Substrate-Induced Liquid Layering: A New Insight into the Heterogeneous Nucleation of Liquid Metals

1
Key Laboratory for Advanced Materials Processing Technology, Ministry of Education, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China
2
Department of Mathematics, University of Leicester, Leicester LE1 7RH, UK
3
Department of Engineering, University of Leicester, Leicester LE1 7RH, UK
*
Authors to whom correspondence should be addressed.
Metals 2018, 8(7), 521; https://doi.org/10.3390/met8070521
Submission received: 31 May 2018
/
Revised: 29 June 2018
/
Accepted: 29 June 2018
/
Published: 6 July 2018
(This article belongs to the Special Issue 5th UK-China Steel Research Forum)
Abstract
:Liquid layering, which is a general phenomenon adjacent to the solid substrates, is less understood for its role in heterogeneous nucleation. In this work, the structural features and dynamics of the liquid Al layers induced by the (0001) sapphire and the (0001) TiB2 substrates, respectively, are quantitatively compared based on the ab initio molecular dynamics simulations. An almost fully ordered liquid Al layer is observed adjacent to the TiB2 substrate above the Al melting point, while the liquid layers near the sapphire substrate are weakly ordered with virtually no in-plane translational symmetry. Further liquid layering is facilitated by the ordered liquid layer near the TiB2 substrate, while impeded by the low in-plane ordering of the liquid layers near the sapphire substrate, resulting in different nucleation behaviors for the two systems. The difference in the liquid layering is caused, in part, by the lower adsorption strength at the sapphire–liquid Al interface than that at the TiB2–liquid Al interface. Additionally, the compressive stress imposed on the liquid layers seriously hinders the sapphire-induced liquid layering. We conclude from this work that the interfacial adsorption strength and mismatch alter the heterogeneous nucleation by influencing the features of the substrate-induced liquid layering.
1. Introduction
The ordered structures in liquid metals play a critical role in influencing wetting and nucleation behaviors [1,2]. These ordered structures may manifest as clusters with unique symmetry (such as the well-assumed icosahedral structure [3]) or quasi-layers [4,5,6,7,8,9]. The former mainly occur in the systems absence of inoculation [3] and the latter have frequently been observed near the atomically smooth surfaces [10,11,12,13]. In this work, we focus on the substrate-induced liquid layers, which have been extensively studied in the field of lubrication, epitaxial growth, superhydrophobic materials [6], energy-storage materials [8,14], etc. Apart from the aforementioned applications, the liquid layers have also been interpreted as pre-nucleation structures [15,16], which can enhance or weaken the nucleation process [7,17] and dictate their structural features. A further understanding of the substrate-induced liquid layers, in particular the in-plane atomic arrangement, can provide new insights into the heterogeneous nucleation mechanisms [17], and is also beneficial to many industrial applications, such as the design of nucleating agents [18].
Heterogeneous nucleation is a central issue in analyzing grain refinement mechanisms. After the well-accepted classical nucleation theory (CNT) was challenged by the fact that the contact angle (between nucleus and substrate) becomes unrealistic when it is less than 20° [19,20], a new adsorption-based heterogeneous model was proposed by Greer et al. [21]. This model was established based on the heterogeneous nucleation behaviors of liquid Al on the (0001) surface of TiB2 substrates, and underlined the precursor role of the substrate-induced liquid layers in triggering the heterogeneous nucleation. The liquid layers at the (0001) TiB2–liquid Al interface could be Al3Ti [22], which eliminates the lattice mismatch f at interface and thus improves the wettability and nucleant potency. The high nucleant potency of the TiB2 substrate with respect to liquid Al makes it an ideal industrial grain refiner for Al alloys [18,23]. On the contrary, the wettability of liquid Al with regard to the sapphire (α-Al2O3) substrate is much poorer, with nucleation undercooling reaching as high as 175K [17]. However, it is interesting to find that substrates with poor nucleant potency can also induce liquid layers at the solid/liquid (S/L) interface, such as the liquid Al layers that emerged at the (0001) sapphire surface [10]. These findings demonstrate that the appearance of liquid layers could be a general phenomenon when liquid metals are in contact with atomically smooth surfaces, and that the structural characteristics of the liquid layers are the factors that determine the wettability and nucleant potency of the substrates. Obviously, the free growth model mentioned above is not suited to the systems with poor nucleant substrates [24,25]. Thus, the heterogeneous nucleation of systems with potent and poor nucleant substrates may show different features, which are presumably related to the structure of the substrate-induced liquid layers. Accordingly, it is imperative and desirable to compare the liquid layers induced by potent and poor nucleant substrates, respectively, and to unveil the influence of the liquid layers on heterogeneous nucleation.
