
Prevalent Variants in the LDLR Gene Impair Responsiveness to Rosuvastatin among Family Members of Patients with Premature Myocardial Infarction
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Patients and Methods
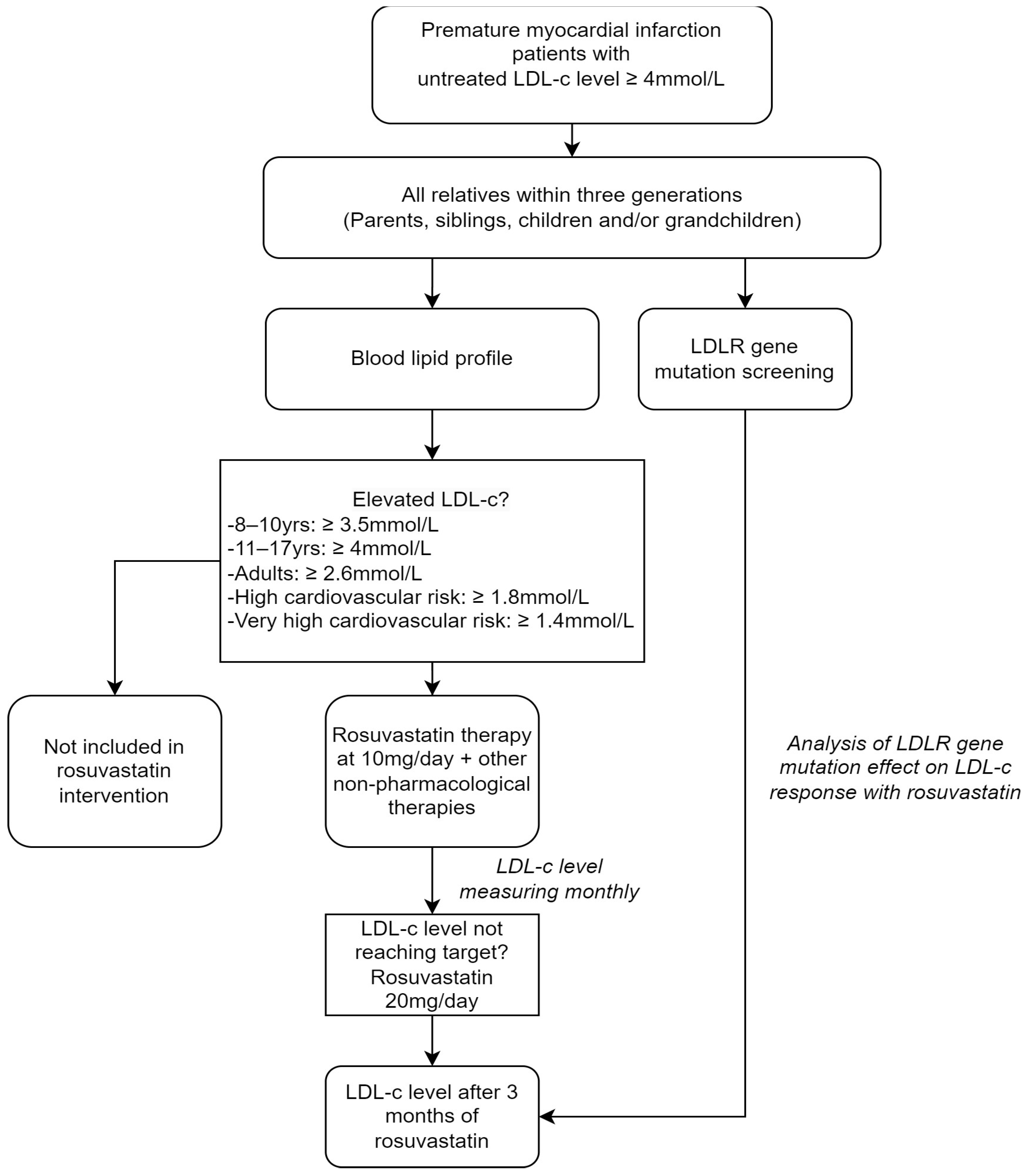
2.1. Subjects and Study Design
2.2. LDLR Gene Variant Screening and Patient Genotyping
2.3. Study Protocol
2.3.1. Outcomes
2.3.2. Statistical Analysis
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Kayikcioglu, M.; Ozkan, H.S.; Yagmur, B. Premature Myocardial Infarction: A Rising Threat. Balk. Med. J. 2022, 39, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Dugani, S.B.; Hydoub, Y.M.; Ayala, A.P.; Reka, R.; Nayfeh, T.; Ding, J.; McCafferty, S.N.; Alzuabi, M.; Farwati, M.; Murad, M.H.; et al. Risk Factors for Premature Myocardial Infarction: A Systematic Review and Meta-analysis of 77 Studies. Mayo Clin. Proc. Innov. Qual. Outcomes 2021, 5, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R. Genetics of premature myocardial infarction. Curr. Atheroscler. Rep. 2008, 10, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R.; O’kane, M.; McGilligan, V.; Watterson, S. The genetics and screening of familial hypercholesterolaemia. J. Biomed. Sci. 2016, 23, 39. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.-X.; Wu, N.-Q.; Sun, D.; Liu, H.-H.; Jin, J.-L.; Li, S.; Guo, Y.-L.; Zhu, C.-G.; Gao, Y.; Dong, Q.-T.; et al. Application of expanded genetic analysis in the diagnosis of familial hypercholesterolemia in patients with very early-onset coronary artery disease. J. Transl. Med. 2018, 16, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Neil, A.; Cooper, J.; Betteridge, J.; Capps, N.; McDowell, I.; Durrington, P.; Seed, M.; Humphries, S.E. Reductions in all-cause, cancer, and coronary mortality in statin-treated patients with heterozygous familial hypercholesterolaemia: A prospective registry study. Eur. Heart J. 2008, 29, 2625–2633. [Google Scholar] [CrossRef]
