
Mitochondrial Genetic Diversity of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) Associated with Cassava in Lao PDR

 , , ,
, , , 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
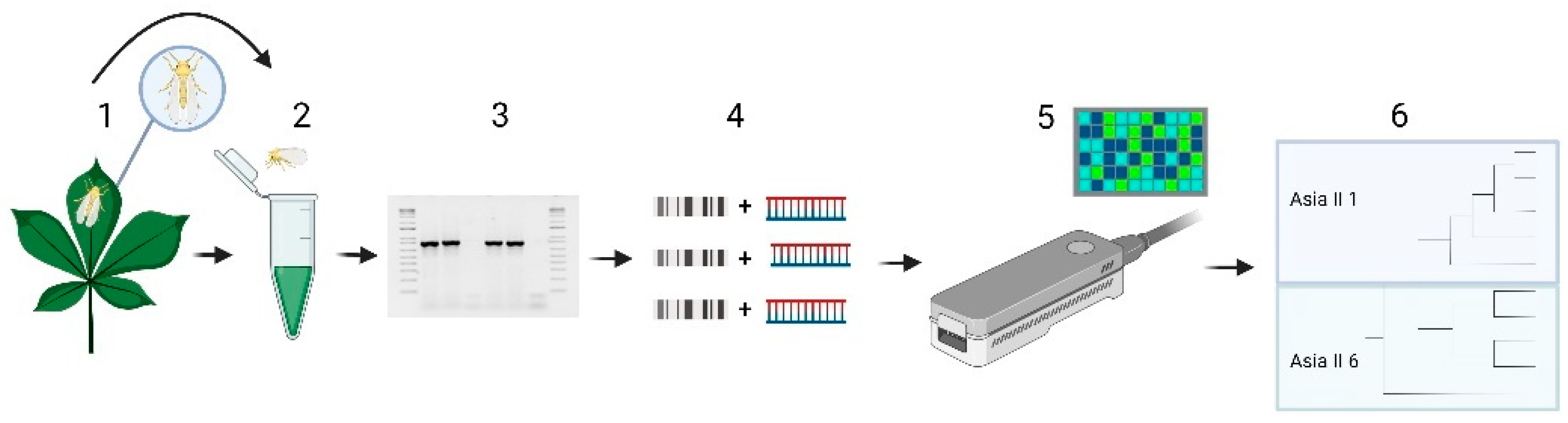
2.1. Plant Material and Whitefly Sampling
2.2. Mitochondrial COI (mtCOI) DNA Amplification
2.3. Nanopore Sequencing
2.4. Bioinformatic Analysis
2.5. Statistical Analysis
3. Results
3.1. Whitefly Abundance and CMD Incidence
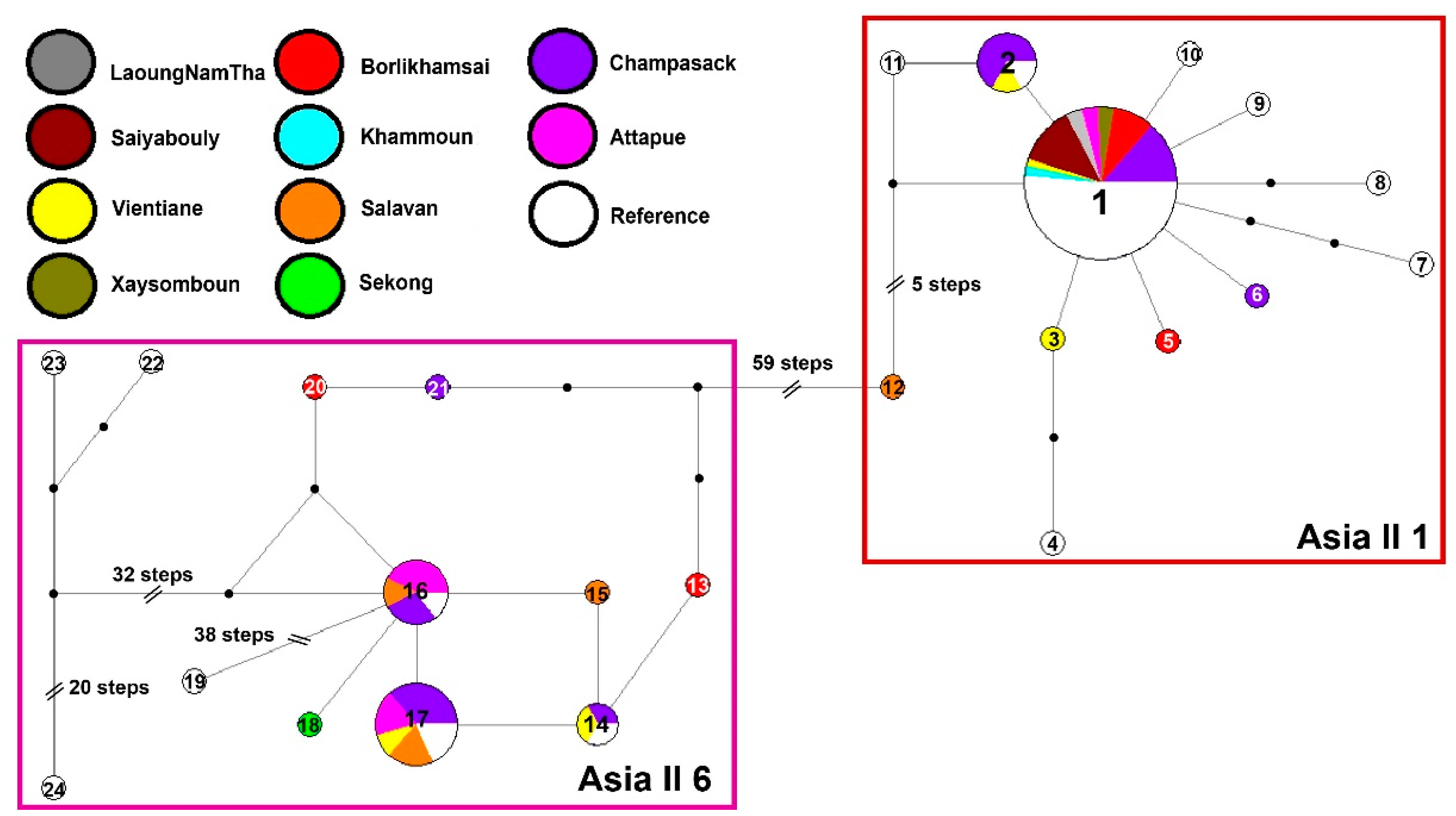
3.2. Genetic Diversity
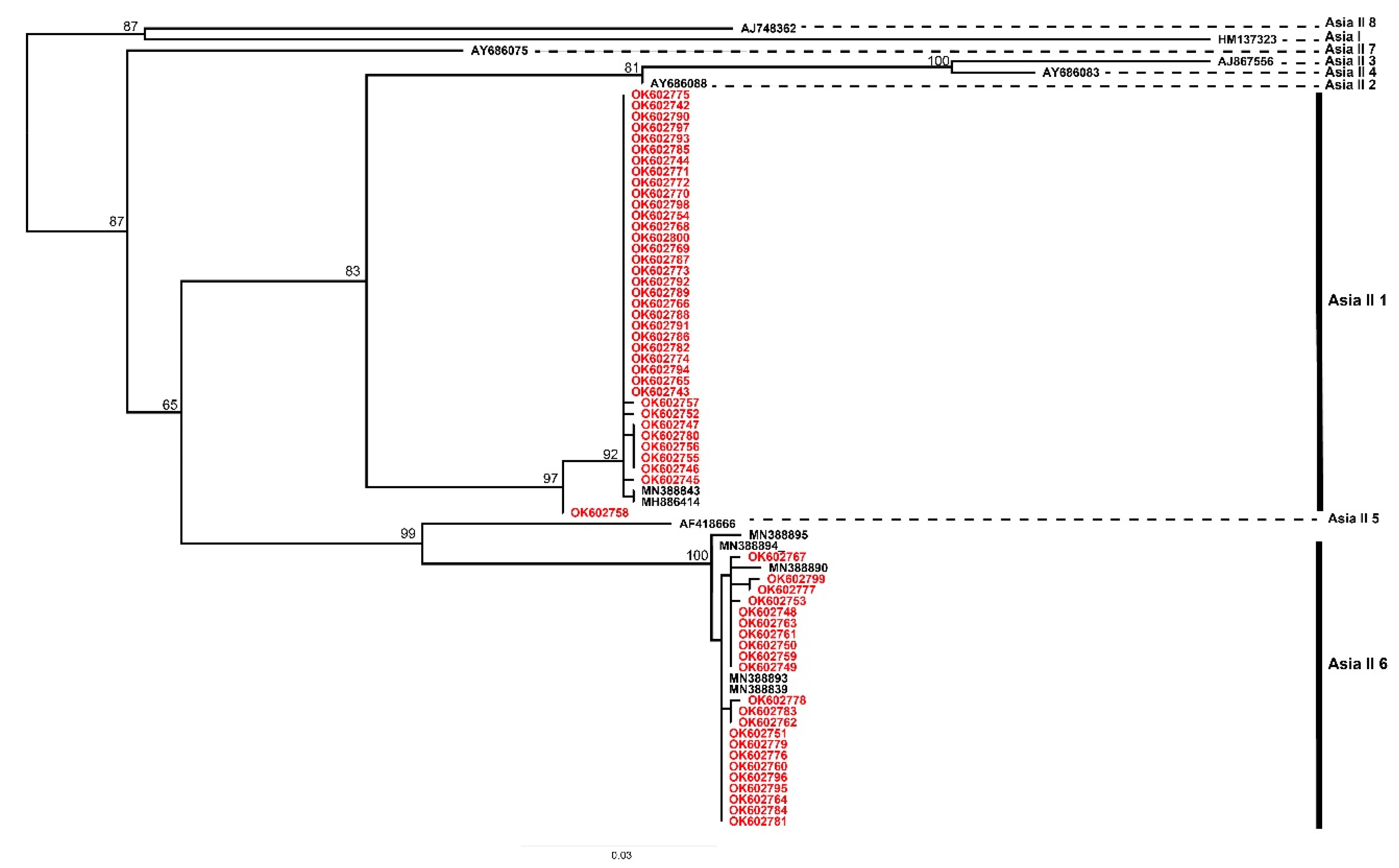
3.3. Phylogenetic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alene, A.D.; Abdoulaye, T.; Rusike, J.; Labarta, R.; Creamer, B.; del Río, M.; Ceballos, H.; Becerra, L.A. Identifying crop research priorities based on potential economic and poverty reduction impacts: The case of cassava in Africa, Asia, and Latin America. PLoS ONE 2018, 13, e0201803. [Google Scholar] [CrossRef] [PubMed]
- Howeler, R. Cassava in Asia: Trends in Cassava Production, Processing and Marketing. In Proceedings of the Partnership in Modern Science to Develop a Strong Cassava Commercial Sector in Africa and Appropriate Varieties by 2020, Bellagio, Italy, 2–6 May 2006; Available online: http://citeseerx.ist.psu.edu/viewdoc/download?doi=10.1.1.497.2007&rep=rep1&type=pdf (accessed on 1 March 2022).
- Malik, A.I.; Kongsil, P.; Nguyễn, V.A.; Ou, W.; Srean, P.; Sheela, M.N.; Becerra, L.A.; Utsumi, Y.; Lu, C.; Kittipadakul, P.; et al. Cassava breeding and agronomy in Asia- 50 years of history and future directions. Breed Sci. 2020, 20, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Legg, J.P.; Kumar, P.L.; Makeshkumar, T.; Tripathi, L.; Ferguson, M.; Kanju, E.; Ntawuruhunga, P.; Cuellar, W. Cassava virus diseases: Biology, epidemiology, and management. Adv. Virus Res. 2015, 91, 85–142. [Google Scholar]