As two typical nucleant substrates of liquid Al, sapphire (poor nucleant substrate) and TiB2 (potent nucleant substrate) substrates have received much attention [22,26,27,28,29]. Liquid layering at the (0001) sapphire-liquid Al interface was first directly observed in experiments by Oh et al. [10] in 2005. Their results suggest that the (0001) sapphire-induced liquid layering occurs above the Al melting point and extends about three layers into the bulk liquid Al. Recently, using aberration-corrected high-resolution transmission electron microscopy (HRTEM), Gandman et al. [9] found that, apart from the (0001) surface of sapphire, liquid Al layering also occurred adjacent to (), (), and () sapphire surfaces, with a decreasing degree of order according to the above order. As for the TiB2 substrate, to our knowledge, no experimental work has been reported to observe the TiB2–Al S/L interface directly. Instead, the solid/solid (S/S) interface of TiB2 and Al has been extensively studied using HRTEM by Fan et al. [22] They observed a monolayer of adsorbed atoms near the (0001) TiB2 substrate, which can be regarded as a sign of TiB2-induced liquid layering.
Because of the difficulty in characterizing the S/L interfaces at the atomic scale, simulations become necessary and imperative to gain further insight into the S/L interfacial structures. Molecular dynamics (MD) [30,31], phase-field-crystal (PFC) [32,33], ab initio molecular dynamics (AIMD) [12,13,14,34,35,36,37], etc., are the popular methods used to simulate the atomic arrangement at the S/L interfaces. Compared with the other methods, AIMD obtains the interatomic potential directly from the electronic interaction, without introducing any empirical parameters. Thus, this method is appropriate to deal with the problems related to the interfaces, where the interatomic forces are more complicated than those with the bulk materials. The AIMD method has been extensively applied to characterize the structures of liquid metals [34,35,36,37], and recently has been successfully used to probe the liquid layers adjacent to the solid substrates. Wang et al. [12] used the AIMD method to characterize the features of liquid Al layering induced by the (0001) TiB2 surface. In their simulation, approximately three liquid Al layers with considerable in-plane ordering formed within 3 ps. Further AIMD study by Zhang et al. [13] suggests these liquid layers to be a rhombohedral-centered hexagonal structure aligning with the TiB2 substrate. For the sapphire–liquid Al interface, Kang et al. performed AIMD simulations to characterize the stratified structures of the liquid Al near the (0001) sapphire surface. Their results reproduced the liquid Al layers reported in Oh’s experiments, and demonstrated the formation mechanism of the transitionary interfacial structures from the perspective of thermodynamics. Although liquid layering is observed near both the (0001) sapphire and (0001) TiB2 substrates, to our knowledge, no work compares the structural or kinetic features between the liquid layers induced by these two substrates. However, such comparisons, as demonstrated above, are of great significance in further understanding the relationship between the substrate-induced liquid layers and heterogeneous nucleation.
In this work, AIMD simulations are performed to study the substrate-induced liquid layers at the S/L interfaces of (0001) sapphire–Al and (0001) TiB2–Al. The structural features and dynamical properties of the liquid layers in these two systems are compared quantitatively. From the simulation results, it can be concluded that the competition between the adsorption strength and the interfacial lattice mismatch determines the final features of the substrate-induced liquid layers, which then significantly influence the heterogeneous nucleation behavior.
2. Materials and Methods
All of the calculations in this work were performed using the Vienna ab initio simulation package (VASP) [38,39]. The interaction of the valence electrons with the ionic cores was treated using the projected-augmented waves (PAW) [40,41]. The exchange-correlation energy was described by the Perdew–Burke–Ernzerhof functional within the generalized gradient approximation (GGA-PBE) [42]. The plane wave energy cutoff was set to 400 eV and 320 eV for the sapphire–Al and TiB2–Al systems, respectively. The ground state charge density was obtained using the Gaussian smearing method, and the smearing width was set at 0.01 eV for all of the simulation systems.
The initial S/L interfacial models for the simulations were prepared using the method proposed by Wang et al. [12]. In the first step, the solid substrates were fully relaxed, until the residual forces fell below 0.01 eV/Å; meanwhile, liquid Al was prepared by melting the solid Al ( supercell) in Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) [43] using the embedded atom model (EAM) potential [44]. In the second step, a region matching the substrate was selected in the bulk liquid Al obtained from the last step, and then the selected liquid Al was re-equilibrated by continuing the molecular dynamics for 1 ps to eliminate the influence of the imposed periodic boundary condition when the surrounding atoms were removed. Finally, the re-equilibrated liquid Al was combined with the substrate. For the (0001) sapphire–liquid Al system, an Al-terminated sapphire substrate in 15 atomic layers (225 atoms) was combined with a liquid region with 192 Al atoms. A vacuum slab with a thickness of 15 Å was inserted above the liquid Al region to eliminate the influence of volume change caused by the liquid layering. For the (0001) TiB2–liquid Al system, the Ti-terminated TiB2 was used as this termination has been shown to be more stable than the B-termination [28,29]. A TiB2 in seven atomic layers (176 atoms) was combined with a liquid Al region and a 15 Å-thick vacuum slab. Both of the two interfaces built above were oriented normally to the z-axis.