- Austin, M.A.; Zimmern, R.L.; Humphries, S.E. High “population attributable fraction” for coronary heart disease mortality among relatives in monogenic familial hypercholesterolemia. Genet Med. 2002, 4, 275–278. [Google Scholar] [CrossRef]
- Zimmerman, J.; Duprez, D.; Veach, P.M.; Zierhut, H.A. Barriers to the identification of familial hypercholesterolemia among primary care providers. J. Community Genet. 2019, 10, 229–236. [Google Scholar] [CrossRef]
- Nordestgaard, B.G.; Chapman, M.J.; Humphries, S.E.; Ginsberg, H.N.; Masana, L.; Descamps, O.S.; Wiklund, O.; Hegele, R.A.; Raal, F.J.; Defesche, J.C.; et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: Guidance for clinicians to prevent coronary heart disease: Consensus Statement of the European Atherosclerosis Society. Eur. Heart J. 2013, 34, 3478–3490. [Google Scholar] [CrossRef]
- Tran, A.V.; Nguyen, T.T.; Tran, L.N.T.; Nguyen, P.M.; Nguyen, T. Efficacy of Rosuvastatin and Atorvastatin in Vietnamese Patients with Acute Coronary Syndrome: A randomized trial. Pharm Sci Asia. 2021, 48, 413–419. [Google Scholar] [CrossRef]
- Truong, T.H.; Kim, N.T.; Nguyen, M.N.T.; Pang, J.; Hooper, A.J.; Watts, G.F.; Do, D.L. Homozygous familial hypercholesterolaemia in Vietnam: Case series, genetics and cascade testing of families. Atherosclerosis 2018, 277, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.-H.; Do, D.-L.; Kim, N.-T.; Nguyen, M.-N.T.; Le, T.-T.; Le, H.-A. Genetics, Screening, and Treatment of Familial Hypercholesterolemia: Experience Gained from the Implementation of the Vietnam Familial Hypercholesterolemia Registry. Front. Genet. 2020, 11, 914. [Google Scholar] [CrossRef] [PubMed]
