
Optical Coherence Tomography in Inherited Macular Dystrophies: A Review

Abstract
:1. Introduction
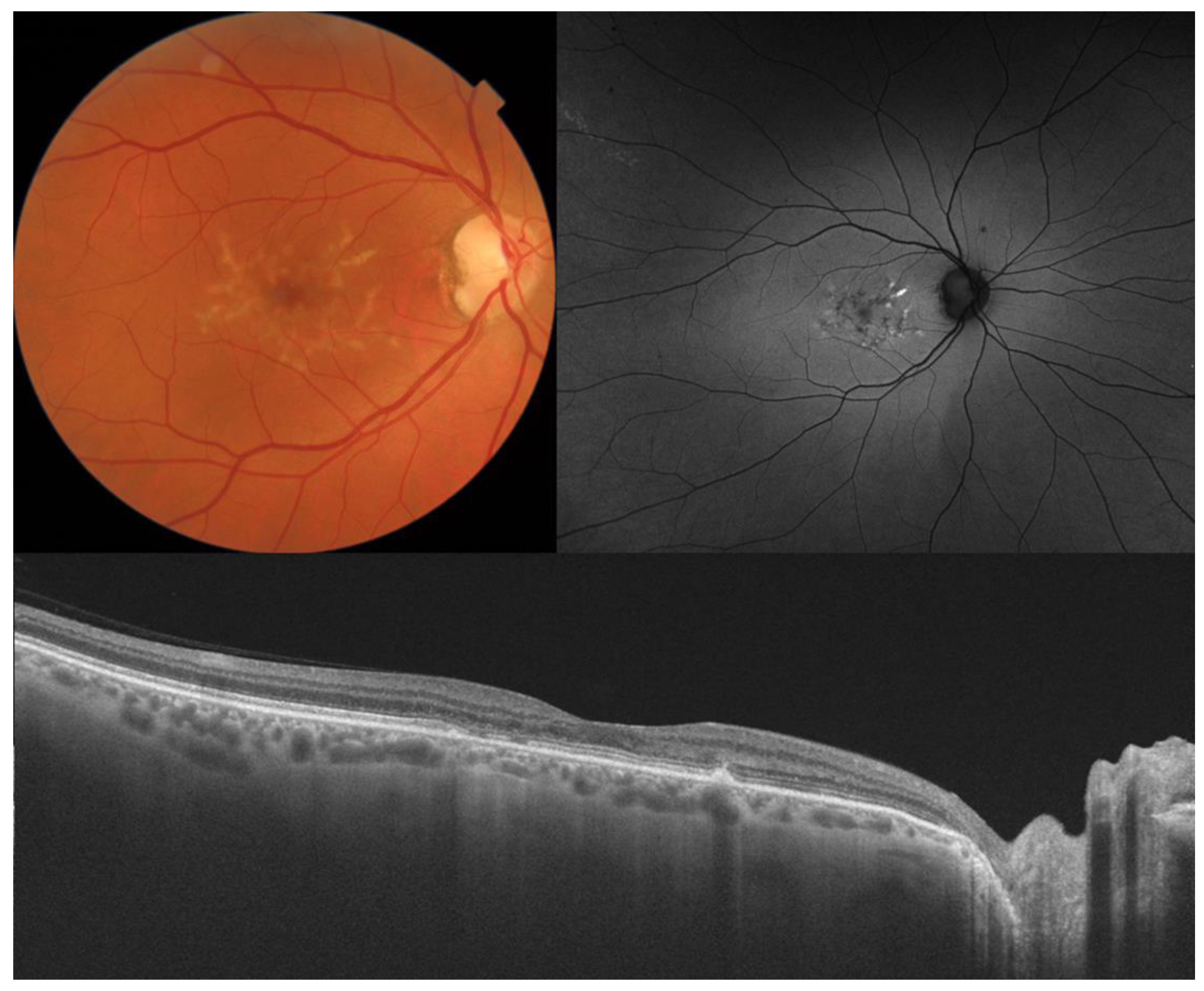
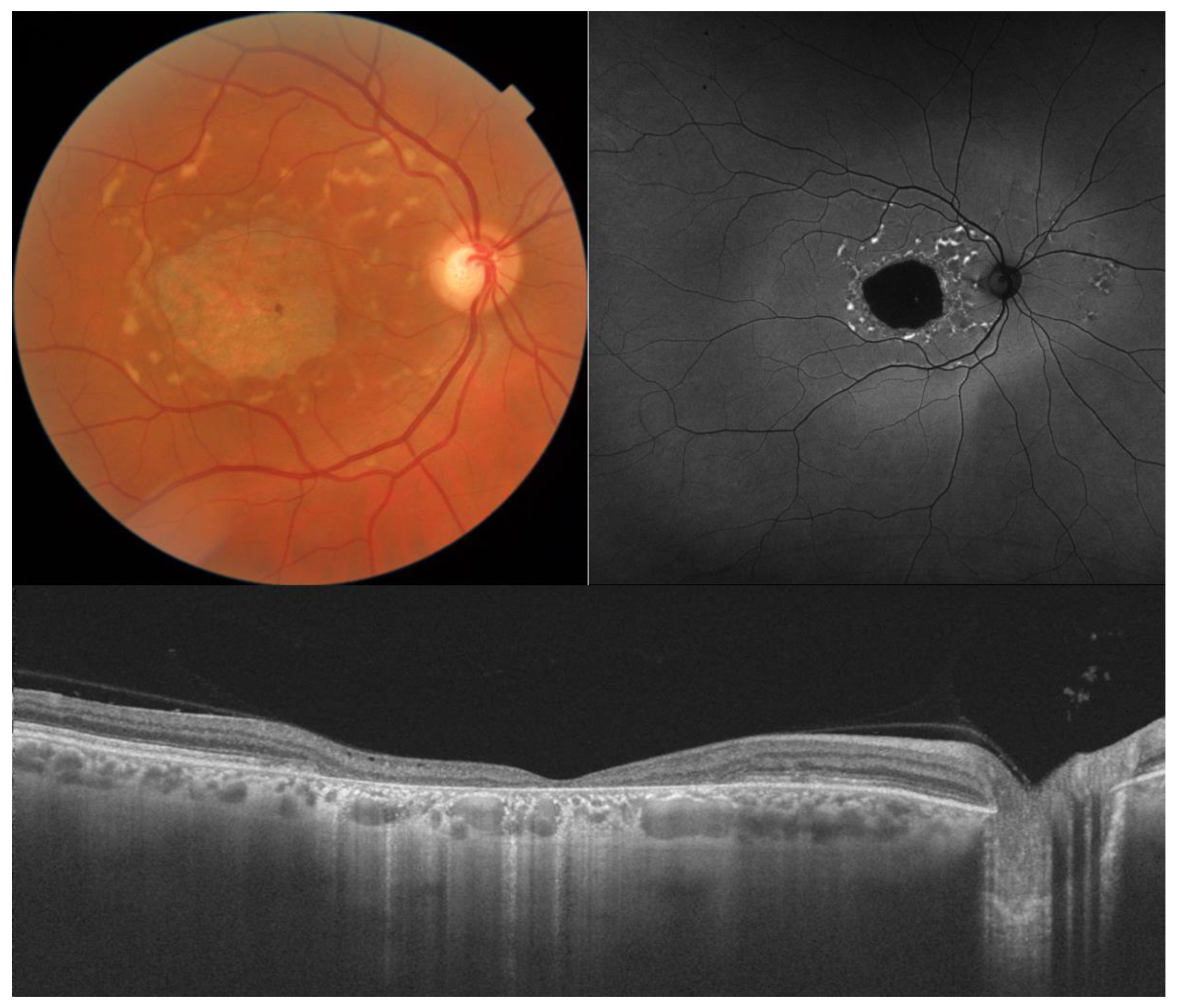
2. Stargardt Disease
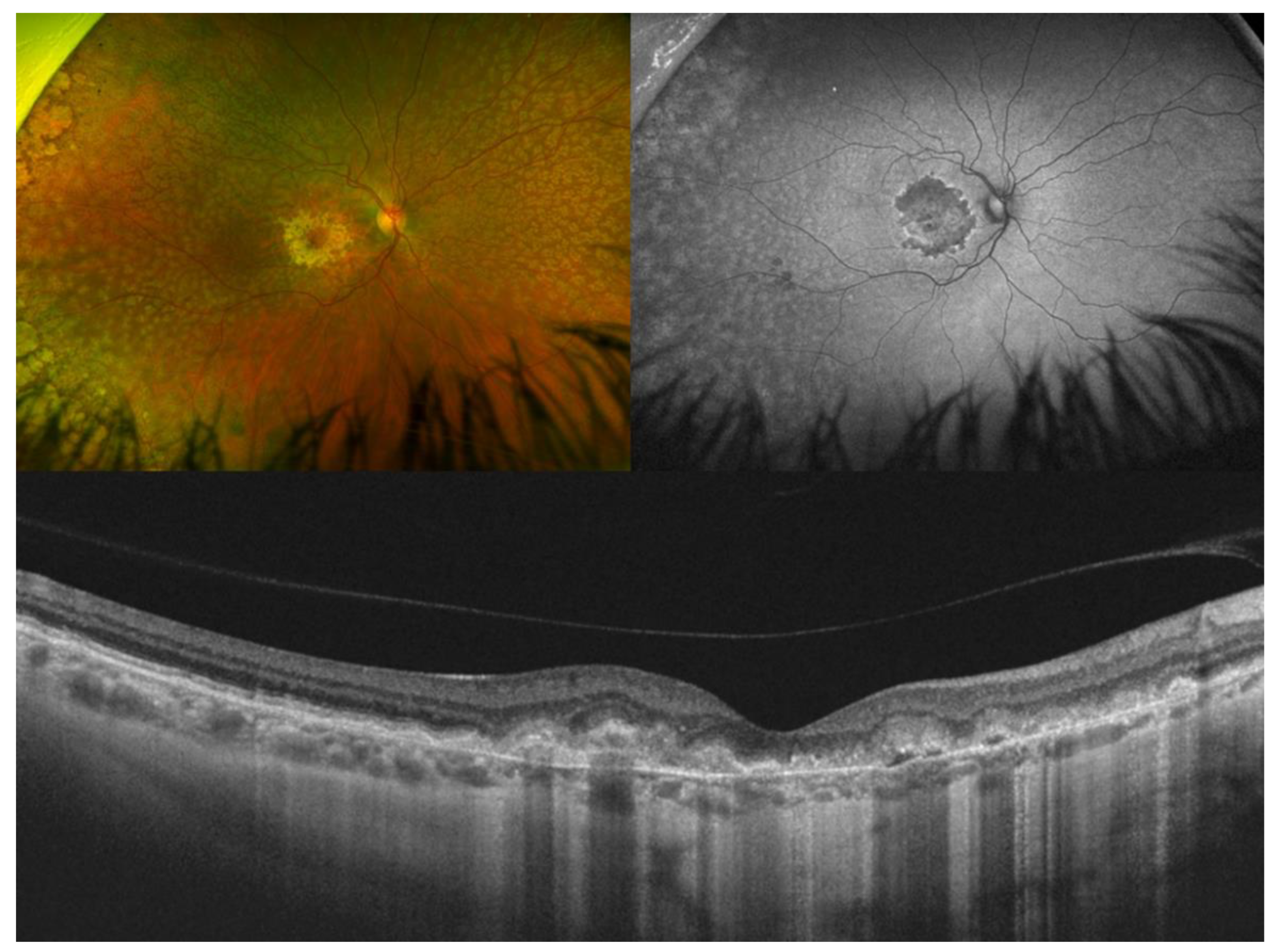
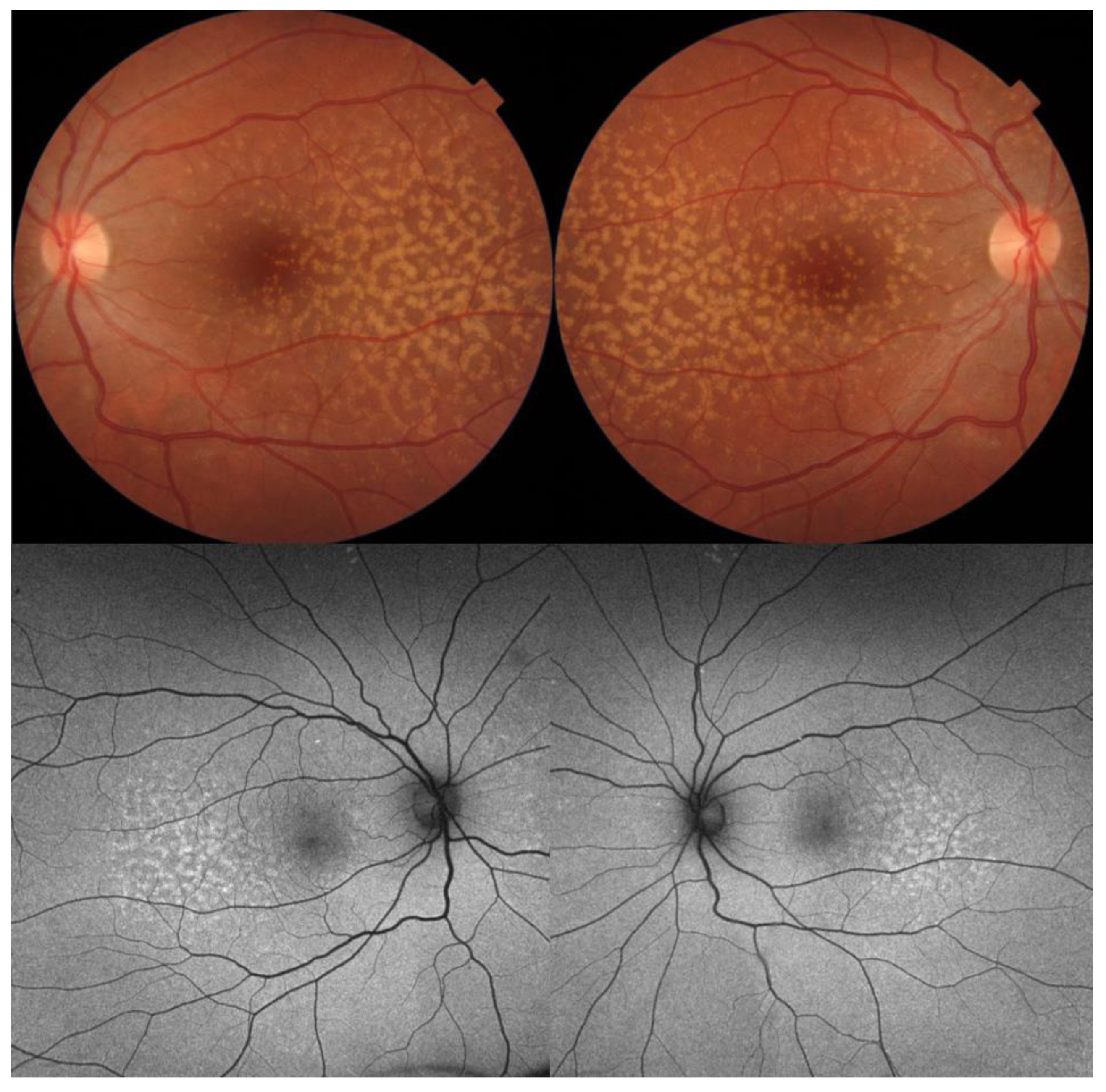
3. Best Vitelliform Macular Dystrophy
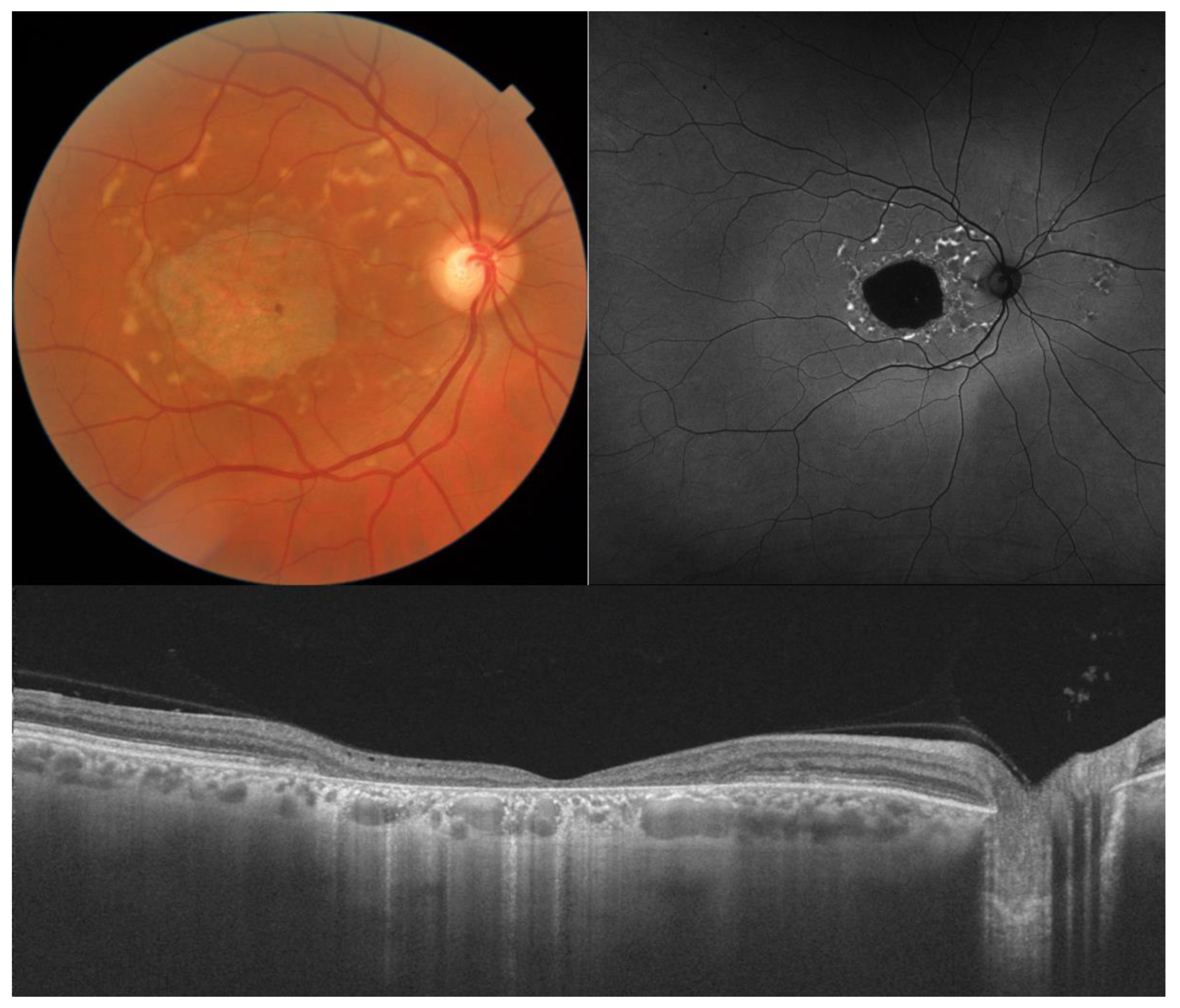
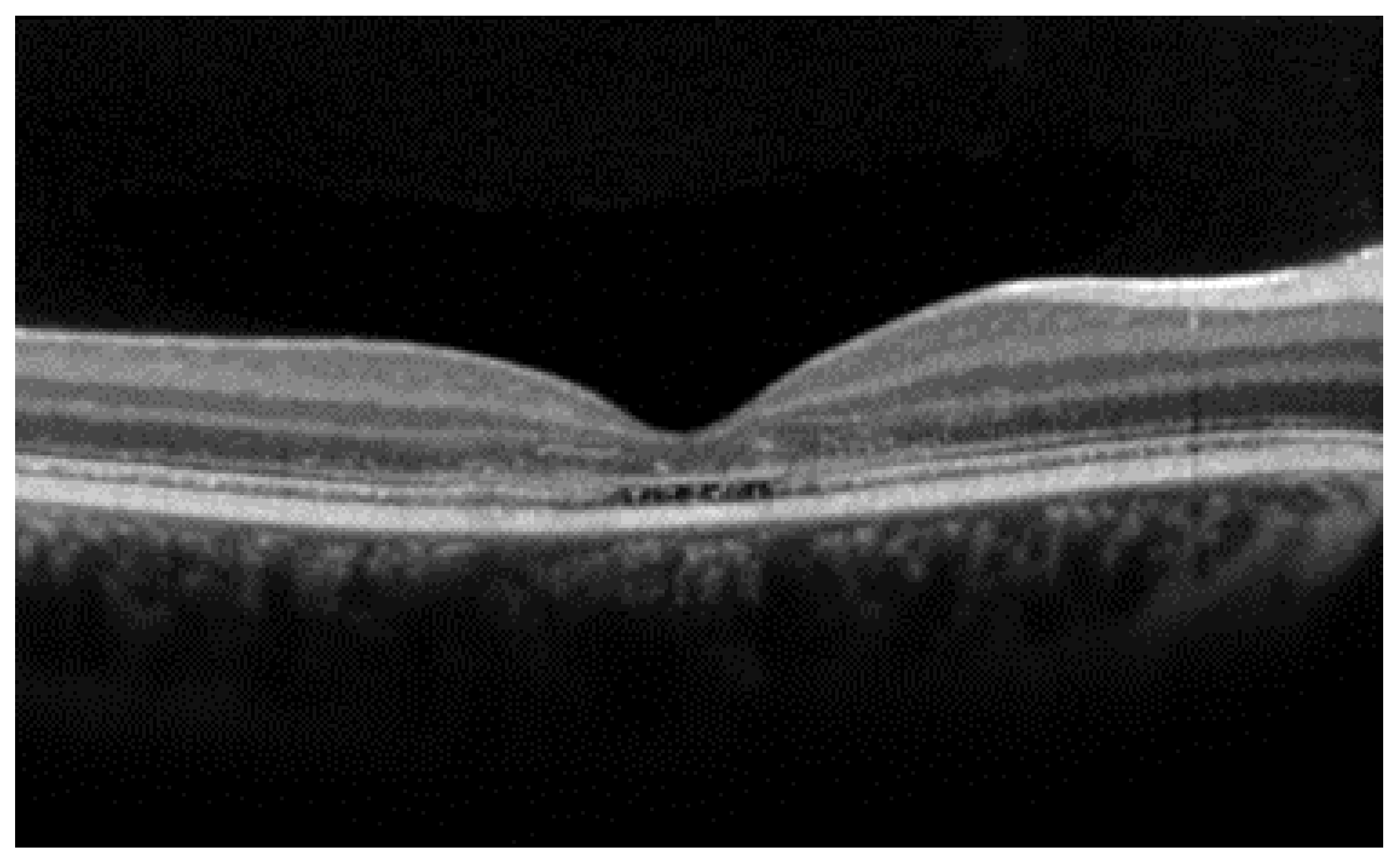
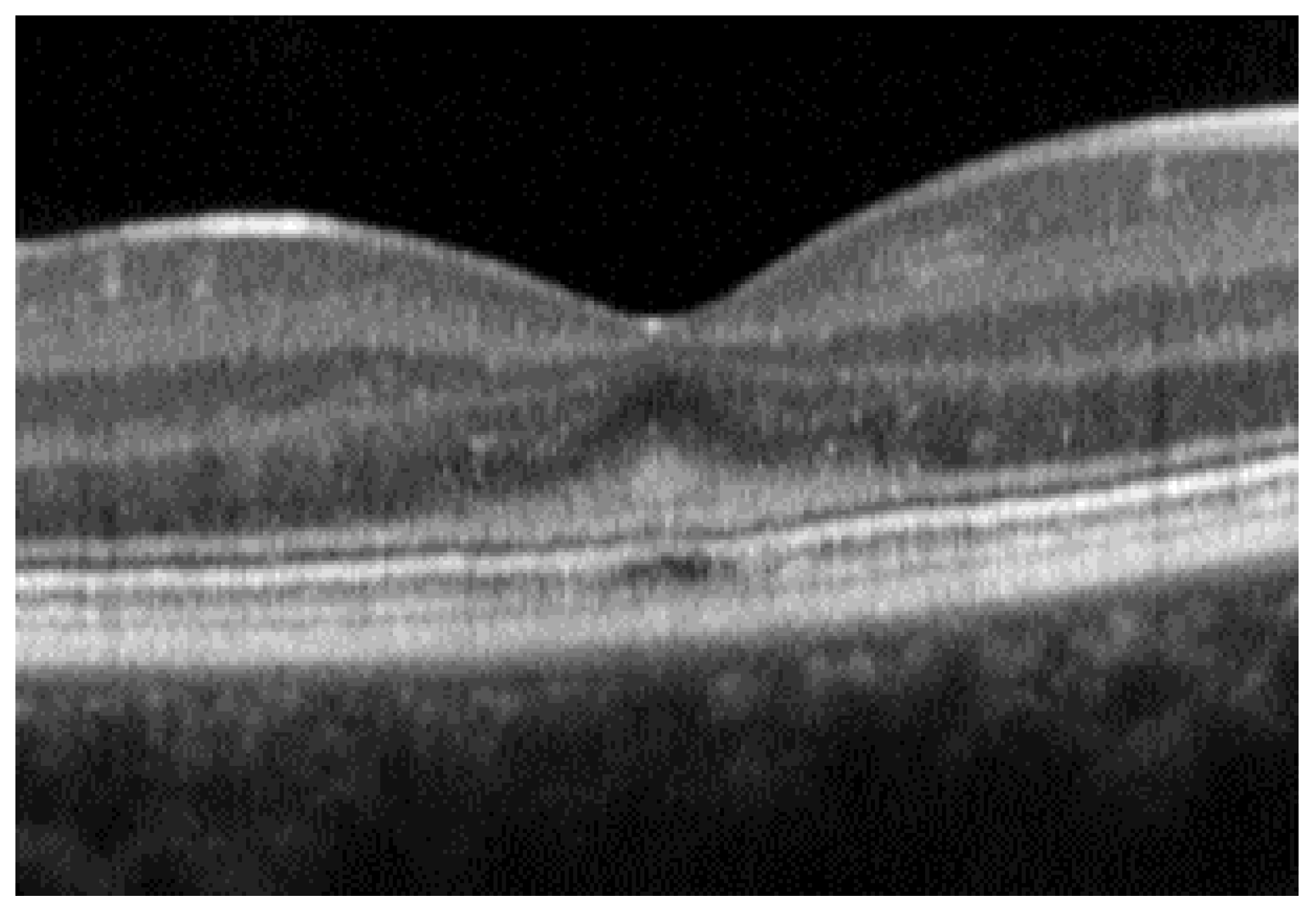