The canonical (NVT) ensemble was employed to evolve the S/L interfaces built above. The time step was set at 1.0 fs for both of the two systems. The Nosé thermostat method was used to keep the system temperature around 950 K, which is just above the Al melting point from the previous AIMD calculations [45,46]. The Brillouin zone was sampled with only the point, which is accurate enough for the systems used in our simulation. The evolution of the S/L interfaces continued for 6 ps (6000 ionic steps), during which the following parameters were calculated to quantify the structural features and the dynamics of the substrate-induced liquid layering.
- Atomic density profile along the z direction , as follows:where is the number of atoms in a bin between and ; denotes the ensemble average, which was calculated by the time average in our simulations; is the volume of a bin with being its cross-sectional area and being the width. This parameter is used to analyze the out-of-plane features of the substrate-induced liquid layers.
- Orientational order parameter [47], as follows:where i, j, and k are three atoms in a bin between and ; atom j and k are the nearest neighbors of atom i; (i,j,k) denotes the xy-plane-projected angle between ij and ik; is the average number of added in one time step; n is an integer; and we used n = 6 in this work as has been shown to clearly distinguish a close-packed layer from a disordered region [48].
- Two-dimensional (2D) structure factor [49,50], as follows:is the scattering vector, which is a vector in the reciprocal space; N is the number of atoms in a bin from to ; and denotes the position of atom i. In this work, was calculated within the substrate-induced liquid layers to show the in-plane structural features.
- Diffusion coefficient D(z), as follows:
In this formula, M is the degree of freedom; denotes the position of atom i at the moment t. When calculating the mean square displacement (MSD) in our work, in addition to averaging over the atoms in a bin, 1000 different start times t0 separated by 1 fs were also averaged. For one bin, the in-plane and out-of-plane diffusion coefficients, Dxy and Dz, were calculated, respectively.
3. Results
3.1. Structural Features and Dynamical Properties of the Liquid Al Layers Induced by Sapphire and TiB2
In this section, the structural (including the out-of-plane and in-plane) features and the dynamical properties of the liquid Al layers induced by the (0001) sapphire substrate and the (0001) TiB2 substrate, respectively, are compared.
3.1.1. Out-Of-Plane Ordering
The initial and evolved (at 6 ps) interfacial structures of the (0001) sapphire–liquid Al and (0001) TiB2–liquid Al systems are exhibited in Figure 1. The liquid Al layers can be observed in the vicinity of the solid substrates in both of the two evolved interfacial structures. The liquid Al layering induced by the two substrates is more clearly depicted in Figure 2 by the atomic density profiles along the z-direction, where z = 0 is set to be the average position of the first liquid layers (LL1-sapphire and LL1-TiB2, in Figure 1) in both of the two systems. From Figure 2, it can be clearly found that the (0001) sapphire–liquid Al interface is characterized by a double-peak sapphire surface (boxed in blue) with two liquid Al layers adjacent to it. And interestingly, such features are consistent with the results from our surface X-ray diffraction experiments [51]. The coincidence between the simulation and experimental results confirms the capability of the AIMD method in faithfully reproducing the substrate-induced liquid layering and validates the simulation models used in this work.
To quantitatively compare the out-of-plane ordering of the substrate-induced liquid layers in different systems, two parameters, layer deviation and layer density peaks , are used (as shown in Table 1). The layer deviation quantifies the standard deviation of the atomic z coordinates within a liquid layer; a smaller means a higher degree of out-of-plane ordering. The layer density peaks are the atomic density peaks of the liquid layers, as shown in Figure 2, and a large is expected for a liquid layer with a high degree of out-of-plane ordering. From Table 1, we can find that layer deviation of the (0001) TiB2-induced liquid layers (LL1-TiB2 and LL2-TiB2) presents smaller values than those of the corresponding (0001) sapphire-induced liquid layers (LL1-sapphire, LL2-sapphire). While, for the layer density peaks , the above trend is the opposite. These comparisons demonstrate that the TiB2 substrate can render more ordered liquid layers in terms of out-of-plane ordering than the sapphire substrate. Moreover, the and of the LL1-sapphire, the most ordered liquid layer near the sapphire substrate, are close to those of the LL3-TiB2, which is the least ordered liquid layer near the TiB2 substrate. This quantitative comparison illustrates again that the degree of out-of-plane ordering of the (0001) TiB2-induced liquid Al layers is much higher than that of the (0001) sapphire-induced liquid layers.