- Couture, P.; Brun, L.D.; Szots, F.; Lelièvre, M.; Gaudet, D.; Després, J.-P.; Simard, J.; Lupien, P.J.; Gagné, C. Association of Specific LDL Receptor Gene Mutations With Differential Plasma Lipoprotein Response to Simvastatin in Young French Canadians With Heterozygous Familial Hypercholesterolemia. Arter. Thromb. Vasc. Biol. 1998, 18, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Roy, G.; Couture, P.; Genest, J.; Ruel, I.; Baass, A.; Bergeron, J.; Brisson, D.; Brunham, L.R.; Cermakova, L.; Gaudet, D.; et al. Influence of the LDL-Receptor Genotype on Statin Response in Heterozygous Familial Hypercholesterolemia: Insights from the Canadian FH Registry. Can. J. Cardiol. 2022, 38, 311–319. [Google Scholar] [CrossRef]
- Miltiadous, G.; Xenophontos, S.; Bairaktari, E.; Ganotakis, M.; Cariolou, M.; Elisaf, M. Genetic and environmental factors affecting the response to statin therapy in patients with molecularly defined familial hypercholesterolaemia. Pharmacogenet. Genom. 2005, 15, 219–225. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk. Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Chora, J.R.; Iacocca, M.A.; Tichy, L.; Wand, H.; Kurtz, C.L.; Zimmermann, H.; Leon, A.; Williams, M.; Humphries, S.E.; Hooper, A.J.; et al. The Clinical Genome Resource (ClinGen) Familial Hypercholesterolemia Variant Curation Expert Panel Consensus Guidelines for LDLR Variant Classification. Genet. Genom. Med. 2021, 24, 293–306. [Google Scholar] [CrossRef]
- Huang, C.-C.; Niu, D.-M.; Charng, M.-J. Genetic Analysis in a Taiwanese Cohort of 750 Index Patients with Clinically Diagnosed Familial Hypercholesterolemia. J. Atheroscler. Thromb. 2022, 29, 639–653. [Google Scholar] [CrossRef]