- Siriwan, W.; Jimenez, J.; Hemniam, N.; Saokham, K.; Lopez-Alvarez, D.; Leiva, A.M.; Martinez, A.; Mwanzia, L.; Becerra, L.A.; Cuellar, W.J. Surveillance and diagnostics of the emergent Sri Lankan cassava mosaic virus (Fam. Geminiviridae) in Southeast Asia. Virus Res. 2020, 285, 197959. [Google Scholar] [CrossRef]
- Wang, H.L.; Cui, X.Y.; Wang, X.W.; Liu, S.S.; Zhang, Z.H.; Zhou, X.P. First Report of Sri Lankan Cassava Mosaic Virus Infecting Cassava in Cambodia. Plant Dis. 2016, 100, 1029. [Google Scholar] [CrossRef]
- Uke, A.; Hoat, T.X.; Quan, M.V.; Liem, N.V.; Ugaki, M.; Natsuaki, K.T. First Report of Sri Lankan Cassava Mosaic Virus Infecting Cassava in Vietnam. Plant Dis. 2018, 102, 2669. [Google Scholar] [CrossRef]
- Wang, D.; Yao, X.M.; Huang, G.X.; Shi, T.; Wang, G.F.; Ye, J. First Report of Sri Lankan Cassava Mosaic Virus Infected Cassava in China. Plant Dis. 2019, 103, 1437. [Google Scholar] [CrossRef]
- Leiva, A.M.; Siriwan, W.; Lopez-Alvarez, D.; Barrantes, I.; Hemniam, N.; Saokham, K.; Cuellar, W.J. Nanopore-based complete genome sequence of a Sri Lankan cassava mosaic virus (Geminivirus) strain from Thailand. Microbiol. Resour. Announc. 2020, 9, 1–3. [Google Scholar] [CrossRef]
- Chittarath, K.; Jimenez, J.; Vongphachanh, P.; Leiva, A.M.; Sengsay, S.; Lopez-Alvarez, D.; Bounvilayvong, T.; Lourido, D.; Vorlachith, V.; Cuellar, W.J. First report of Sri Lankan cassava mosaic virus and Cassava Mosaic Disease in Laos. Plant Dis. 2021, 105, 1861. [Google Scholar] [CrossRef]
- Legg, J.; Somado, E.A.; Barker, I.; Beach, L.; Ceballos, H.; Cuellar, W.; Elkhoury, W.; Gerling, D.; Helsen, J.; Hershey, C. A global alliance declaring war on cassava viruses in Africa. Food Secur. 2014, 6, 231–248. [Google Scholar] [CrossRef]
- Delaquis, E.; Andersen, K.F.; Minato, N.; Cu, T.T.L.; Karssenberg, M.E.; Sok, S.; Karssenberg, M.E.; Sok, S.; Wyckhuys, K.A.; Newby, J.C.; et al. Raising the stakes: Cassava seed networks at multiple scales in Cambodia and Vietnam. Front. Sustain. Food Syst. 2018, 2, 73. [Google Scholar] [CrossRef]
