as a good proxy for the ultimate fate and speciation of engineered nanoparticles that evolve in heterogenous natural systems. Further work should continue to focus on the speciation, biogeochemical behavior and ecotoxicological impacts of both natural and engineered Fe nanoparticles existing in Earth’s various subsystems (e.g., hydrosphere, biosphere, etc.).
Author Contributions
Conceptualization, S.M., A.R., B.v.d.H.; methodology, S.M., B.v.d.H.; formal analysis, investigation and data curation, B.v.d.H.; writing—original draft preparation, B.v.d.H., A.R.; writing—review and editing, S.M.; validation, supervision, project administration and funding acquisition, A.R., S.M.
Funding
This research is supported by grants from NRF, South Africa, the Stellenbosch University VR(R) fund, the National Science Foundation (chemical sciences), the U.S. Department of Energy (BES and SBR), and the Princeton-in-Africa program.
Acknowledgments
The authors would like to thank T. Tyliszczak (beamline scientist) for assistance with data collection at the Molecular Environmental Sciences beamline 11.0.2. at Lawrence Berkeley National Laboratory (Berkeley, CA, USA). Colleagues who assisted with sample collection from lacustrine and marine sampling sites are also duly acknowledged. Finally, the authors wish to thank the two anonymous reviewers and the editors for the comments which served to strengthen the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Lead, J.R.; Wilkinson, K.J. Aquatic colloids and nanoparticles: Current knowledge and future trends. Environ. Chem. 2006, 3, 159–171. [Google Scholar] [CrossRef]
- Hochella, M.F.J.; Mogk, D.W.; Ranville, J.; Allen, I.C.; Luther, G.W.; Marr, L.C.; Mcgrail, B.P.; Murayama, M.; Qafoku, N.P.; Rosso, K.M.; et al. Natural, incidental, and engineered nanomaterials and their impacts on the Earth system. Science 2019, 363, eaau8299. [Google Scholar] [CrossRef]
- Nowack, B.; Bucheli, T.D. Occurrence, behavior and effects of nanoparticles in the environment. Environ. Pollut. 2007, 150, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Gottschalk, F.; Nowack, B. The release of engineered nanomaterials to the environment. J. Environ. Monit. 2011, 13, 1145–1155. [Google Scholar] [CrossRef]
- Hassellöv, M.; von der Kammer, F. Iron oxides as geochemical nanovectors for metal transport in soil-river systems. Elements 2008, 4, 401–406. [Google Scholar] [CrossRef]
- Hochella, M.F.; Moore, J.N.; Putnis, C.V.; Putnis, A.; Kasama, T.; Eberl, D.D. Direct observation of heavy metal-mineral association from the Clark Fork River Superfund Complex: Implications for metal transport and bioavailability. Geochim. Cosmochim. Acta 2005, 69, 1651–1663. [Google Scholar] [CrossRef]
- Yan, W.L.; Lien, H.-L.; Koel, B.E.; Zhang, W.-X. Iron nanoparticles for environmental clean-up: Recent developments and future outlook. Environ. Sci. Process. Impacts 2013, 15, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Tagliabue, A.; Bowie, A.R.; Boyd, P.W.; Buck, K.N.; Johnson, K.S.; Saito, M.A. The integral role of iron in ocean biogeochemistry. Nature 2017, 543, 51. [Google Scholar] [CrossRef]
- Boyd, P.W.; Ellwood, M.J. The biogeochemical cycle of iron in the ocean. Nat. Geosci. 2010, 3, 675–682. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Fan, M.; Brown, R.C.; Van Leeuwen, J.H.; Wang, W.; Song, Y.; Zhang, P. Synthesis, Properties, and Environmental Applications of Nanoscale Iron-Based Materials: A Review Applications of Nanoscale Iron-Based Materials: A Review. Crit. Rev. Environ. Sci. Technol. 2006, 36, 405–431. [Google Scholar] [CrossRef]