As another important parameter to describe the out-of-plane structural features, the interlayer distances between the consecutive liquid layers are also calculated, as shown in Table 1. The time-averaged positions of the liquid layers in the simulations are used when is calculated. Different from and , the interlayer distances d show similar values and trends for both the (0001) sapphire–liquid Al and the (0001) TiB2–liquid Al systems. The interlayer distances d of the LL1-sapphire and the LL1-TiB2 to the substrates (2.19 Å and 2.22 Å, respectively) are close, which are slightly smaller than that of the (111) Al planes (2.33 Å). In spite of the increase in with increasing distance to the substrates, their values are still close to the interlayer distance of the Al (111) planes. This finding indicates that the liquid layers induced by both the (0001) sapphire substrate and the (0001) TiB2 substrate may have the tendency of becoming the close-packed Al (111) layers. However, only the analysis of the out-of-plane ordering is inadequate to draw the conclusion about the stacking mode of the liquid layers. A detailed study of the in-plane structure is also required.
3.1.2. In-Plane Atomic Arrangement
In-plane atomic distribution, which is a direct way to characterize the in-plane structure, is calculated for the substrate-induced liquid layers and depicted in Figure 3. It is clearly shown that the LL1-TiB2 is almost fully ordered, with atoms closely packed within the layer. Then, for the LL2-TiB2, it also looks like a close-packed layer, but its in-plane ordering seems weakened compared with the LL1-TiB2. The stacking sequence of the LL1-TiB2 and the LL2-TiB2 is similar to the stacking mode of two consecutive Al (111) layers, which confirms our above speculation that the liquid Al layers tend to form the Al (111) planes based on the analysis of the interlayer distances in Section 3.1.1. For the LL3-TiB2, the in-plane atomic arrangement is almost disordered, so this layer cannot be regarded as the continuation of the Al (111) planes. In the (0001) sapphire–liquid Al system, both the in-plane structures of the LL1-sapphire and the LL2-sapphire are almost disordered. Thus, although the interlayer distances of the (0001) sapphire-induced liquid Al layers are close to the Al (111) planes, the in-plane structures of these layers are far from close-packed layers. On this ground, it can be deduced that the in-plane ordering seems to be more difficult to develop compared with the out-of-plane ordering. To have a further understanding of the in-plane atomic arrangement of the liquid Al layers, the 2D structural factor is calculated by Fourier transformation of the atomic arrangement from the real space into the reciprocal space, as shown in Figure 3. The 2D structural factor distributions of the LL1-TiB2 and the LL2-TiB2 manifest as discrete spots with hexagonal symmetry (Figure 3), indicating that the ordered real space atomic arrangement is also hexagonally symmetrical. This finding is consistent with the analysis based on the in-plane atomic distribution. The 2D structural factor distributions for the LL3-TiB2, the LL1-sapphire, and the LL2-sapphire are all ring-like instead of spot-like, which means the in-plane atomic arrangement of these layers is not well ordered, which is also in accords with the findings from the in-plane atomic distribution.
The above analysis has shown that the in-plane structure of the (0001) TiB2-induced liquid layers is much more ordered compared with the (0001) sapphire-induced liquid layers. However, such a qualitative analysis may miss the important structural features particularly for the weakly ordered in-plane structure, such as the LL1-sapphire. In the above analysis, such layers are regarded as disordered layers because the partially ordered in-plane structure is difficult to be distinguished from the fully disordered structure using the above method (in-plane atomic density and 2D structural factor). Next, to have a quantitative analysis of the in-plane atomic arrangement, two parameters, the orientational order parameter and the in-plane coordination number (CN), are calculated within bins (bin size Å) separating the liquid Al regions (z > 0) for the two S/L systems. From Equation (2), we can find that quantifies the degree of in-plane ordering using the bond angles within each liquid layer. Thus, it can reflect the orientational symmetry [52] of the in-plane structure. indicates the layer to be 6-fold symmetrical, which is the feature of the Al (111) planes; while means a liquid-like disordered in-plane structure with infinite symmetry axes. In Figure 4a, it is illustrated that of the LL1-TiB2 and the LL2-TiB2 shows evidently larger values than those of the bulk liquid. For the LL1-TiB2, reaches as large as ~0.7, which suggests that the in-plane symmetry of this layer is nearly 6-fold. For the (0001) sapphire–liquid Al system, although of the LL1-sapphire seems larger than that of the bulk liquid, its value is still quite small (less than 0.3), which means the in-plane symmetry of the LL1-sapphire is much weaker than the LL1-TiB2. To have a further look at the in-plane symmetry of the LL1-sapphire, the bond angle distribution of this layer, together with LL2-sapphire and the bulk liquid Al, is depicted in Figure 4c. It is shown that the values of the bond angles have the tendency of evolving into or during the formation of substrate-induced liquid Al layers. As bond angles with values of or are the sign of in-plane six-fold symmetry, the LL1-sapphire is deduced to be weakly 6-fold symmetrical. By comparing and the bond angle distribution of (0001) sapphire–liquid Al and the (0001) TiB2–liquid Al systems, it can be concluded that the liquid Al layers with in-plane 6-fold symmetry are induced for both of the two systems, while the (0001) sapphire-induced 6-fold symmetry is much weaker than that induced by the (0001) TiB2 substrate.