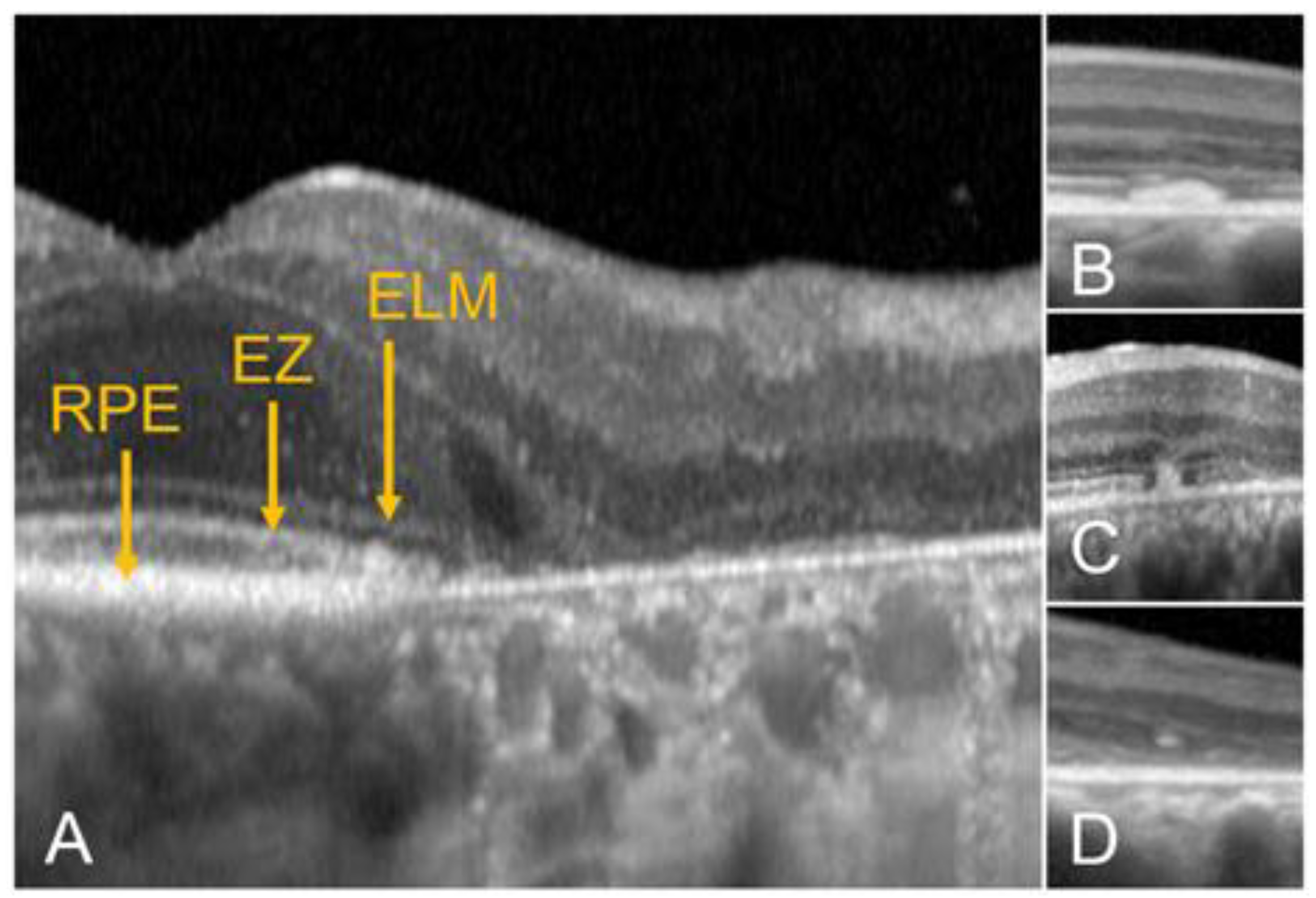
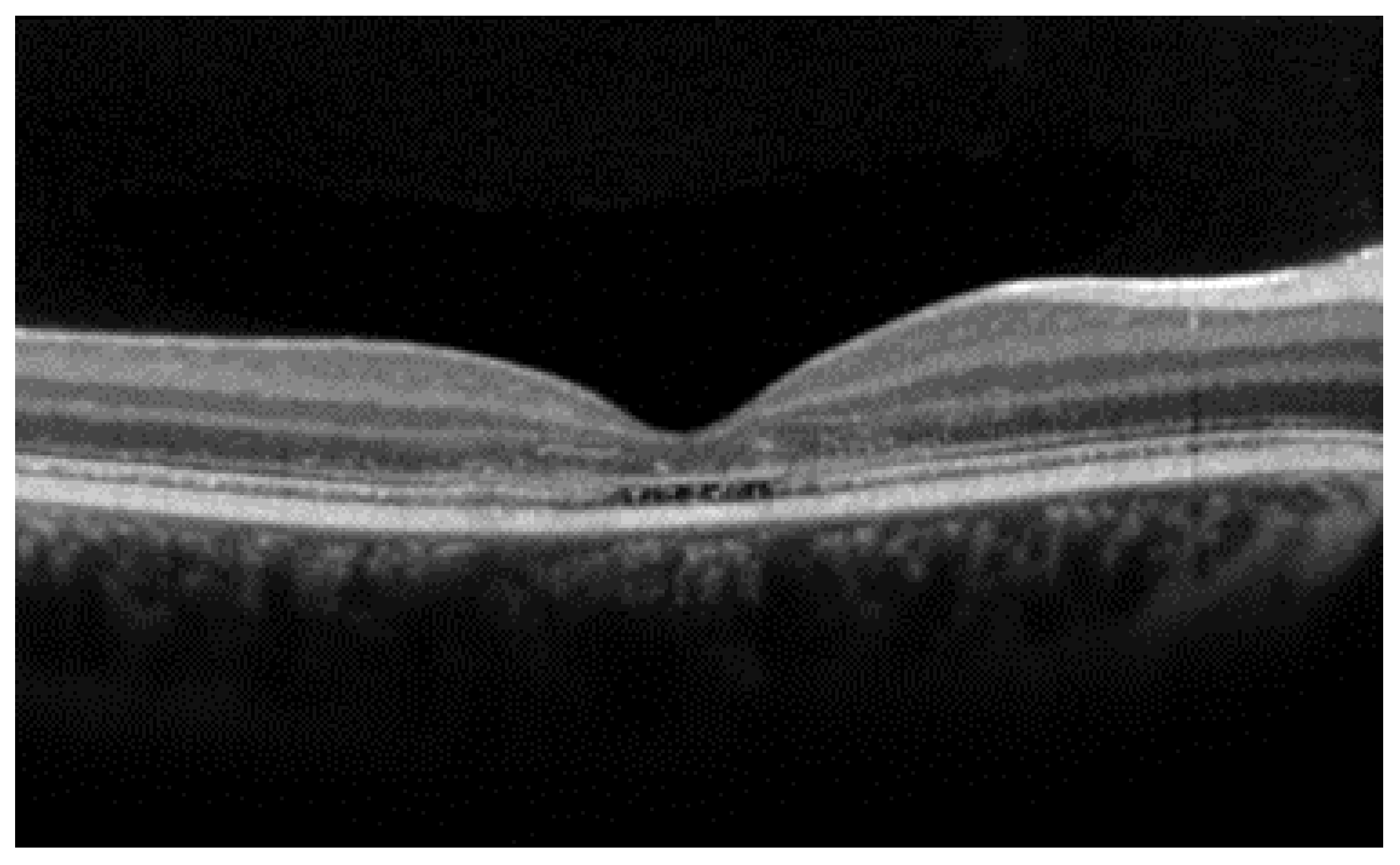
- The integrity of the EZ is the most closely linked factor to a decline in visual acuity. Its intensity, both in the affected areas and the surrounding regions, is attenuated in patients who show disease progression, and expansion of the disrupted EZ region is linked to a higher probability of visual acuity loss. Additionally, it is worth noting that many patients exhibit a central region with a preserved EZ, referred to as an optically preserved islet or OPI, which is associated with improved visual outcomes [29]. However, it is essential to recognize that EZ loss tends to occur in later stages and is typically associated with already significant visual impairment.
- Another noteworthy OCT biomarker is the measurement of ONL thickness, which can serve as an indicator of early photoreceptor degeneration. Irrespective of the clinical stage, BVMD patients exhibit reduced ONL thickness compared to healthy individuals [30]. Also, there is a correlation between ONL thickness and both BCVA and retinal sensitivity. Therefore, ONL thickness could prove to be a more valuable quantitative measure for assessing the anatomical loss of photoreceptors and the decline in macular function.