- Maruthi, M.N.; Colvin, J.; Seal, S.; Gibson, G.; Cooper, J. Co-adaptation between cassava mosaic geminiviruses and their local vector populations. Virus Res. 2002, 86, 71–85. [Google Scholar] [CrossRef]
- Navas-Castillo, J.; Fiallo-Olive, E.; Sanchez-Campos, S. Emerging virus diseases transmitted by whiteflies. Annu. Rev. Phytopathol. 2011, 49, 219–248. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kanakala, S.; Lebedev, G.; Kontsedalov, S.; Silverman, D.; Alon, T.; Mor, N.; Sela, N.; Luria, N.; Dombrovsky, A.; et al. Transmission of a New Polerovirus Infecting Pepper by the Whitefly Bemisia tabaci. J. Virol. 2019, 93, e00488-19. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.M.; Inoue-Nagata, A.K.; Vidal, A.H.; Ribeiro, S.G.; Nagata, T. The recombinant isolate of cucurbit aphid-borne yellows virus from Brazil is a polerovirus transmitted by whiteflies. Plant Pathol. 2020, 69, 1042–1050. [Google Scholar] [CrossRef]
- Acharya, R.; Shrestha, Y.K.; Khatun, M.F.; Lee, K.Y. Identification of Begomoviruses from Three Cryptic Species of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) in Nepal. Agronomy 2021, 11, 2032. [Google Scholar] [CrossRef]
- Hogenhout, S.A.; Ammar, E.D.; Whitfield, A.E.; Redinbaugh, M.G. Insect vector interactions with persistently transmitted viruses. Annu. Rev. Phytopathol. 2008, 46, 327–359. [Google Scholar] [CrossRef]
- Wang, X.; Blanc, S. Insect transmission of plant single-stranded DNA viruses. Annu. Rev. Phytopathol. 2021, 66, 389–405. [Google Scholar] [CrossRef]
- Kanakala, S.; Ghanim, M. Whitefly-transmitted begomoviruses and advances in the control of their vectors. In Genes, Genetics and Transgenics for Virus Resistance in Plants; Pali, B., Ed.; Caister Academic Press: Poole, UK, 2018; pp. 201–220. [Google Scholar]