- Setia, N.; Saxena, R.; Arora, A.; Verma, I.C. Spectrum of mutations in homozygous familial hypercholesterolemia in India, with four novel mutations. Atherosclerosis 2016, 255, 31–36. [Google Scholar] [CrossRef]
- Grenkowitz, T.; Kassner, U.; Wühle-Demuth, M.; Salewsky, B.; Rosada, A.; Zemojtel, T.; Hopfenmüller, W.; Isermann, B.; Borucki, K.; Heigl, F.; et al. Clinical characterization and mutation spectrum of German patients with familial hypercholesterolemia. Atherosclerosis 2016, 253, 88–93. [Google Scholar] [CrossRef]
- Khoo, K.; Van Acker, P.; Defesche, J.; Tan, H.; Van De Kerkhof, L.; Eijk, S.H.; Kastelein, J.; Deslypere, J. Low-density lipoprotein receptor gene mutations in a Southeast Asian population with familial hypercholesterolemia. Clin. Genet. 2000, 58, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Mollaki, V.; Drogari, E. Genetic causes of monogenic familial hypercholesterolemia in the Greek population: Lessons, mistakes, and the way forward. J. Clin. Lipidol. 2016, 10, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Moradi, A.; Maleki, M.; Ghaemmaghami, Z.; Khajali, Z.; Noohi, F.; Moghadam, M.H.; Kalyinia, S.; Mowla, S.J.; Seidah, N.G.; Malakootian, M. Mutational Spectrum of LDLR and PCSK9 Genes Identified in Iranian Patients With Premature Coronary Artery Disease and Familial Hypercholesterolemia. Front. Genet. 2021, 12, 625959. [Google Scholar] [CrossRef] [PubMed]
- Esser, V.; Limbird, L.E.; Brown, M.S.; Goldstein, J.L.; Russell, D.W. Mutational analysis of the ligand binding domain of the low density lipoprotein receptor. J. Biol. Chem. 1988, 263, 13282–13290. [Google Scholar] [CrossRef] [PubMed]
- Wiesbauer, F.; Blessberger, H.; Azar, D.; Goliasch, G.; Wagner, O.; Gerhold, L.; Huber, K.; Widhalm, K.; Abdolvahab, F.; Sodeck, G.; et al. Familial-combined hyperlipidaemia in very young myocardial infarction survivors (< or =40 years of age). Eur. Heart J. 2009, 30, 1073–1079. [Google Scholar] [CrossRef]