In addition to the orientational symmetry, the translational symmetry is also an important factor influencing the liquid ordering and crystallization [7,52]. The in-plane coordination number (CN) is used to quantify the translational symmetry as well as the in-plane ordering. The above analysis has demonstrated that the substrate-induced liquid Al layers tend to become close-packed layers. On this ground, the in-plane CN ratio of a liquid layer to a close-packed layer (in-plane CN = 6) is calculated. As shown in Figure 4b, more than 60% of the atoms are fully coordinated in the LL1-TiB2, whose is close to 1.0, indicating a nearly perfect in-plane translational symmetry. As decreases, more atoms in the close-packed structure are substituted by vacancies, resulting in the decrease of the in-plane translational symmetry. The lower limit of is defined by the bulk liquid Al whose translational symmetry is negligible. For the LL1-sapphire, is between the values of the bulk liquid Al and the LL1-TiB2, which means that the translational symmetry of the LL1-sapphire is higher than the bulk liquid Al, while being much lower than the LL1-TiB2. A detailed look at the in-plane CN distribution (Figure 4e) shows that the appearance of more 5-fold and fully coordinated atoms in the LL1-sapphire contributes to the translational symmetry improvement compared with the bulk liquid Al. Furthermore, by comparing the in-plane CN distributions of the LL1-sapphire and the LL1-TiB2, we can find that the in-plane ordering process of the LL1-sapphire encounters more resistance than that of the LL1-TiB2, because only ~10% of the atoms evolve into the fully coordinated state in the LL1-sapphire, compared with the ~60% in the LL1-TiB2.
3.1.3. Dynamical Properties of the Substrate-Induced Liquid Al Layers
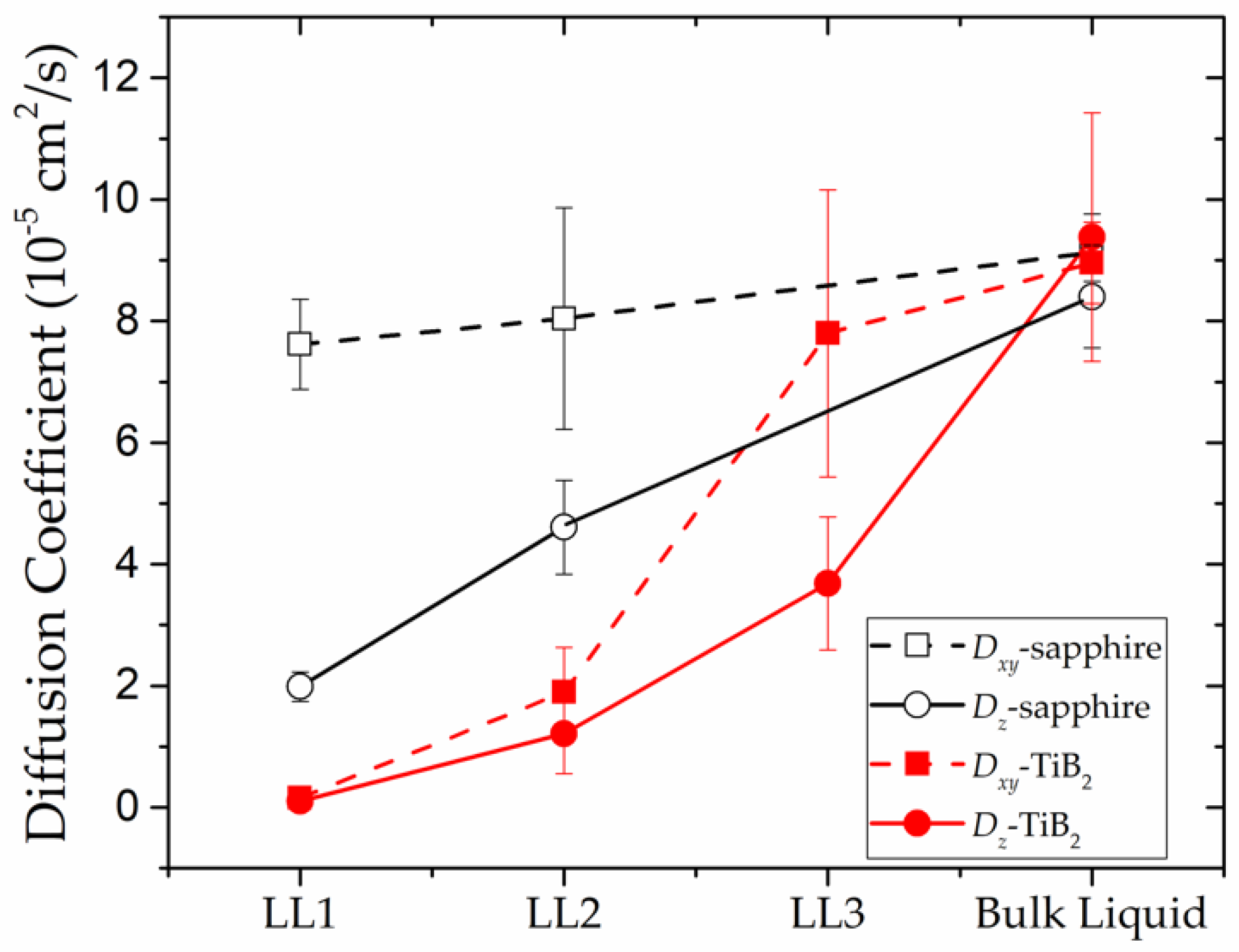
The in-plane and out-of-plane diffusion coefficients are calculated to quantify the dynamical properties of the substrate-induced layers. As shown in Figure 5, both the and of the LL1-TiB2 are almost zero, which indicates that a nearly stable structure has formed with atoms in this layer vibrating around their equilibrium positions. Different from LL1-TiB2, the LL1-sapphire presents relatively larger and , particularly the , whose value is at the same level as the bulk liquid’s. This result illustrates that the diffusive-like atomic motion contributes a lot to the dynamics of the