- Munguti, F.M.; Kilalo, D.C.; Nyaboga, E.N.; Wosula, E.N.; Macharia, I.; Mwango’mbe, A.W. Distribution and molecular diversity of whitefly species colonizing cassava in Kenya. Insects 2021, 12, 875. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, X.; Jiang, Z.; Zhang, F.; Liu, Y.; Li, Z.; Zhang, Z. New putative cryptic species detection and genetic network analysis of Bemisia tabaci (hempitera: Aleyrodidae) in China based on mitochondrial COI sequences. Mitochondrial DNA A 2018, 29, 474–484. [Google Scholar] [CrossRef] [PubMed]
- Ellango, R.; Singh, S.T.; Rana, V.S.; Priya, N.G.; Raina, H.; Chaubey, R.; Naveen, N.C.; Mahmood, R.; Ramamurthy, V.V.; Asokan, R.; et al. Distribution of Bemisia tabaci genetic groups in India. Environ. Entomol. 2015, 44, 1258–1264. [Google Scholar] [CrossRef] [PubMed]
- Ovalle, T.M.; Parsa, S.; Hernández, M.P.; Lopez-Lavalle, L.A.B. Reliable molecular identification of nine tropical whitefly species. Ecol. Evol. 2014, 4, 3778–3787. [Google Scholar] [CrossRef] [PubMed]
- Dinsdale, A.; Cook, L.; Riginos, C.; Buckley, Y.M.; de Barro, P.J. Refined global analysis of Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) mitochondrial cytochrome oxidase 1 to identify species level genetic boundaries. Ann. Entomol. Soc. Am. 2010, 103, 196–208. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Shatters, R.G.; Powell, C.A.; Boykin, L.; Liansheng, H.; McKenzie, C.L.; Shatters, R.G.; Powell, C.A.; Boykin, L.; Liansheng, H.; McKenzie, C.L. Improved DNA barcoding method for Bemisia tabaci and related Aleyrodidae: Development of universal and Bemisia tabaci biotype-specific mitochondrial cytochrome c oxidase I polymerase chain reaction primers. J. Econ. Entomol. 2009, 102, 750–758. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Yuan, Y.A.O.; Rongjiang, W.; Fengming, Y.; Dunxiao, H.; Zhili, Z. The use of mitochondrial cytochrome oxidase I (mt COI) gene sequences for the identification of biotypes of Bemisia tabaci (Gennadius). Acta Entomol. Sin. 2022, 45, 757–763. [Google Scholar]