- Raychoudhury, T.; Scheytt, T. Potential of Zerovalent iron nanoparticles for remediation of environmental organic contaminants in water: A review. Water Sci. Technol. 2013, 68, 1425–1439. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Filip, J.; Zboril, R.; Varma, R.S. Natural inorganic nanoparticles—Formation, fate, and toxicity in the environment. Chem. Soc. Rev. 2015, 44, 8410–8423. [Google Scholar] [CrossRef]
- Neubauer, E.; Schenkeveld, W.D.; Plathe, K.L.; Rentenberger, C.; Von Der Kammer, F.; Kraemer, S.M.; Hofmann, T. The influence of pH on iron speciation in podzol extracts: Iron complexes with natural organic matter, and iron mineral nanoparticles. Sci. Total Environ. 2013, 461, 108–116. [Google Scholar] [CrossRef]
- Carbone, C.; Di Benedetto, F.; Marescotti, P.; Sangregorio, C. Natural Fe-oxide and -oxyhydroxide nanoparticles: An EPR and SQUID investigation. Mineral. Petrol. 2005, 85, 19–32. [Google Scholar] [CrossRef]
- Von der Heyden, B.P.; Roychoudhury, A.N.; Tyliszczak, T.; Myneni, S.C.B. Investigating nanoscale mineral compositions: Iron L3-edge spectroscopic evaluation of iron oxide and oxy-hydroxide coordination. Am. Mineral. 2017, 102, 674–685. [Google Scholar] [CrossRef]
- Tagliabue, A.; Aumont, O.; DeAth, R.; Dunne, J.P.; Dutkiewicz, S.; Galbraith, E.; Misumi, K.; Moore, J.K.; Ridgwell, A.; Sherman, E.; et al. How well do global ocean biogeochemistry models simulate dissolved iron distributions? Global Biogeochem. Cycles 2016, 32, 149–174. [Google Scholar] [CrossRef]
- Upadhyay, S.; Parekh, K.; Pandey, B. Influence of crystallite size on the magnetic properties of Fe3O4 nanoparticles. J. Alloys Compd. 2016, 678, 478–485. [Google Scholar] [CrossRef]
- Marcus, M.A.; Manceau, A.; Kersten, M. Mn, Fe, Zn and As speciation in a fast-growing ferromanganese marine nodule. Geochim. Cosmochim. Acta 2004, 68, 3125–3136. [Google Scholar] [CrossRef] [Green Version]
- Toner, B.M.; Santelli, C.M.; Marcus, M.A.; Wirth, R.; Chan, C.S.; McCollom, T.; Bach, W.; Edwards, K.J. Biogenic iron oxyhydroxide formation at mid-ocean ridge hydrothermal vents: Juan de Fuca Ridge. Geochim. Cosmochim. Acta 2009, 73, 388–403. [Google Scholar] [CrossRef] [Green Version]
- Křížek, M.; Pechoušek, J.; Tuček, J.; Šafářová, K.; Medřík, I.; Machala, L. Iron oxide nanoparticle powders with high surface area. AIP Conf. Proc. 2012, 1489, 88–94. [Google Scholar]
- Kenouche, S.; Larionova, J.; Bezzi, N.; Guari, Y.; Bertin, N.; Zanca, M.; Lartigue, L.; Cieslak, M.; Godin, C.; Morrot, G.; et al. NMR investigation of functionalized magnetic nanoparticles Fe3O4 as T1–T2 contrast agents. Powder Technol. 2014, 255, 60–65. [Google Scholar] [CrossRef]
- Sabale, S.; Jadhav, V.; Khot, V.; Zhu, X.; Xin, M.; Chen, H. Superparamagnetic MFe2O4 (M= Ni, Co, Zn, Mn) nanoparticles: Synthesis, characterization, induction heating and cell viability studies for cancer hyperthermia applications. J. Mater. Sci. Mater. Med. 2015, 26, 127. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.; Flores, D.; Ward, C.R.; Silva, L.F. Identification of nanominerals and nanoparticles in burning coal waste piles from Portugal. Sci. Total Environ. 2010, 408, 6032–6041. [Google Scholar] [CrossRef] [PubMed]