- Pirazzi, C.; Håkansson, L.; Gustafsson, C.; Omerovic, E.; Wiklund, O.; Mancina, R.M. High prevalence of genetic determined familial hypercholesterolemia in premature coronary artery disease. Appl. Clin. Genet. 2019, 12, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Dedoussis, G.V.Z.; Genschel, J.; Bochow, B.; Pitsavos, C.; Skoumas, J.; Prassa, M.; Lkhagvasuren, S.; Toutouzas, P.; Vogt, A.; Kassner, U.; et al. Molecular characterization of familial hypercholesterolemia in German and Greek patients. Hum. Mutat. 2004, 23, 285–286. [Google Scholar] [CrossRef] [PubMed]
- Molfetta, G.A.; Zanette, D.L.; E Santos, J.; Silva, W.A. Research Article Mutational screening in the LDLR gene among patients presenting familial hypercholesterolemia in the Southeast of Brazil. Genet. Mol. Res. 2017, 16, gmr16039226. [Google Scholar] [CrossRef]
- Amsellem, S.; Briffaut, D.; Carrié, A.; Rabès, J.; Girardet, J.; Fredenrich, A.; Moulin, P.; Krempf, M.; Reznik, Y.; Vialettes, B.; et al. Intronic mutations outside of Alu-repeat-rich domains of the LDL receptor gene are a cause of familial hypercholesterolemia. Hum. Genet. 2002, 111, 501–510. [Google Scholar] [CrossRef]
- Leigh, S.; Futema, M.; Whittall, R.; Taylor-Beadling, A.; Williams, M.; Dunnen, J.T.D.; Humphries, S.E. The UCL low-density lipoprotein receptor gene variant database: Pathogenicity update. J. Med. Genet. 2017, 54, 217–223. [Google Scholar] [CrossRef]
- Santos, P.C.J.L.; Morgan, A.C.; Jannes, C.E.; Turolla, L.; Krieger, J.E.; Santos, R.D.; Pereira, A.C. Presence and type of low density lipoprotein receptor (LDLR) mutation influences the lipid profile and response to lipid-lowering therapy in Brazilian patients with heterozygous familial hypercholesterolemia. Atherosclerosis 2014, 233, 206–210. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient’s Characteristics | Number of Cases (%) |