LL1-sapphire, which also means that the in-plane structure of this layer is not stable as the atoms are not sitting at their equilibrium positions. Conclusions can be drawn from the above comparison, that the atoms in the (0001) TiB2-induced liquid Al layers stay in a more stable state than those in the (0001) sapphire-induced liquid Al layers. This conclusion is consistent with the results of the in-plane CN (discussed in Section 3.1.2), as the large number of vacancies in the LL1-sapphire provide more space for the atoms to move, thus resulting in the low stability of the in-plane structure. In addition, the adsorption strength between the substrates—(0001) sapphire and (0001) TiB2—and the liquid Al also plays an important role in determining the dynamics of the liquid Al layers, which will be discussed in detail in Section 3.2.
3.2. Factors Influencing Substrate-Induced Liquid Al Layering
The above analysis has demonstrated the structural and dynamical differences between the liquid Al layers induced by two substrates, the (0001) sapphire substrate and the (0001) TiB2 substrate, respectively. It is necessary to explore the factors that might contribute to these differences. The surface roughness of the substrate is a vital factor that greatly influences the behaviors of the near-surface layering or the self-assembly of atoms (or molecules) [8,53,54]. In this work, we focus on the atomically smooth surfaces, and our results show that when liquid Al is in contact with the atomically smooth surfaces of (0001) sapphire and (0001) TiB2, stratified liquid structures develop spontaneously. It is necessary, in our future work, to examine whether the liquid layers can form near a rough surface and to unveil the influence of surface roughness on the evolution of liquid structures near the surface. For the atomically smooth surfaces used in our simulations, we analyze the factors that determine the structural features of the substrate-induced liquid layers from the perspective of thermodynamics. As demonstrated by Kaplan et al. [5], the driving force for liquid layering at the S/L interface is the reduction of interfacial energy. Meanwhile, the result of liquid layering is to form several partially ordered layers, which may not well be matched with the substrate, thus causing interfacial strain to form. Accordingly, interfacial lattice mismatch f is the barrier impeding the formation of liquid layers.