- Pyrgiotakis, G.; Blattmann, C.O.; Pratsinis, S.; Demokritou, P. Nanoparticle–nanoparticle interactions in biological media by atomic force microscopy. Langmuir 2013, 29, 11385–11395. [Google Scholar] [CrossRef] [PubMed]
- Raj, X.J.; Nishimura, T. Investigation of the Surface Potential on Iron Nanoparticles During the Corrosion by Atomic Force Microscopy (AFM) and Kelvin Probe Force Microscopy (KFM). Int. J. Electrochem. Sci. 2014, 9, 2090–2100. [Google Scholar]
- Poulton, S.W.; Raiswell, R. Chemical and physical characteristics of iron oxides in riverine and glacial meltwater sediments. Chem. Geol. 2005, 218, 203–221. [Google Scholar] [CrossRef]
- Allard, T.; Menguy, N.; Salomon, J.; Calligaro, T.; Weber, T.; Calas, G.; Benedetti, M.F. Revealing forms of iron in river-borne material from major tropical rivers of the Amazon Basin (Brazil). Geochim. Cosmochim. Acta 2004, 68, 3079–3094. [Google Scholar] [CrossRef]
- Allard, T.; Weber, T.; Bellot, C.; Damblans, C.; Bardy, M.; Bueno, G.; Nascimento, N.R.; Fritsch, E.; Benedetti, M.F. Tracing source and evolution of suspended particles in the Rio Negro Basin (Brazil) using chemical species of iron. Chem. Geol. 2011, 280, 79–88. [Google Scholar] [CrossRef]
- Liu, T.; Guo, L.; Tao, Y.; Hu, T.D.; Xie, Y.N.; Zhang, J. Bondlength alternation of nanoparticles Fe2O3 coated with organic surfactants probed by EXAFS. Nanostruct. Mater. 1999, 11, 1329–1334. [Google Scholar] [CrossRef]
- Sundman, A.; Karlsson, T.; Laudon, H.; Persson, P. XAS study of iron speciation in soils and waters from a boreal catchment. Chem. Geol. 2014, 364, 93–102. [Google Scholar] [CrossRef]
- Sun, Y.P.; Li, X.Q.; Cao, J.; Zhang, W.X.; Wang, H.P. Characterization of zero-valent iron nanoparticles. Adv. Colloid Interface Sci. 2006, 120, 47–56. [Google Scholar] [CrossRef]
- Hirst, C.; Andersson, P.S.; Shaw, S.; Burke, I.T.; Kutscher, L.; Murphy, M.J.; Maximov, T.; Pokrovsky, O.S.; Mörth, C.M.; Porcelli, D. Characterisation of Fe-bearing particles and colloids in the Lena River basin, NE Russia. Geochemica Cosmochim. Acta 2017, 213, 553–573. [Google Scholar] [CrossRef]
- von der Heyden, B.P.; Roychoudhury, A.N.; Mtshali, T.N.; Tyliszczak, T.; Myneni, S.C.B. Chemically and geographically distinct solid-phase iron pools in the Southern Ocean. Science 2012, 338, 1199–1201. [Google Scholar] [CrossRef]
- Von Der Heyden, B.P.; Frith, M.G.; Bernasek, S.; Tylizszak, T.; Roychoudhury, A.N.; Myneni, S.C.B. Geochemistry of Al and Fe in freshwater and coastal water colloids from the west coast of Southern Africa. Geochim. Cosmochim. Acta 2018, 241, 56–68. [Google Scholar] [CrossRef]
- Sheng, G.; Yang, P.; Tang, Y.; Hu, Q.; Li, H.; Ren, X.; Hu, B.; Wang, X.; Huang, Y. New insights into the primary roles of diatomite in the enhanced sequestration of UO22+ by zerovalent iron nanoparticles: An advanced approach utilizing XPS and EXAFS. Appl. Catal. B Environ. 2016, 193, 189–197. [Google Scholar] [CrossRef]
- Mourdikoudis, S.; Pallares, R.M.; Thanh, N.T. Characterization techniques for nanoparticles: Comparison and complementarity upon studying nanoparticle properties. Nanoscale 2018, 10, 12871–12934. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, M.; Srivastav, A.; Gandhi, S.; Rao, S.; Roychoudhury, A.; Kumar, A.; Singhal, R.K.; Kumar, S.; Singh, S.D. Monitoring of engineered nanoparticles in soil-plant system: A review. Environ. Nanotechnol. Monit. Manag. 2019, 11, 100218. [Google Scholar] [CrossRef]