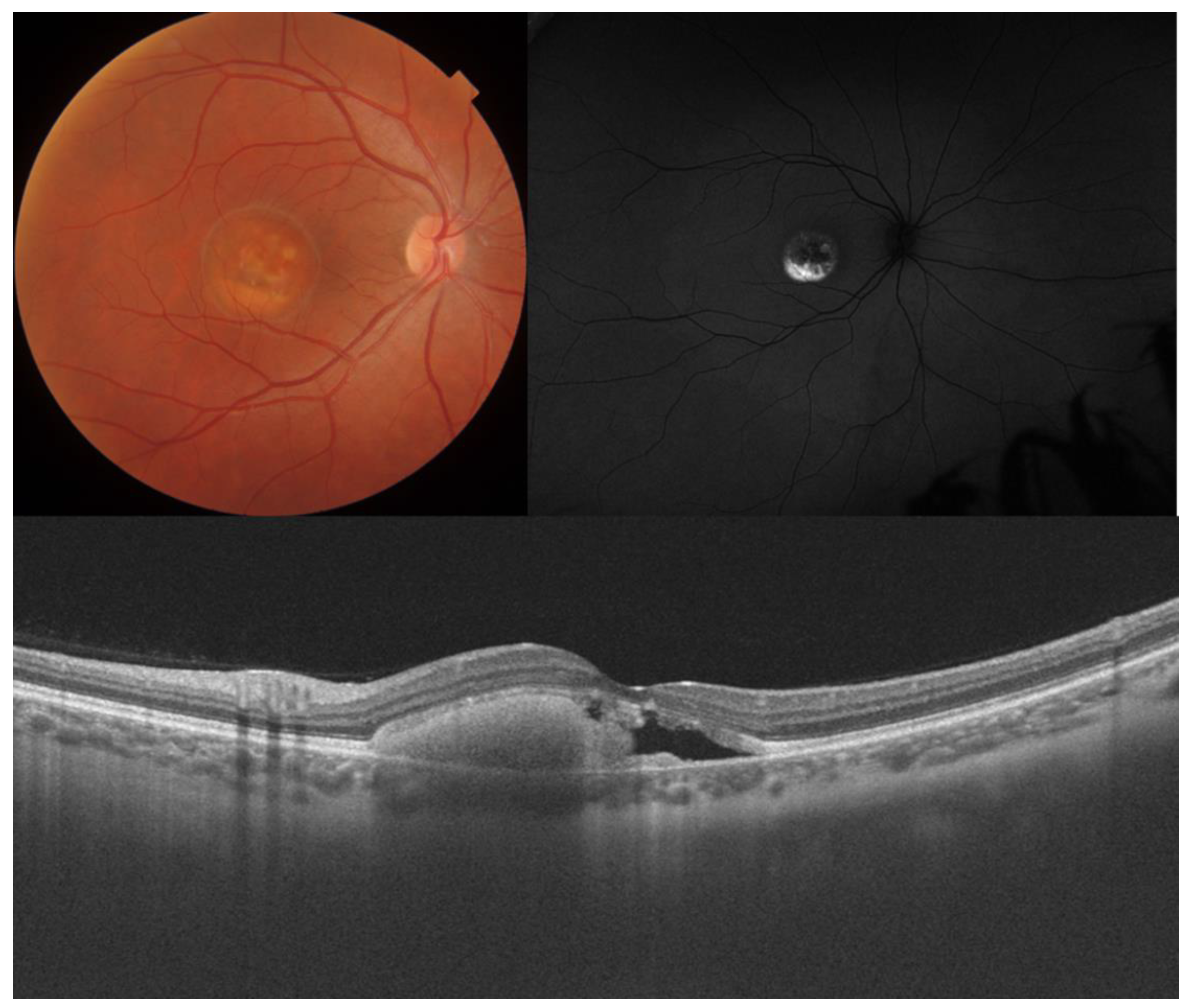
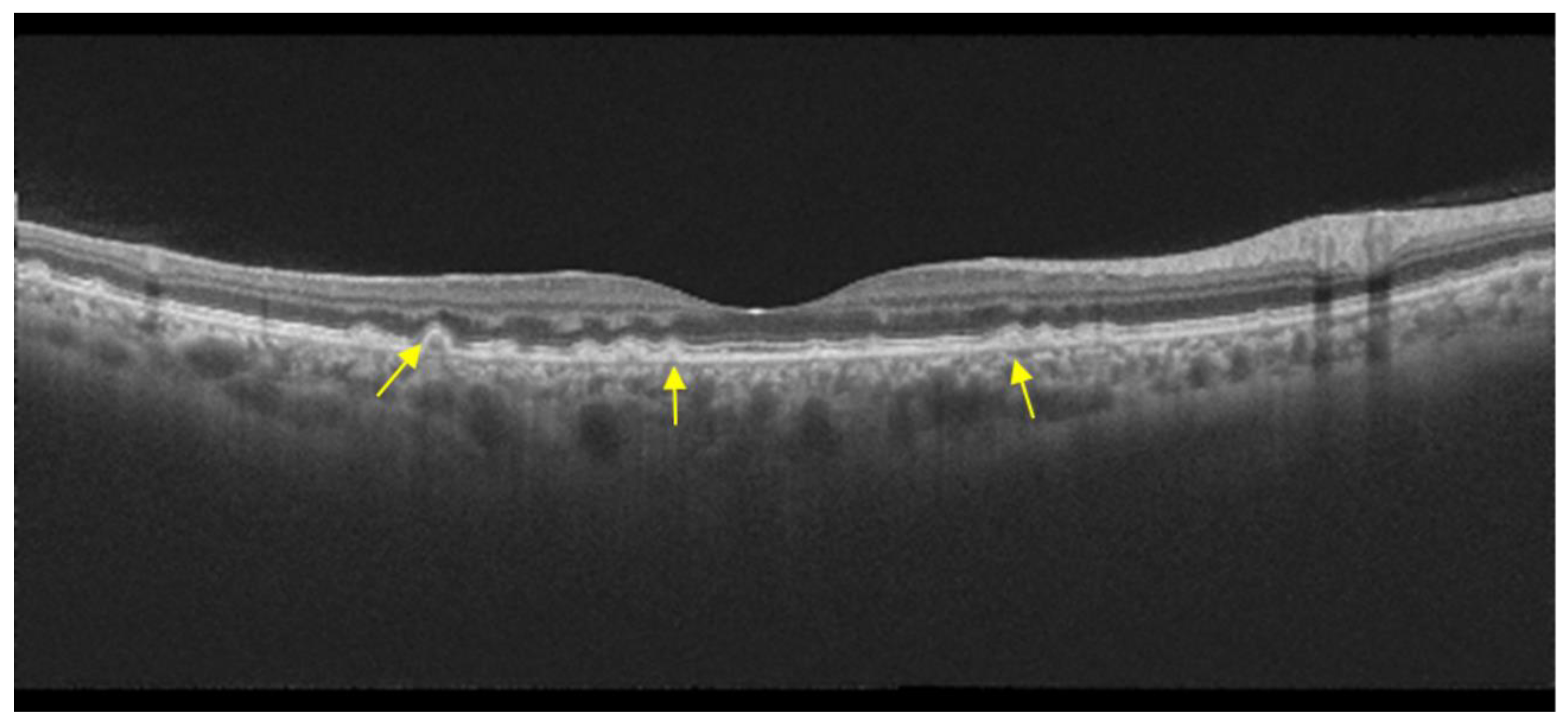
- Visual acuity is directly impacted by the presence of both vitelliform material and subretinal fluid. Eyes exhibiting vitelliform material typically demonstrate superior visual function, while the gradual reabsorption of this material, accompanied by the detection of subretinal fluid, is associated with a decline in visual acuity. Additionally, sensitivity, as measured by microperimetry, declines as vitelliform material is reabsorbed and subretinal fluid accumulates [31].
- Retinal hyperreflective foci (HRF) are punctiform lesions, typically smaller than 30 μm, primarily located in the outer nuclear and plexiform layers of the neuroretina. Battaglia Parodi et al. identified the presence of HRF in BVMD and noted an increase in their number as the disease progressed [32]. HRF may constitute accumulations of microglia or RPE cells detached from Bruch’s membrane (BM) and migrated into the subretinal space, eventually reaching the neuroretina [33]. Recent data obtained using polarization-sensitive OCT, which provides tissue-specific contrast, has lent support to the theory by demonstrating the presence of RPE cells within the vitelliform material and the neuroretina. Consequently, HRF appears to reflect the progression of the disease and may serve as a valuable biomarker for monitoring BVMD.