Firstly, we analyze the driving forces of the liquid layering in the two systems. Because of the difficulty in accurately calculating S/L interfacial energy using the AIMD method, another parameter, adsorption energy W, is used to quantify the driving force. Adsorption energy W is calculated for an Al atom adsorbing onto the (0001) sapphire and (0001) TiB2 substrates according to the following formula:
where , and are the total energy of the substrate-Al system, the substrate surface slab and the isolated Al, respectively. is the number of Al atoms adsorbed onto the substrate, and in this work, only one Al adsorbate is used to avoid the influence of the interfacial strain energy. The calculated adsorption energy is listed in Table 2, together with the vibrational frequencies of the modes involving significant adsorbate in-plane or out-of-plane motion for the substrate-Al systems. An analysis of the vibrational frequencies (no imaginary frequency) suggests that the Al adsorbates have reached local minimums instead of the transition states or the higher-order saddle points for both the two systems [55]. The results of the adsorption energy show a larger adsorption strength for the Al adsorbate–(0001) TiB2 system than the Al adsorbate–(0001) sapphire system. A larger adsorption strength means a stronger driving force for the liquid layering, which could be the intrinsic factor for the (0001) TiB2 substrate to induce a more ordered liquid Al structure than the (0001) sapphire substrate.
Additionally, the interfacial lattice mismatches f between the substrates and the solid Al are calculated according to the following equation:
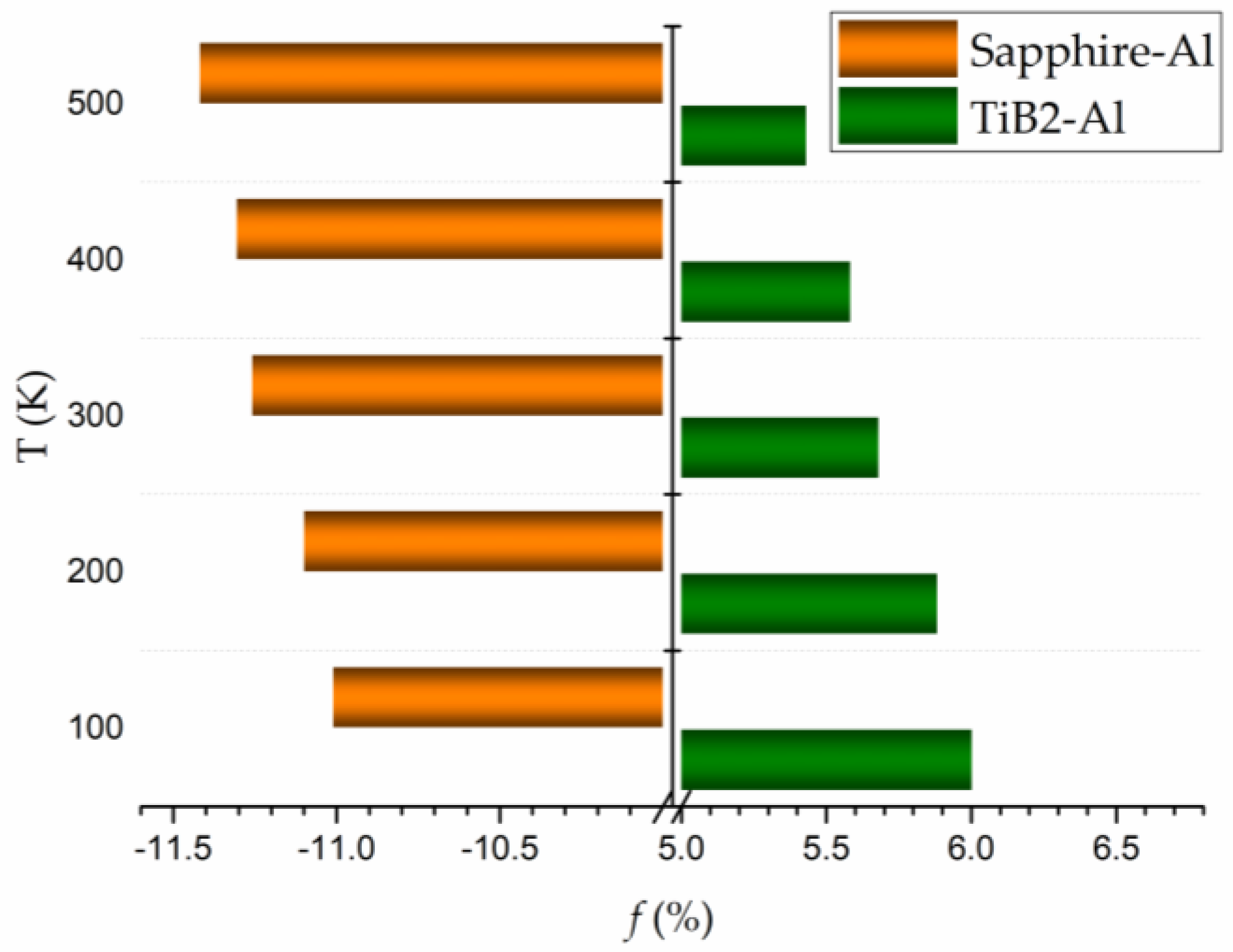
where and are the interatomic distances along the close-packed directions of the substrates (, , and in Figure 6a,b) and the solid Al. The well-accepted orientation relationships [28,56] (OR) of sapphire Al and TiB2 Al are applied for the interfacial lattice mismatch analysis. According to Equation (6), a positive f can cause in-plane tensile stress in the solid Al, rendering the shrinkage and densification of the Al atomic layers along the out-of-plane direction. While a negative f indicates a compressive state for the solid Al, which may cause the expansion or depletion of the Al atomic layers along the z-direction. A large negative lattice mismatch of ~−10.74% and a relatively small positive lattice mismatch of ~5.43% are found for the sapphire–Al and TiB2–Al S/S interfaces, respectively. Correspondingly, the relaxed interfacial structures for these two systems are depicted in Figure 6c,d. The Al atomic layers at the sapphire–Al interface is buckled, with about 1/3 of the atoms of the Al layer immediately next to the substrate moving inward, as shown in Figure 6c. While perfectly flat Al atomic layers are observed at the TiB2–Al interface. The different features of the Al atomic layers at these two interfaces are likely attributed to the positive or negative value of the interfacial lattice mismatch f. For the sapphire–Al interface, the buckled Al layers can eliminate the compressive stress caused by the negative lattice mismatch, while for the TiB2–Al interface, the tensile stress will keep the Al layers flat. In terms of the liquid layering, it can be deduced that the in-plane tensile stress in a liquid layer may make it more ordered, while the in-plane compressive stress in a liquid layer may cause it to break down. Therefore, liquid layering can be more difficult to develop when the liquid layers are under the compressive stress than under the same level of tensile stress. In addition, f, discussed above, is based on the lattice parameters at 0 K, obtained from