- De Groot, F.M.F.; Glatzel, P.; Bergmann, U.; Van Aken, P.A.; Barrea, R.A.; Klemme, S.; Haevecker, M.; Knop-Gericke, A.; Heijboer, W.M.; Weckhuysen, B.M. 1s2p Resonant Inelastic X-ray Scattering of Iron Oxides. J. Phys. Chem. B 2005, 109, 20751–20762. [Google Scholar] [CrossRef] [PubMed]
- Bluhm, H.; Andersson, K.; Araki, T.; Benzerara, K.; Brown, G.E.; Dynes, J.J.; Ghosal, S.; Gilles, M.K.; Hansen, H.C.; Hemminger, J.C.; et al. Soft X-ray microscopy and spectroscopy at the molecular environmental science beamline at the Advanced Light Source. J. Electron. Spectros. Relat. Phenom. 2006, 150, 86–104. [Google Scholar] [CrossRef]
- Von Der Heyden, B.P.; Hauser, E.J.; Mishra, B.; Martinez, G.A.; Bowie, A.R.; Tyliszczak, T.; Mtshali, T.N.; Roychoudhury, A.N.; Myneni, S.C.B. Ubiquitous Presence of Fe(II) in Aquatic Colloids and Its Association with Organic Carbon. Environ. Sci. Technol. Lett. 2014, 1, 387–392. [Google Scholar] [CrossRef] [Green Version]
- Cressey, G.; Henderson, C.M.B.; van der Laan, G. Use of L-edge X-ray absorption spectroscopy to characterize multiple valence states of 3d transition metals; a new probe for mineralogical and geochemical research. Phys. Chem. Miner. 1993, 20, 111–119. [Google Scholar] [CrossRef]
- van Aken, P.A.; Liebscher, B. Quantification of ferrous/ferric ratios in minerals: New evaluation schemes of Fe L23 electron energy-loss near-edge spectra. Phys. Chem. Miner. 2002, 28, 188–200. [Google Scholar] [CrossRef]
- Calvert, C.C.; Brown, A.; Brydson, R. Determination of the local chemistry of iron in inorganic and organic materials. J. Electron. Spectros. Relat. Phenom. 2005, 143, 173–187. [Google Scholar] [CrossRef]
- Peak, D.; Regier, T. Direct observation of tetrahedrally coordinated Fe(III) in ferrihydrite. Environ. Sci. Technol. 2012, 46, 3163–3168. [Google Scholar] [CrossRef] [PubMed]
- Miot, J.; Benzerara, K.; Morin, G.; Kappler, A.; Bernard, S.; Obst, M.; Férard, C.; Skouri-Panet, F.; Guigner, J.M.; Posth, N.; et al. Iron biomineralization by anaerobic neutrophilic iron-oxidizing bacteria. Geochim. Cosmochim. Acta 2009, 73, 696–711. [Google Scholar] [CrossRef]
- Chan, C.S.; De Stasio, G.; Welch, S.A.; Girasole, M.; Frazer, B.H.; Nesterova, M.V.; Fakra, S.; Banfield, J.F. Microbial polysaccharides template assembly of nanocrystal fibers. Science 2004, 303, 1656–1658. [Google Scholar] [CrossRef]
- Chen, C.; Sparks, D.L. Multi-elemental scanning transmission X-ray microscopy-near edge X-ray absorption fine structure spectroscopy assessment of organo-mineral associations in soils from reduced environments. Environ. Chem. 2015, 12, 64–73. [Google Scholar] [CrossRef]
- Toner, B.M.; Fakra, S.C.; Manganini, S.J.; Santelli, C.M.; Marcus, M.A.; Moffett, J.W.; Rouxel, O.; German, C.R.; Edwards, K.J. Preservation of iron(II) by carbon-rich matrices in a hydrothermal plume. Nat. Geosci. 2009, 2, 197–201. [Google Scholar] [CrossRef] [Green Version]
- Todd, E.C.; Sherman, D.M.; Purton, J.A. Surface oxidation of chalcopyrite (CuFeS2) under ambient atmospheric and aqueous (pH 2-10) conditions: Cu, Fe L- and O K-edge X-ray spectroscopy. Geochim. Cosmochim. Acta 2003, 67, 2137–2146. [Google Scholar] [CrossRef]