- Götz, M.; Winter, S. Diversity of Bemisia tabaci in Thailand and Vietnam and indications of species replacement. J. Asia Pac. Entomol. 2016, 19, 537–543. [Google Scholar] [CrossRef]
- Shadmany, M.; Boykin, L.M.; Muhamad, R.; Omar, D. Genetic diversity of Bemisia tabaci (Hemiptera: Aleyrodidae) species complex across Malaysia. J. Econ. Entomol. 2019, 112, 75–84. [Google Scholar] [CrossRef]
- Khatun, M.; Jahan, S.; Lee, S.; Lee, K.-Y. Genetic diversity and geographic distribution of the Bemisia tabaci species complex in Bangladesh. Acta Trop. 2018, 187, 28–36. [Google Scholar] [CrossRef]
- Chi, Y.; Pan, L.-L.; Bouvaine, S.; Fan, Y.-Y.; Liu, Y.-Q.; Liu, S.-S.; Seal, S.; Wang, X.-W. Differential transmission of Sri Lankan cassava mosaic virus by three cryptic species of the whitefly Bemisia tabaci complex. Virology 2020, 540, 141–149. [Google Scholar] [CrossRef]
- FAOSTAT Production Data. Available online: https://www.fao.org/faostat/es/#data/QCL (accessed on 1 March 2022).
- Gautam, S.; Crossley, M.S.; Dutta, B.; Coolong, T.; Simmons, A.M.; da Silva, A.; Snyder, W.E.; Srinivasan, R. Low Genetic Variability in Bemisia tabaci MEAM1 Populations within Farmscapes of Georgia, USA. Insects 2020, 11, 834. [Google Scholar] [CrossRef] [PubMed]
- Arias, B. Estudio Sobre el Comportamiento de la "Mosca Blanca" Aleurotrachelus Socialis Bondar (Homoptera: Aleyrodidae) en Diferentes Genotipos de Yuca Manihot Escalenta Crantz. Tesis de Maestría; Universidad Nacional de Colombia: Palmira, Colombia, 1995; p. 181. [Google Scholar]
- Cuellar, W.J.; Mwanzia, L.; Lourido, D.; Martinez, A.F.; Rodriguez, R.; Garcia, C. PestDisPlace, version 3.0; Monitoring the Distribution of Pests and Diseases; International Center for Tropical Agriculture (CIAT): Palmira, Colombia, 2018; Available online: https://pestdisplace.org (accessed on 25 July 2022).