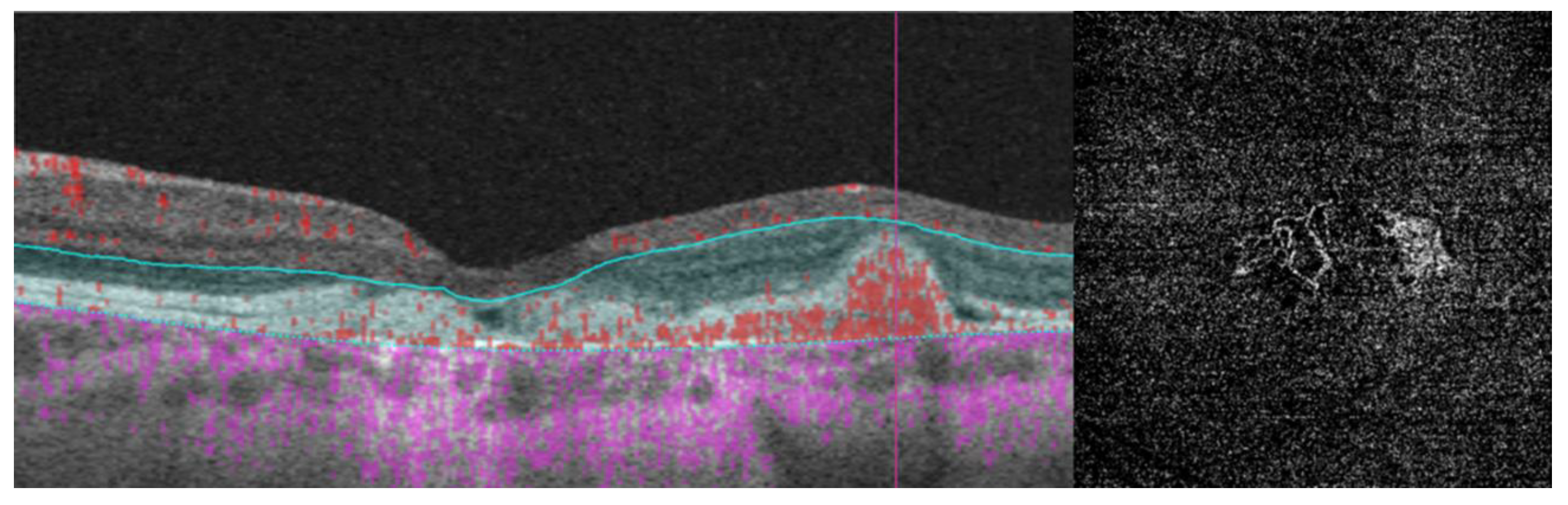
- Researchers have also explored biomarkers related to choroidal angioarchitecture in BVMD. One study examined changes in choroidal thickness across various BVMD stages, revealing that stages featuring vitelliform deposits showed a thicker choroid, while atrophic/fibrotic stages exhibited a thinner choroid compared to healthy controls. The underlying hypothesis was that the metabolic strain on the RPE and photoreceptor layers, due to the accumulation of vitelliform material, could lead to increased blood flow and the dilation of choroidal vessels, consequently resulting in greater choroidal thickness. Conversely, in later stages marked by outer retinal degeneration and reduced metabolic demands, choroidal thickness would be expected to decrease [34]. However, a more recent investigation observed no significant difference in subfoveal choroidal thickness between BVMD and control eyes [35]. On a different note, the choroidal vascularity index, a biomarker reflecting the relative ratio of vascular to stromal components in the choroid, exhibited a noteworthy decrease in BVMD irrespective of its stage [35].
- Additional OCT findings that may be detected in the later stages of BVMD include choroidal excavation and full-thickness macular holes. Choroidal excavation has been documented in individuals with BVMD and could be regarded as an indication of heightened degeneration in the outer retinal layers and the choroid [36]. Full-thickness macular holes, though rare, can be a potential complication of BVMD, as suggested by recent OCT-based research. Progressive retinal atrophy may be the most likely cause of the development of these holes.
4. Pattern Dystrophies
5. Sorsby Fundus Dystrophy
6. Autosomal Dominant Drusen
7. Occult Macular Dystrophy
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huang, D.; Swanson, E.A.; Lin, C.P.; Schuman, J.S.; Stinson, W.G.; Chang, W.; Hee, M.R.; Flotte, T.; Gregory, K.; Puliafito, C.A.; et al. Optical coherence tomography. Science 1991, 254, 1178–1181. [Google Scholar] [CrossRef] [PubMed]
- Bhende, M.; Shetty, S.; Parthasarathy, M.K.; Ramya, S. Optical coherence tomography: A guide to interpretation of common macular diseases. Indian J. Ophthalmol. 2018, 66, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Taubitz, T.; Tschulakow, A.V.; Tikhonovich, M.; Illing, B.; Fang, Y.; Biesemeier, A.; Julien-Schraermeyer, S.; Schraermeyer, U. Ultrastructural alterations in the retinal pigment epithelium and photoreceptors of a Stargardt patient and three Stargardt mouse models: Indication for the central role of RPE melanin in oxidative stress. PeerJ 2018, 6, e5215. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Tschulakow, A.; Taubitz, T.; Illing, B.; Biesemeier, A.; Julien-Schraermeyer, S.; Radu, R.A.; Jiang, Z.; Schraermeyer, U. Fundus autofluorescence, spectral-domain optical coherence tomography, and histology correlations in a Stargardt disease mouse model. FASEB J. 2020, 34, 3693–3714. [Google Scholar] [CrossRef] [PubMed]
- Cremers, F.P.M.; Lee, W.; Collin, R.W.J.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef] [PubMed]
- Fishman, G.A. Fundus flavimaculatus. A clinical classification. Arch. Ophthalmol. 1976, 94, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Lois, N.; Mukherjee, R.; McBain, V.A.; Tsunoda, K.; Tsubota, K.; Stone, E.M.; Fitzke, F.W.; Bunce, C.; Moore, A.T.; et al. A longitudinal study of Stargardt disease: Quantitative assessment of fundus autofluorescence, progression, and genotype correlations. Investig. Ophthalmol. Vis. Sci. 2013, 54, 8181–8190. [Google Scholar] [CrossRef]
- Lois, N.; Holder, G.E.; Bunce, C.; Fitzke, F.W.; Bird, A.C. Phenotypic subtypes of Stargardt macular dystrophy-fundus flavimaculatus. Arch. Ophthalmol. 2001, 119, 359–369. [Google Scholar] [CrossRef] [PubMed]
- Burke, T.R.; Yzer, S.; Zernant, J.; Smith, R.T.; Tsang, S.H.; Allikmets, R. Abnormality in the external limiting membrane in early Stargardt disease. Ophthalmic Genet. 2013, 34, 75–77. [Google Scholar] [CrossRef]
- Sadda, S.R.; Guymer, R.; Holz, F.G.; Schmitz-Valckenberg, S.; Curcio, C.A.; Bird, A.C.; Blodi, B.A.; Bottoni, F.; Chakravarthy, U.; Chew, E.Y.; et al. Consensus Definition for Atrophy Associated with Age-Related Macular Degeneration on OCT: Classification of Atrophy Report 3. Ophthalmology 2018, 125, 537–548. [Google Scholar] [CrossRef]
- Ervin, A.M.; Strauss, R.W.; Ahmed, M.I.; Birch, D.; Cheetham, J.; Ferris, F.L.; Ip, M.S.; Jaffe, G.J.; Maguire, M.G.; Schönbach, E.M.; et al. A workshop on measuring the progression of atrophy secondary to stargardt disease in the progstar studies: Findings and lessons learned. Transl. Vis. Sci. Technol. 2019, 8, 16. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.L.; Sun, M.; Grossetta Nardini, H.K.; Del Priore, L.V. Natural history of autosomal recessive Stargardt Disease in untreated eyes: A systematic review and meta-analysis of study- and individual-level data. Ophthalmology 2019, 126, 1288–1296. [Google Scholar] [CrossRef] [PubMed]
- Berisha, F.; Feke, G.T.; Aliyeva, S.; Hirai, K.; Pfeiffer, N.; Hirose, T. Evaluation of macular abnormalities in Stargardt’s disease us-ing optical coherence tomography and scanning laser ophthalmoscope microperimetry. Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 247, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Noupuu, K.; Lee, W.; Zernant, J.; Tsang, S.H.; Allikmets, R. Structural and genetic assessment of the ABCA4-associated optical gap phenotype. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7217–7226. [Google Scholar] [CrossRef] [PubMed]
- Heath Jeffery, R.C.; Lo, J.; Thompson, J.A.; Lamey, T.M.; McLaren, T.L.; De Roach, J.N.; Ayton, L.N.; Vincent, A.L.; Sharma, A.; Chen, F.K. Analysis of the outer retinal bands in ABCA4 and PRPH2-associated retinopathy using OCT. Ophthalmol. Retin. 2024, 8, 174–183. [Google Scholar] [CrossRef]
- Thiele, S.; Wu, Z.; Isselmann, B.; Pfau, M.; Guymer, R.H.; Luu, C.D. Natural history of the relative ellipsoid zone reflectivity in age-related macular degeneration. Ophthalmol. Retin. 2022, 6, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.; Grazioli, A.; Romano, F.; Aragona, E.; Bordato, A.; di Nunzio, C.; Sperti, A.; Bandello, F.; Parodi, M.B. Choroidal patterns in Stargardt disease: Correlations with visual acuity and disease progression. J. Clin. Med. 2019, 8, 1388. [Google Scholar] [CrossRef] [PubMed]
- Piri, N.; Nesmith, B.L.W.; Schaal, S. Choroidal hyperreflective foci in Stargardt disease shown by spectral-domain optical coherence tomography imaging: Correlation with disease severity. JAMA Ophthalmol. 2015, 133, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Heath Jeffery, R.C.; Chen, F.K. Macular neovascularization in inherited retinal diseases: A review. Surv. Ophthalmol. 2024, 69, 1–23. [Google Scholar] [CrossRef]
- Mastropasqua, R.; Toto, L.; Borrelli, E.; Di Antonio, L.; Mattei, P.A.; Senatore, A.; Di Nicola, M.; Mariotti, C. Optical coherence tomography angiography findings in Stargardt disease. PLoS ONE 2017, 12, e0170343. [Google Scholar] [CrossRef]