- Hawkings, J.R.; Benning, L.G.; Raiswell, R.; Kaulich, B.; Araki, T.; Abyaneh, M.; Stockdale, A.; Koch-müller, M.; Wadham, J.L.; Tranter, M. Biolabile ferrous iron bearing nanoparticles in glacial sediments. Earth Planet. Sci. Lett. 2018, 493, 92–101. [Google Scholar] [CrossRef]
- Schwertmann, U.; Carlson, L. Aluminum Influence on Iron Oxides: XVII. Unit-Cell Parameters and Aluminum Substitution of Natural Goethites. Soil Sci. Soc. Am. J. 1994, 58, 256–261. [Google Scholar] [CrossRef]
- Myneni, S.; Hay, M.; Mishra, B. Applications of scanning transmission X-ray microscopy in studying clays and their chemical interactions. In Special Volume, Advanced Applications of Synchrotron Radiation in Clay Science; Clay Minerals Society: Chantilly, VA, USA, 2013; pp. 231–258. [Google Scholar]
- Baalousha, M.; Manciulea, A.; Cumberland, S.; Kendall, K.; Lead, J.R. Aggregation and surface properties of iron oxide nanoparticles: Influence of pH and natural organic matter. Environ. Toxicol. Chem. 2008, 27, 1875–1882. [Google Scholar] [CrossRef] [PubMed]
- Wigginton, N.S.; Haus, K.L.; Hochella, M.F. Aquatic environmental nanoparticles. J. Environ. Monit. 2007, 9, 1306–1316. [Google Scholar] [CrossRef] [PubMed]
- Potter, H.A.B.; Yong, R.N. Influence of iron/aluminium ratio on the retention of lead and copper by amorphous iron–aluminium oxides. Appl. Clay Sci. 1999, 14, 1–26. [Google Scholar] [CrossRef]
- Jentzsch, T.L.; Penn, R.L. Effects of aluminum doping on the reactivity of ferrihydrite nanoparticles. J. Phys. Chem. B 2006, 110, 11746–11750. [Google Scholar] [CrossRef]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurrences and Uses, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003; ISBN 3527302743. [Google Scholar]
- Lefevre, C.T.; Bazylinski, D.A. Ecology, diversity, and evolution of magnetotactic bacteria. Microbiol. Mol. Biol. Rev. 2013, 77, 497–526. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Gao, J.; Ai, H.; Chen, X. Applications and Potential Toxicity of Magnetic Iron Oxide Nanoparticles. Small 2013, 9, 1533–1545. [Google Scholar] [CrossRef]
- Phogat, N.; Khan, S.A.; Shankar, S.; Ansary, A.A.; Uddin, I. Fate of inorganic nanoparticles in agriculture. Adv. Mater. Lett. 2016, 7, 3–12. [Google Scholar] [CrossRef]
- Keller, A.A.; Lazareva, A. Predicted Releases of Engineered Nanomaterials: From Global to Regional to Local. Environ. Sci. Technol. Lett. 2013, 1, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Liu, A.; Liu, J.; Han, J.; Zhang, W. Evolution of nanoscale zero-valent iron (nZVI) in water: Microscopic and spectroscopic evidence on the formation of nano- and micro-structured iron oxides. J. Hazard. Mater. 2017, 322, 129–135. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(A) The Fe L2,3 X-ray Absorption Near-Edge Structure (XANES) spectrum for three standard iron oxide minerals (goethite (α-FeOOH); lepidocrocite (γ-FeOOH); and hematite (α-Fe2O3)) clearly illustrating the peak splitting that occurs in both the L2-edge and the L3-edge on account of ligand field splittingeffects. (B) The fine structure of the L3-edge region of the spectrum (i.e., the degree of L3-edge peak splitting (ΔeV) and the intensity ratio (i) between the split peaks) reflects the differences in chemistry and mineralogy of the three minerals. These differences can be quantified and represent a useful tool for Fe nanoparticle characterisation (e.g., Figure 2; Reference [15]).