|---|---|
| Age (years) | |
| ≥60 | 14 (14.1%) |
| 50–59 | 9 (9.1%) |
| 40–49 | 25 (25.3%) |
| 30–39 | 20 (20.2%) |
| 20–29 | 5 (5.1%) |
| <20 | 26 (26.3%) |
| Mean age (years) | 37.3 ± 18.5 * |
| Male sex (yes) | 56 (56.6%) |
| BMI classification | |
| Obese (yes) | 8 (8.1%) |
| Overweight (yes) | 36 (36.4%) |
| Normal (yes) | 55 (55.6%) |
| Increased waist length (yes) | 19 (19.2%) |
| Increased waist to hip ratio (Yes) | 31 (31.3%) |
| Cardiovascular risk factors | |
| Smoking (yes) | 11 (11.1%) |
| Sedentary lifestyle (yes) | 62 (62.6%) |
| Hypertension (yes) | 10 (10.1%) |
| Diabetes (yes) | 7 (7.1%) |
| Previous myocardial infarction (yes) | 8 (8.1%) |
| Lipid profile | |
| Hyperlipidemia (yes) | 71 (71.7%) |
| Increased LDL-c levels (yes) | 47 (47.5%) |
| Increased triglyceride levels (yes) | 51 (51.5%) |
| Increased cholesterol levels (yes) | 24 (24.3%) |
| Decreased HDL-C levels (yes) | 22 (22.2%) |
| Increased LDL-c + triglyceride levels (yes) | 28 (28.3%) |
| Increased LDL-c + decreased HDL-c levels (yes) | 12 (12.1%) |
| Increased LDL-c + triglyceride + decreased HDL-c levels (yes) | 12 (12.1%) |
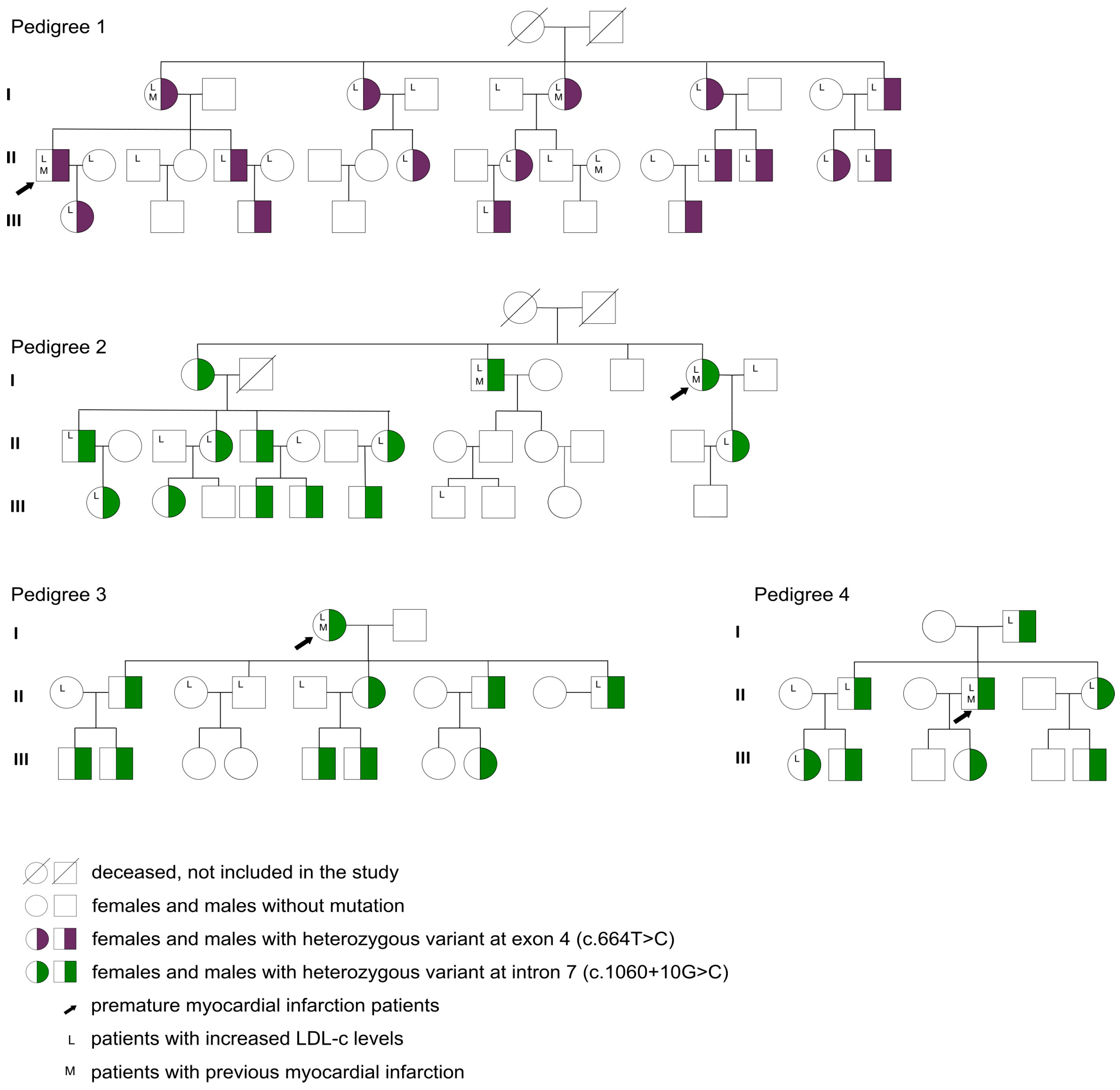
| LDLR gene variant (yes) | 49/99 (49.5%) |
| Heterozygous genotype (yes) | 49/49 (100%) |
| Exon 4 (c.664T>C) (yes) | 17/49 (34.7%) |
| Intron 7 (c.1060+10G>C) (yes) | 32/49 (65.3%) |
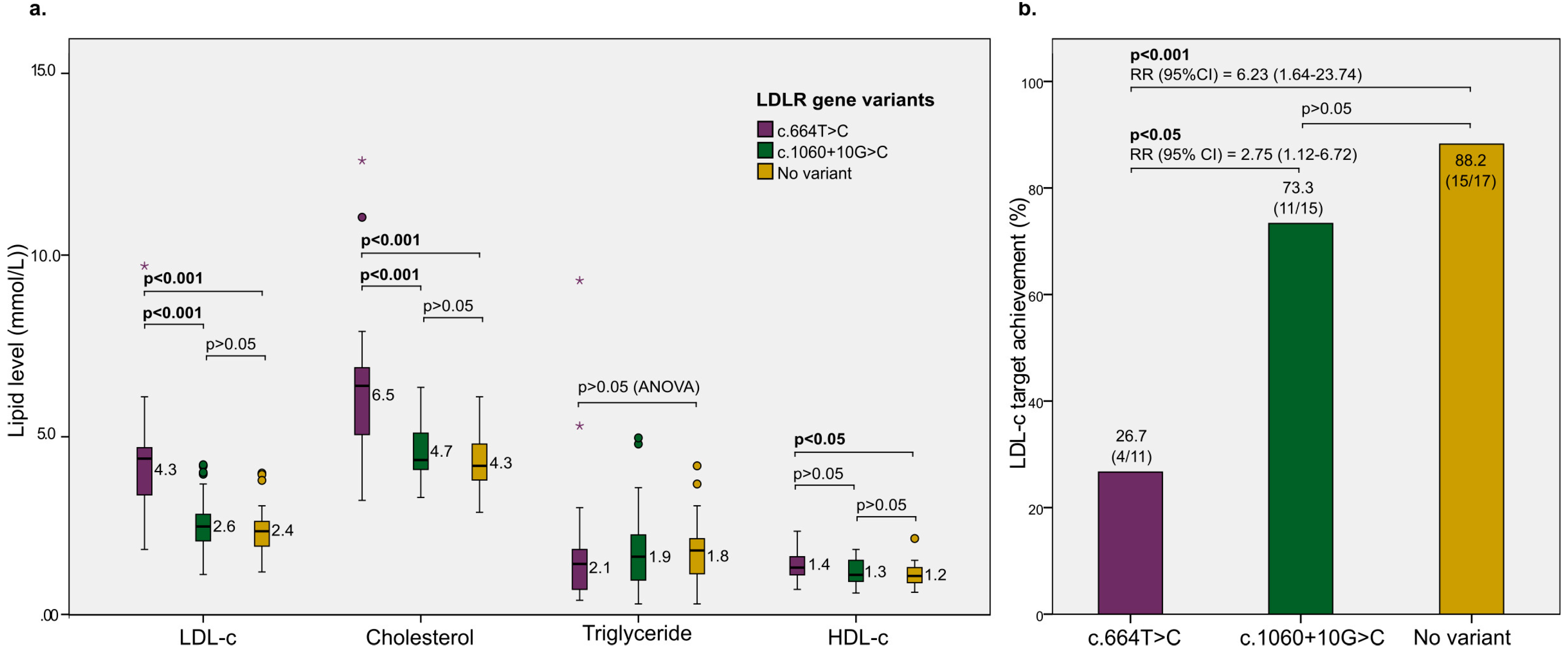
| Patients enrolled in rosuvastatin intervention (yes) | 47 (47.5%) |
| Patients achieving LDL-c target after 3 months (yes) | 30/47 (63.8%) |
| Characteristics | LDLR Gene Variant | OR (CI 95%) | p-Value; χ2 | |
|---|---|---|---|---|
| Yes | No | |||
| Male sex (Yes) | 28 | 28 | 1.04 (0.47–2.32) | 0.91; 0.013 |