- Strauss, R.W.; Ho, A.; Munoz, B.; Cideciyan, A.V.; Sahel, J.A.; Sunness, J.S.; Birch, D.G.; Bernstein, P.S.; Michaelides, M.; Traboulsi, E.I.; et al. The natural history of the progression of atrophy secondary to Stargardt disease (ProgStar) studies: Design and baseline characteristics: ProgStar report no. 1. Ophthalmology 2016, 123, 817–828. [Google Scholar] [CrossRef] [PubMed]
- Greenstein, V.C.; Nunez, J.; Lee, W.; Schuerch, K.; Fortune, B.; Tsang, S.H.; Allikmets, R.; Sparrow, J.R.; Hood, D.C. A comparison of en face optical coherence tomography and fundus autofluorescence in Stargardt disease. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5227–5236. [Google Scholar] [CrossRef]
- Boon, C.J.F.; Klevering, B.J.; Leroy, B.P.; Hoyng, C.B.; Keunen, J.E.E.; den Hollander, A.I. The spectrum of ocular phenotypes caused by mutations in the BEST1 gene. Prog. Retin. Eye Res. 2009, 28, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Dalvin, L.A.; Pulido, J.S.; Marmorstein, A.D. Vitelliform dystrophies: Prevalence in Olmsted County, Minnesota, United States. Ophthalmic Genet. 2017, 38, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Lima de Carvalho, J.R.; Paavo, M.; Chen, L.; Chiang, J.; Tsang, S.H.; Sparrow, J.R. Multimodal imaging in Best vitelliform macular dystrophy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Rahman, N.; Georgiou, M.; Khan, K.N.; Michaelides, M. Macular dystrophies: Clinical and imaging features, molecular genetics and therapeutic options. Br. J. Ophthalmol. 2020, 104, 451–460. [Google Scholar] [CrossRef] [PubMed]
- Parodi, M.B.; Iacono, P.; Romano, F.; Bolognesi, G.; Fasce, F.; Bandello, F. Optical coherence tomography in Best vitelliform macular dystrophy. Eur. J. Ophthalmol. 2017, 27, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.X.; Charran, D.; Strong, C.R.; Steffens, T.J.; Jayasundera, T.; Heckenlively, J.R. Optical coherence tomography examination of the retinal pigment epithelium in Best Vitelliform Macular Dystrophy. Ophthalmology 2017, 124, 456–463. [Google Scholar] [CrossRef]
- Romano, F.; Arrigo, A.; Leone, P.P.; Bandello, F.; Battaglia Parodi, M. Short-term modifications of ellipsoid zone in best vitelliform macular dystrophy. Retina 2021, 41, 1010–1017. [Google Scholar] [CrossRef]
- Augstburger, E.; Orès, R.; Mohand-Said, S.; Mrejen, S.; Keilani, C.; Antonio, A.; Condroyer, C.; Andrieu, C.; Sahel, J.-A.; Zeitz, C.; et al. Outer retinal alterations associated with visual outcomes in Best Vitelliform Macular Dystrophy. Am. J. Ophthalmol. 2019, 208, 429–437. [Google Scholar] [CrossRef]
- Battaglia Parodi, M.; Bianco, L.; Arrigo, A.; Saladino, A.; Antropoli, A.; Pina, A.; Marchese, A.; Aragona, E.; Rashid, H.F.; Bandello, F. Clinical correlation between optical coherence tomography biomarkers and retinal sensitivity in Best Vitelliform Macular Dystrophy. Transl. Vis. Sci. Technol. 2022, 11, 24. [Google Scholar] [CrossRef] [PubMed]
- Parodi, M.B.; Romano, F.; Sacconi, R.; Casati, S.; Marchini, G.; Bandello, F.; Iacono, P. Intraretinal hyperreflective foci in best vitelliform macular dystrophy. Retina 2018, 38, 2379–2386. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.C.; Jung, J.J.; Curcio, C.A.; Balaratnasingam, C.; Gallego-Pinazo, R.; Dolz-Marco, R.; Freund, K.B.; Yannuzzi, L.A. Intraretinal hyperreflective foci in acquired vitelliform lesions of the macula: Clinical and histologic study. Am. J. Ophthalmol. 2016, 164, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Battaglia Parodi, M.; Sacconi, R.; Iacono, P.; Del Turco, C.; Bandello, F. Choroidal thickness in best vitelliform macular dystrophy. Retina 2016, 36, 764–769. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Roy, R.; Saurabh, K.; Sen, P.; Bhende, M.; Shelke, K.; Finocchio, L.; Sodi, A.; Virgili, G.; Invernizzi, A.; et al. Optical coherence tomography-based choroidal structural analysis and vascularity index in Best vitelliform macular dystrophy. Ophthalmol. Ther. 2022, 11, 2141–2152. [Google Scholar] [CrossRef] [PubMed]
- Coussa, R.G.; Fortenbach, C.R.; Critser, D.B.; Collins, M.M.; Tucker, B.A.; Mullins, R.F.; Sohn, E.H.; Stone, E.M.; Han, I.C. Correlation of features on OCT with visual acuity and Gass lesion type in Best vitelliform macular dystrophy. BMJ Open Ophthalmol. 2021, 6, e000860. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.N.; You, Q.S.; Li, Q.; Li, Y.; Mao, Y.; Hu, F.; Zhao, H.Y.; Tsai, F.F.; Peng, X.Y. Findings of optical coherence tomography angiography in Best vitelliform macular dystrophy. Ophthalmic Res. 2018, 60, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Garibaldi, D.C.; Li, Y.; Richard Green, W.; Zack, D.J. Butterfly-shaped pattern dystrophy: A genetic, clinical, and histopathological report. Arch. Ophthalmol. 2002, 120, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Francis, P.J.; Schultz, D.W.; Gregory, A.M.; Schain, M.B.; Barra, R.; Majewski, J.; Ott, J.; Acott, T.; Weleber, R.G.; Klein, M.L. Genetic and phenotypic heterogeneity in pattern dystrophy. Br. J. Ophthalmol. 2005, 89, 1115–1119. [Google Scholar] [CrossRef]
- Hannan, S.R.; De Salvo, G.; Stinghe, A.; Shawkat, F.; Lotery, A.J. Common spectral domain OCT and electrophysiological findings in different pattern dystrophies. Br. J. Ophthalmol. 2013, 97, 605–610. [Google Scholar] [CrossRef]
- Querques, G.; Forte, R.; Querques, L.; Massamba, N.; Souied, E.H. Natural course of adult-onset foveomacular vitelliform dystrophy: A spectral-domain optical coherence tomography analysis. Am. J. Ophthalmol. 2011, 152, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Casillo, L.; Tricarico, S.; Contento, L.; Vingolo, E.M. Clinical Features, Prognosis, and Long-Term Response to Ranibizumab of Macular CNVs in Pattern Dystrophies Spectrum: A Pilot Study. J. Ophthalmol. 2021, 16, 6698522. [Google Scholar] [CrossRef] [PubMed]
- Mimoun, G.; Caillaux, V.; Querques, G.; Rothschild, P.R.; Puche, N.; Souied, E.H. Ranibizumab for choroidal neovascularization associated with adult-onset foveomacular vitelliform dystrophy: One-year results. Retina 2013, 33, 513–521. [Google Scholar] [CrossRef] [PubMed]
- Weber, B.H.F.; Vogt, G.; Pruett, R.C.; Stöhr, H.; Felbor, U. Mutations in the tissue inhibitor of met-alloproteinases-3 (TIMP3) in patients with Sorsby’s fundus dystrophy. Nat. Genet. 1994, 8, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, S.G.; Cideciyan, A.V.; Regunath, G.; Rodriguez, F.J.; Vandenburgh, K.; Sheffield, V.C.; Stone, E.M. Night blindness in Sorsby’s fundus dystrophy reversed by vitamin A. Nat. Genet. 1995, 11, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Anand-Apte, B.; Chao, J.R.; Singh, R.; Stöhr, H. Sorsby fundus dystrophy: Insights from the past and looking to the future. J. Neurosci. Res. 2019, 97, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Raming, K.; Gliem, M.; Charbel Issa, P.; Birtel, J.; Herrmann, P.; Holz, F.G.; Pfau, M.; Hess, K. Visual dysfunction and structural correlates in Sorsby fundus dystrophy. Am. J. Ophthalmol. 2022, 234, 274–284. [Google Scholar] [CrossRef]