Figure 1.
(A) The Fe L2,3 X-ray Absorption Near-Edge Structure (XANES) spectrum for three standard iron oxide minerals (goethite (α-FeOOH); lepidocrocite (γ-FeOOH); and hematite (α-Fe2O3)) clearly illustrating the peak splitting that occurs in both the L2-edge and the L3-edge on account of ligand field splittingeffects. (B) The fine structure of the L3-edge region of the spectrum (i.e., the degree of L3-edge peak splitting (ΔeV) and the intensity ratio (i) between the split peaks) reflects the differences in chemistry and mineralogy of the three minerals. These differences can be quantified and represent a useful tool for Fe nanoparticle characterisation (e.g., Figure 2; Reference [15]).

Figure 2.
Plot of the spectral parameters ΔeV versus intensity ratio clearly illustrating the discrete fields occupied by synthetic Fe mineral standards (black squares and associated bars representing standard deviation (modified after Reference [15])). Colored point data represent the spectral signatures for individual Fe nanoparticles from fluvial (orange) and marine (green) systems, whereas the shaded region represents the density distribution of data points for sub-micrometer (larger than 100 nm) and micrometer sized particles collected from marine and fluvial environments (generated using ArcGIS point density function).
Figure 2.
Plot of the spectral parameters ΔeV versus intensity ratio clearly illustrating the discrete fields occupied by synthetic Fe mineral standards (black squares and associated bars representing standard deviation (modified after Reference [15])). Colored point data represent the spectral signatures for individual Fe nanoparticles from fluvial (orange) and marine (green) systems, whereas the shaded region represents the density distribution of data points for sub-micrometer (larger than 100 nm) and micrometer sized particles collected from marine and fluvial environments (generated using ArcGIS point density function).

Figure 3.
(A,B) X-ray absorption maps of marine nanoparticles collected at 709.8 eV using Scanning Transmission X-ray Microscopy (STXM)). (C,D) Fe distribution maps generated by subtracting the X-ray absorption map collected below the Fe L-edge (i.e., 703 eV) from the X-ray absorption map collected at the Fe L-edge (e.g., Figure 3A,B). Colour bar shows relative Fe concentration. (E,F) Fe L2,3-edge spectra for identified Fe-rich nanoparticles. Spectrum E reflects magnetite chemistry, whereas Spectrum F is indicative of Fe(III)—likely in the form of an amorphous Fe oxy-hydroxide phase.
Figure 3.
(A,B) X-ray absorption maps of marine nanoparticles collected at 709.8 eV using Scanning Transmission X-ray Microscopy (STXM)). (C,D) Fe distribution maps generated by subtracting the X-ray absorption map collected below the Fe L-edge (i.e., 703 eV) from the X-ray absorption map collected at the Fe L-edge (e.g., Figure 3A,B). Colour bar shows relative Fe concentration. (E,F) Fe L2,3-edge spectra for identified Fe-rich nanoparticles. Spectrum E reflects magnetite chemistry, whereas Spectrum F is indicative of Fe(III)—likely in the form of an amorphous Fe oxy-hydroxide phase.

Figure 4.