| Increased LDL-c level (Yes) | 30 | 17 | 3.06 (1.35–6.96) | 0.007; 7.356 |
| Increased cholesterol level (Yes) | 19 | 5 | 5.79 (1.92–16.92) | 0.001; 11.15 |
| Hypertension (Yes) | 6 | 4 | 1.6 (0.42–6.07) | 0.357; 0.494 |
| Diabetes (Yes) | 5 | 2 | 2.72 (0.50–14.78) | 0.210; 1.492 |
| Covariates | LDL-c Target After 3 Months | Crude OR (CI 95%) | Crude p-Value | Adjusted OR (CI 95%) | Adjusted p-Value | |
|---|---|---|---|---|---|---|
| Achieved | Not Achieved | |||||
| LDLR gene variant (yes) | 15/30 | 15/17 | 0.13 (0.03–0.69) | 0.02 | 0.13 (0.02–0.68) | 0.02 |
| Sex (females) | - | - | - | - | 0.33 (0.07–1.52) | 0.15 |
| Age (+1 year) | - | - | - | - | 1.02 (0.97–1.07) | 0.42 |
| BMI (+1 kg/m2) | - | - | - | - | 0.99 (0.64–1.52) | 0.95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kien, N.T.; Nghia, T.T.; Hoang, N.M.; Phu, T.N.T.; Nga, P.T.N.; Mai, H.T.T.; Espinoza, J.L. Prevalent Variants in the LDLR Gene Impair Responsiveness to Rosuvastatin among Family Members of Patients with Premature Myocardial Infarction. J. Pers. Med. 2023, 13, 1725. https://doi.org/10.3390/jpm13121725
Kien NT, Nghia TT, Hoang NM, Phu TNT, Nga PTN, Mai HTT, Espinoza JL. Prevalent Variants in the LDLR Gene Impair Responsiveness to Rosuvastatin among Family Members of Patients with Premature Myocardial Infarction. Journal of Personalized Medicine. 2023; 13(12):1725. https://doi.org/10.3390/jpm13121725
Chicago/Turabian StyleKien, Nguyen Trung, Tran Tin Nghia, Nguyen Minh Hoang, Tran Nguyen Trong Phu, Pham Thi Ngoc Nga, Ha Thi Thao Mai, and J. Luis Espinoza. 2023. "Prevalent Variants in the LDLR Gene Impair Responsiveness to Rosuvastatin among Family Members of Patients with Premature Myocardial Infarction" Journal of Personalized Medicine 13, no. 12: 1725. https://doi.org/10.3390/jpm13121725