- Mugerwa, H.; Seal, S.; Wang, H.L.; Patel, M.V.; Kabaalu, R.; Omongo, C.A.; Alicai, T.; Tairo, F.; Ndunguru, J.; Sseruwagi, P.; et al. African ancestry of New World, Bemisia tabaci-whitefly species. Sci. Rep. 2018, 8, 2734. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, A.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.H.; Zhao, Y.; Bollas, A.; Wang, Y.R.; Au, K.F. Nanopore Sequencing Technology, Bioinformatics and Applications. Nat. Biotechnol. 2021, 39, 1348–1365. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 2015, 32, 292–294. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Sseruwagi, P.; Sserubombwe, W.S.; Legg, J.P.; Ndunguru, J.; Thresh, J.M. Methods of surveying the incidence and severity of cassava mosaic disease and whitefly vector populations on cassava in Africa: A review. Virus Res. 2004, 100, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Tajebe, L.S.; Boni, S.B.; Guastella, D.; Cavalieri, V.; Lund, O.S.; Rugumamu, C.P.; Rapisarda, C.; Legg, J.P. Abundance, diversity and geographic distribution of cassava mosaic disease pandemic-associated Bemisia tabaci in Tanzania. J. Appl. Entomol. 2015, 139, 627–637. [Google Scholar] [CrossRef]
- Nwezeobi, J.; Onyegbule, O.; Nkere, C.; Onyeka, J.; van Brunschot, S.; Seal, S.; Colvin, J. Cassava whitefly species in eastern Nigeria and the threat of vector-borne pandemics from East and Central Africa. PLoS ONE 2020, 15, e0232616. [Google Scholar] [CrossRef]
- Macfayden, S.; Paull, C.; Boykin, L.M.; de Barro, P.; Maruthi, M.N.; Otim, M.; Kalyebi, A.; Vassão, D.G.; Sseruwagi, P.; Tay, W.T.; et al. Cassava whitefly, Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) in East African farming landscapes: A review of the factors determining abundance. Bull. Entomol Res. 2018, 108, 565–582. [Google Scholar] [CrossRef]
- Ally, H.M.; el Hamss, H.; Simiand, C.; Maruthi, M.N.; Colvin, J.; Omongo, C.A.; Delatte, H. What has changed in the outbreaking populations of the severe crop pest whitefly species in cassava in two decades? Sci. Rep. 2019, 9, 14796. [Google Scholar] [CrossRef]
- Mugerwa, H.; Rey, M.E.; Alicai, T.; Ateka, E.; Atuncha, H.; Ndunguru, J.; Sseruwagi, P. Genetic diversity and geographic distribution of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) genotypes associated with cassava in East Africa. Ecol. Evol. 2012, 2, 2749–2762. [Google Scholar] [CrossRef] [PubMed]
- Szyniszewska, A.M.; Busungu, C.; Boni, S.B.; Shirima, R.R.; Bouwmeester, H.; Legg, J.P. Spatial Analysis of Temporal Changes in the Pandemic of Severe Cassava Mosaic Disease in North-Western Tanzania. Phytopathology 2017, 107, 1229–1242. [Google Scholar] [CrossRef] [PubMed]
- Milenovic, M.; Wosula, E.N.; Rapisarda, C.; Legg, J.P. Impact of Host Plant Species and Whitefly Species on Feeding Behavior of Bemisia tabaci. Front. Plant Sci. 2019, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Vereecke, N.; Bokma, J.; Haesebrouck, F.; Nauwynck, H.; Boyen, F.; Pardon, B.; Theuns, S. High Quality Genome Assemblies of Mycoplasma Bovis Using a Taxon-Specific Bonito Basecaller for MinION and Flongle Long-Read Nanopore Sequencing. BMC Bioinform. 2020, 21, 517. [Google Scholar] [CrossRef]
- Grädel, C.; Miani, M.A.T.; Barbani, M.T.; Leib, S.L.; Suter-Riniker, F.; Ramette, A. Rapid and Cost-Efficient Enterovirus Genotyping from Clinical Samples Using Flongle Flow Cells. Genes 2019, 10, 659. [Google Scholar] [CrossRef]
- Cao, M.D.; Ganesamoorthy, D.; Cooper, M.A.; Coin, L.J. Realtime analysis and visualization of MinION sequencing data with npReader. Bioinformatics 2016, 32, 764–766. [Google Scholar] [CrossRef] [PubMed]