Box-and-whisker diagram comparing the Al/Fe ratio measured in nanoparticles versus the range measured in larger sub-micrometer (>100 nm) and micrometer sized particles (data extracted from [34]). Side panels show the diversity in the number of hydroxyl ligands coordinated to the Al metal centers in the two size fractions (from interpretations of Al K-edge XANES [34,52]; ligand positions are not absolute). These side panels highlight that hydroxyl ligands predominate over oxygen ligands in the Fe-rich nanoparticles, whereas larger-sized natural Fe particles are characterized by a prevalence of both oxygen and hydroxyl ligand bonding.
Figure 4.
Box-and-whisker diagram comparing the Al/Fe ratio measured in nanoparticles versus the range measured in larger sub-micrometer (>100 nm) and micrometer sized particles (data extracted from [34]). Side panels show the diversity in the number of hydroxyl ligands coordinated to the Al metal centers in the two size fractions (from interpretations of Al K-edge XANES [34,52]; ligand positions are not absolute). These side panels highlight that hydroxyl ligands predominate over oxygen ligands in the Fe-rich nanoparticles, whereas larger-sized natural Fe particles are characterized by a prevalence of both oxygen and hydroxyl ligand bonding.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Overview of techniques used to study Fe nanoparticles and nanominerals.
| Technique | Acronym | Signal Analyzed | Property/Information Retrieved | Relevant References |
|---|---|---|---|---|
| Micro-focused X-ray diffraction | µXRD | Diffraction pattern of X-rays | Crystal structure, Grain size, Lattice parameters | [17,18,19] |
| Brunauer-Emmett-Teller Surface area analysis | BET | Gas sorption | Surface area, Pore analysis | [20] |
| Nuclear Magnetic Resonance Spectroscopy | NMR | Electromagnetic signal | Purity, Molecular structure | [21] |
| Fourier Transform Infrared Spectroscopy | FTIR | Absorpiton of electromagnetic radiations | Surface composition, Ligand binding | [22] |
| Scanning Electron Microscopy - Energy dispersive spectrometry | SEM-EDS | Elemental X-ray flourescence | Size, Shape, Composition | [23] |
| Atomic Force Microscopy | AFM | Force spectroscopy, imaging | Atomic structure, Crystal latice defects, Youngs modulus, Surface potential | [24,25] |
| Transmission Electron Microscopy | TEM | Electron beam flourescence | Size, Shape, Composition, Aggregation, Size distribution, Bonding environment | [26,27,28] |
| 3-D Tomography | µCT | X-ray Irradiation | Size, Shape, Aggregation, Porosity | - |
| Extended X-ray Absorption Fine Structure | EXAFS | X-ray absorption spectrum | X-ray absorption coefficent, Chemical state of species | [29,30] |
| X-ray Absorption Near Edge Structure | XANES | X-ray absorption spectrum, excitation of an inner shell electron | Vacant orbitals, Oxidation state, Chemical bonding environment | [31,32] |
| Scanning Transmission X-ray Microscopy | STXM | Elemental X-ray absroption and spectrum | Elemental concentration and speciation map, valency, coordination number | [33,34] |
| X-ray Photoelectron Spectroscopy | XPS | Kinetic energy of ejected photoelectron | Elemental concentration and speciation on particle surfaces | [31,35] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
von der Heyden, B.; Roychoudhury, A.; Myneni, S. Iron-Rich Nanoparticles in Natural Aquatic Environments. Minerals 2019, 9, 287. https://doi.org/10.3390/min9050287
AMA Style
von der Heyden B, Roychoudhury A, Myneni S. Iron-Rich Nanoparticles in Natural Aquatic Environments. Minerals. 2019; 9(5):287. https://doi.org/10.3390/min9050287
Chicago/Turabian Stylevon der Heyden, Bjorn, Alakendra Roychoudhury, and Satish Myneni. 2019. "Iron-Rich Nanoparticles in Natural Aquatic Environments" Minerals 9, no. 5: 287. https://doi.org/10.3390/min9050287
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.