- Sivaprasad, S.; Webster, A.R.; Egan, C.A.; Bird, A.C.; Tufail, A. Clinical course and treatment outcomes of Sorsby fundus dystrophy. Am. J. Ophthalmol. 2008, 146, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Gliem, M.; Muller, P.L.; Mangold, E.; Holz, F.G.; Bolz, H.J.; Stohr, H.; Weber, B.H.F.; Issa, P.C. Sorsby fundus dystrophy: Novel mutations, novel phenotypic characteristics, and treatment outcomes. Investig. Ophthalmol. Vis. Sci. 2015, 56, 2664–2676. [Google Scholar] [CrossRef]
- Iyer, P.G.; Zhou, H.; Zhang, Q.; Chu, Z.; Shen, M.; Shi, Y.; Liu, J.B.; Trivizki, O.; Lam, B.L.; Wang, R.K.; et al. Swept-source optical coherence tomography detection of bruch membrane and choriocapillaris abnormalities in sorsby macular dystrophy. Retina 2022, 42, 1645–1654. [Google Scholar] [CrossRef]
- Hess, K.; Raming, K.; Gliem, M.; Charbel Issa, P.; Herrmann, P.; Holz, F.G.; Pfau, M. Choriocapillaris flow signal impairment in sorsby fundus dystrophy. Ophthalmologica 2022, 245, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.N.; Borooah, S.; Lando, L.; Dans, K.; Mahroo, O.A.; Meshi, A.; Kalitzeos, A.; Agorogiannis, G.; Moghimi, S.; Freeman, W.R.; et al. Quantifying the separation between the retinal pigment epithelium and bruch’s membrane using optical coherence tomography in patients with inherited macular degeneration. Transl. Vis. Sci. Technol. 2020, 9, 26. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.; Giralt, J.; Alforja, S.; Casaroli-Marano, R.P. Altering the clinical course of Sorsby fundus dystrophy with the use of anti-vascular endothelial growth factor intraocular therapy. Retin. Cases Brief. Rep. 2015, 9, 104–105. [Google Scholar] [CrossRef] [PubMed]
- Mohla, A.; Khan, K.; Kasilian, M.; Michaelides, M. OCT angiography in the management of choroidal neovascular membrane secondary to Sorsby fundus dystrophy. BMJ Case Rep. 2016, 2016, 216453. [Google Scholar] [CrossRef] [PubMed]
- Chatziralli, I.; Theodossiadis, G.; Panagiotidis, D.; Pousoulidi, P.; Theodossiadis, P. Choriocapillaris’ alterations in the presence of reticular pseudodrusen compared to drusen: Study based on OCTA findings. Int. Ophthalmol. 2018, 38, 1887–1893. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Lotery, A.J.; Munier, F.L.; Héon, E.; Piguet, B.; Guymer, R.H.; Vandenburgh, K.; Cousin, P.; Nishimura, D.; Swiderski, R.E.; et al. A single EFEMP1 mutation associated with both Malattia Leventinese and Doyne honeycomb retinal dystrophy. Nat. Genet. 1999, 22, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Gerber, D.M.; Munier, F.L.; Niemeyer, G. Cross-sectional study of visual acuity and electroretinogram in two types of dominant drusen. Investig. Ophthalmol. Vis. Sci. 2003, 44, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Souied, E.H.; Leveziel, N.; Querques, G.; Darmon, J.; Coscas, G.; Soubrane, G. Indocyanine green angiography features of Malattia leventinese. Br. J. Ophthalmol. 2006, 90, 296–300. [Google Scholar] [CrossRef]
- Querques, G.; Guigui, B.; Leveziel, N.; Querques, L.; Bandello, F.; Souied, E.H. Multimodal morphological and functional characterization of Malattia Leventinese. Graefe’s Arch. Clin. Exp. Ophthalmol. 2013, 251, 705–714. [Google Scholar] [CrossRef]
- Zweifel, S.A.; Maygar, I.; Berger, W.; Tschuor, P.; Becker, M.; Michels, S. Multimodal imaging of autosomal dominant drusen. Klin. Monbl. Augenheilkd. 2012, 229, 399–402. [Google Scholar] [CrossRef]
- Serra, R.; Coscas, F.; Messaoudi, N.; Srour, M.; Souied, E. Choroidal neovascularization in malattia leventinese diagnosed using optical coherence tomography angiography. Am. J. Ophthalmol. 2017, 176, 108–117. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Kameya, S.; Kikuchi, S.; Ueno, S.; Kondo, M.; Hayashi, T.; Shinoda, K.; Machida, S.; Kuniyoshi, K.; Kawamura, Y.; et al. Novel RP1L1 variants and genotype-photoreceptor microstructural phenotype associations in cohort of japanese patients with occult macular dystrophy. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4837–4846. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Yang, L.; Joo, K.; Tsunoda, K.; Kameya, S.; Hanazono, G.; Fujinami-Yokokawa, Y.; Arno, G.; Kondo, M.; Nakamura, N.; et al. Clinical and genetic characteristics of east Asian patients with occult macular dystrophy (Miyake disease): East Asia occult macular dystrophy studies report number 1. Ophthalmology 2019, 126, 1432–1444. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Tsunoda, K.; Mizuno, Y.; Usui, T.; Hatase, T.; Ueno, S.; Kuniyoshi, K.; Hayashi, T.; Katagiri, S.; Kondo, M.; et al. Clinical stages of occult macular dystrophy based on optical coherence tomographic findings. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4691–4700. [Google Scholar] [CrossRef]
- Kato, Y.; Hanazono, G.; Fujinami, K.; Hatase, T.; Kawamura, Y.; Iwata, T.; Miyake, Y.; Tsunoda, K. Parafoveal photoreceptor abnormalities in asymptomatic patients with RP1L1 mutations in families with occult macular dystrophy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6020–6029. [Google Scholar] [CrossRef]
- Chen, C.J.; Scholl, H.P.; Birch, D.G.; Iwata, T.; Miller, N.R.; Goldberg, M.F. Characterizing the phenotype and genotype of a family with occult macular dystrophy. Arch. Ophthalmol. 2012, 130, 1554–1559. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of OCT | Image Acquisition | Scanning Speed | Axial Resolution | Transverse Resolution | Range of Imaging |
|---|---|---|---|---|---|
| Time domain | Superluminescent diode (810 nm) single photon detector, moving mirror | 400 A-scans per second | 10 µm | 20 µm | Vitreoretinal interface to RPE |
| Spectral domain | Broadband superluminescent diode source (840 nm), array of detectors, fixed mirror | 27,000–70,000 A-scans per second | 5–7 µm | 14–20 µm | Posterior cortical vitreous to sclera using EDI mode |
| Swept source | Swept-source tunable laser (1050 nm), single detector | 100,000–400,000 A-scans per second | 5 µm | 20 µm | Posterior cortical vitreous to sclera using EDI mode |
| Stargardt disease |
|
| Best vitelliform macular dystrophy |
|
| Pattern dystrophies |
|
| Sorsby fundus dystrophy |
|
| Autosomal dominant drusen |
|
| Occult macular dystrophy |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gómez-Benlloch, A.; Garrell-Salat, X.; Cobos, E.; López, E.; Esteve-Garcia, A.; Ruiz, S.; Vázquez, M.; Sararols, L.; Biarnés, M. Optical Coherence Tomography in Inherited Macular Dystrophies: A Review. Diagnostics 2024, 14, 878. https://doi.org/10.3390/diagnostics14090878
Gómez-Benlloch A, Garrell-Salat X, Cobos E, López E, Esteve-Garcia A, Ruiz S, Vázquez M, Sararols L, Biarnés M. Optical Coherence Tomography in Inherited Macular Dystrophies: A Review. Diagnostics. 2024; 14(9):878. https://doi.org/10.3390/diagnostics14090878
Chicago/Turabian StyleGómez-Benlloch, Alba, Xavier Garrell-Salat, Estefanía Cobos, Elena López, Anna Esteve-Garcia, Sergi Ruiz, Meritxell Vázquez, Laura Sararols, and Marc Biarnés. 2024. "Optical Coherence Tomography in Inherited Macular Dystrophies: A Review" Diagnostics 14, no. 9: 878. https://doi.org/10.3390/diagnostics14090878