- Whitford, W.; Hawkins, V.; Moodley, K.S.; Grant, M.J.; Lehnert, K.; Snell, R.G.; Jacobsen, J.C. Proof of concept for multiplex amplicon sequencing for mutation identification using the MinION nanopore sequencer. Sci. Rep. 2022, 12, 8572. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.; Berghaus, S.; Blessing, F.; Herbeck, H.; Blessing, J.; Schierack, P.; Rödiger, S.; Roggenbuck, D.; Wenzel, F. Genotyping of familial Mediterranean fever gene (MEFV)—Single nucleotide polymorphism—Comparison of Nanopore with conventional Sanger sequencing. PLoS ONE 2022, 17, e0265622. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Chen, Y.-D.; Jiang, Z.-L.; Nardi, F.; Yang, T.-Y.; Jin, J.; Zhang, Z.-K. Global haplotype analysis of the whitefly Bemisia tabaci cryptic species Asia I in Asia. Mitochondrial DNA A 2015, 26, 232–241. [Google Scholar] [CrossRef] [PubMed]
- Kranthi, S.; Kumar, R.; Suke, R.; Chawla, S.; Kranthi, K.R. Mitochondrial COI based genetic diversity and phylogeographic structure of whitefly Bemisia tabaci (Gennadius) on cotton in India. Int. J. Trop. Insect Sci. 2021, 41, 1543–1554. [Google Scholar]
- De Barro, P.J.; Liu, S.-S.; Boykin, L.M.; Dinsdale, A.B. Bemisia tabaci: A Statement of Species Status. Annu. Rev. Entomol. 2011, 56, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Moya, A.; Guirao, P.; Cifuentes, D.; Beitia, F.; Cenis, J.L. Genetic diversity of Iberian populations of Bemisia tabaci (Hemiptera: Aleyrodidae) based on random amplified polymorphic DNA-polymerase chain reaction. Mol. Ecol. 2001, 10, 891–897. [Google Scholar] [CrossRef]
- Liu, S.-S.; de Barro, P.J.; Xu, J.; Luan, J.-B.; Zang, L.-S.; Ruan, Y.-M.; Wan, F.-H. Asymmetric mating interactions drive widespread invasion and displacement in a whitefly. Science 2007, 318, 1769–1772. [Google Scholar] [CrossRef]
- Prasanna, H.C.; Kanakala, S.; Archana, K.; Jyothsna, P.; Varma, R.K.; Malathi, V.G. Cryptic species composition and genetic diversity within Bemisia tabaci complex in soybean in India revealed by mtCOI DNA sequence. J. Integr. Agric. 2015, 14, 1786–1795. [Google Scholar] [CrossRef]
- Mahmood, M.A.; Ahmed, N.; Hussain, S.; Muntaha, S.T.; Amin, I.; Mansoor, S. Dominance of Asia II 1 species of Bemisia tabaci in Pakistan and beyond. Sci. Rep. 2022, 12, 1528. [Google Scholar] [CrossRef]
- Gentile, G.; Torre, A.D.; Maegga, B.; Powell, J.R.; Caccone, A. Genetic differentiation in the African malaria vector, Anopheles gambiae ss, and the problem of taxonomic status. Genetics 2002, 161, 1561–1578. [Google Scholar] [CrossRef] [PubMed]
- Hart, M.W.; Sunday, J. Things fall apart: Biological species form unconnected parsimony networks. Biol. Lett. 2007, 3, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Strand, M.; Norenburg, J.L.; Sun, S.; Kajihara, H.; Chernyshev, A.V.; Maslakova, S.A.; Sundberg, P. Statistical parsimony networks and species assemblages in cephalotrichid nemerteans (Nemertea). PLoS ONE 2010, 5, e12885. [Google Scholar] [CrossRef]
- De Barro, P.J.; Ahmed, M.Z. Genetic networking of the Bemisia tabaci cryptic species complex reveals pattern of biological invasions. PLoS ONE 2011, 6, e25579. [Google Scholar] [CrossRef]
- Pan, L.-L.; Cui, X.-Y.; Chen, Q.-F.; Wang, X.-W.; Liu, S.-S. Cotton leaf curl disease: Which whitefly is the vector? Phytopathology 2018, 108, 1172–1183. [Google Scholar] [CrossRef]
- Ahmed, M.Z.; de Barro, P.J.; Greeff, J.M.; Ren, S.X.; Naveed, M.; Qiu, B.L. Genetic identity of the Bemisia tabaci species complex and association with high cotton leaf curl disease (CLCuD) incidence in Pakistan. Pest Manag. Sci. 2011, 67, 307–317. [Google Scholar] [CrossRef]
- Naveen, N.C.; Chaubey, R.; Kumar, D.; Rebijith, K.B.; Rajagopal, R.; Subrahmanyam, B.; Subramanian, S. Insecticide resistance status in the whitefly, Bemisia tabaci genetic groups Asia-I, Asia-II-1 and Asia-II-7 on the Indian subcontinent. Sci. Rep. 2017, 7, 40634. [Google Scholar] [CrossRef]
- Thangavel, T. First report of Asia II 6 cryptic species from India and Asia I cryptic species of whiteflies, Bemisia tabaci (Gennadius), in Ash gourd. In Proceedings of the ESA Confex Entomology, Vancouver, BC, Canada, 11–14 November 2018; Available online: https://esa.confex.com/esa/2018/meetingapp.cgi/Paper/131026 (accessed on 1 March 2022).
- Kijima, K.; Ohno, S.; Ganaha-Kikumura, T. Effect of several commercial pesticides on the survival of the Nauru biotype of Bemisia tabaci (Hemiptera: Aleyrodidae). Jpn. J. Appl. Entomol. Zool. 2012, 56, 9–12. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Country | Province | No. of Fields | Abundance (Per Leaf) | SD | Cryptic Species | Haplotype | Ref. |
|---|---|---|---|---|---|---|---|
| Lao PDR | LaoungNamTha | 2 | 0.150 | 0.012 | Asia II 1 | H1 | This work |
| Saiyabouly | 9 | 0.041 | 0.021 | Asia II 1 | H1 | ||
| Vientiane | 5 | 0.014 | 0.012 | Asia II 1/Asia II 6 | H1, H2, H3, H14, H17 | ||
| Xaysomboun | 3 | 0.078 | 0.040 | Asia II 1 | H1 | ||
| Borlikhamsai | 11 | 0.145 | 0.034 | Asia II 1/Asia II 6 | H1, H5, H20, H13 | ||
| Khammoun | 1 | 0.166 | 0.000 | Asia II 1 | H1 | ||
| Salavan | 7 | 0.129 | 0.031 | Asia II 1/Asia II 6 | H12, H17, H15, H16 | ||
| Sekong | 1 | 0.167 | 0.000 | Asia II 6 | H18 | ||
| Champasack * | 25 | 0.177 | 0.043 | Asia II 1/Asia II 6 | H1, H2, H6, H17, H16, H14, H21 | ||
| Attapue * | 6 | 0.233 | 0.072 | Asia II 1/Asia II 6 | H1, H17, H16 | ||
| Tanzania | 2.35–71.99 (14.3) | 0.86–22.07 | SSA1-SG1-SG2-SG3 | ND | [47] | ||
| Nigeria | 24 | 2.34–265.5 | SSA1-SG5, SSA3, SSA1-SG1-Bemisia afer, MED-ASL | ND | [48] |
| Cryptic Species | n | S | NS | Pi | K | H | π | Tajima’s D |
|---|---|---|---|---|---|---|---|---|
| Asia II 1 | 37 | 10 | 9 | 4 | 0.727 | 0.417 | 0.00109 | −2.168 |
| Asia II 6 | 22 | 6 | 2 | 1 | 1.433 | 0.775 | 0.00214 | −0.644 |
| Total | 59 | 73 | 2 | 71 | 33.281 | 0.743 | 0.04975 | 3.923 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leiva, A.M.; Chittarath, K.; Lopez-Alvarez, D.; Vongphachanh, P.; Gomez, M.I.; Sengsay, S.; Wang, X.-W.; Rodriguez, R.; Newby, J.; Cuellar, W.J. Mitochondrial Genetic Diversity of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) Associated with Cassava in Lao PDR. Insects 2022, 13, 861. https://doi.org/10.3390/insects13100861
Leiva AM, Chittarath K, Lopez-Alvarez D, Vongphachanh P, Gomez MI, Sengsay S, Wang X-W, Rodriguez R, Newby J, Cuellar WJ. Mitochondrial Genetic Diversity of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) Associated with Cassava in Lao PDR. Insects. 2022; 13(10):861. https://doi.org/10.3390/insects13100861
Chicago/Turabian StyleLeiva, Ana M., Khonesavanh Chittarath, Diana Lopez-Alvarez, Pinkham Vongphachanh, Maria Isabel Gomez, Somkhit Sengsay, Xiao-Wei Wang, Rafael Rodriguez, Jonathan Newby, and Wilmer J. Cuellar. 2022. "Mitochondrial Genetic Diversity of Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) Associated with Cassava in Lao PDR" Insects 13, no. 10: 861. https://doi.org/10.3390/insects13100861