the ab initio calculations. To analyze the influence of temperature on f, the lattice parameters of sapphire, TiB2, and Al at different temperatures are calculated using the constant temperature constant pressure (NPT) ensemble in the AIMD calculations, as listed in Table 3. The temperature influence on f is shown in Figure 7, which illustrates that, as temperature increases, f becomes larger for the sapphire–Al interface, while it gets smaller for the TiB2–Al interface. This result suggests that the thermal expansion effect causes more resistance for the liquid Al layering near the (0001) sapphire substrate.
According to the above discussion, for substrate-induced liquid layering, the adsorption strength (or reduction of interfacial energy) at the S/L interface is the driving force, and the interfacial lattice mismatch f between the substrate and the partially ordered liquid layers is the barrier. The final characteristics of the liquid layers are determined by the competition between these two factors. In addition, the negative interfacial lattice mismatch f, which causes compressive stress in the liquid layers, will cause the liquid layers more difficulty in forming compared with a positive f with the same value.
4. Discussion
For the sapphire and TiB2 substrates, the chemical bonds are mainly covalent; while for the liquid Al, the metallic bonds dominate. Thus, sapphire–liquid Al and TiB2-liquid Al interfaces become the transition regions of chemical bonds. Accordingly, it is inferred that the liquid layers may be connected to the substrates through a mixture of metallic and covalent bonds, and this inference can be verified by the ab initio calculations [56,57]. The existence of covalent bonds between at the S/L interfaces renders a strong stability of the liquid layers. The covalently bonded liquid layers, which form spontaneously when in contact with TiB2 and sapphire substrates, are analogous to the layered water [6] near solid substrates and the self-assembled monolayers (SAMs) grown above the metal substrates [53,54]. Hence, the theories in these systems can be borrowed to analyze the liquid metal layering near the nucleant substrates (normally ceramics).
With interactions between the liquid atoms and substrates, more and more liquid atoms are bound to the substrates until the equilibrium of the driving force (reduction of interfacial energy) and the barrier (interfacial lattice mismatch) is reached. According to the research of Duan et al. [58], the increase of liquid atomic density is the origin of liquid layering. Duan et al.’s study highlights the influence of atomic density on the phase transformation in liquid, and is consistent with the landscape inversion phase transformation (LIPT) proposed by Alert and co-workers [59], which underlines the influence of particle density on the transformation of the in-plane structures of particles constrained on a periodic substrate. In our case, because of the in-plane compressive stress (discussed in Section 3.2), the atomic density of the liquid layers near the sapphire substrate is lower than that near the TiB2 substrate. This causes the nearly disordered in-plane structure of the sapphire-induced liquid layers. If the compressive stress in these liquid layers can be reduced or increased (for example, by adding specific solute atoms), or the interatomic forces at the interface can be changed, the substrate-induced liquid layers then become tunable.
Tuning the substrate-induced liquid layers would be helpful to the control of heterogeneous nucleation, because the substrate-induced liquid layers generally appear above the metals’ melting points, so the features of such liquid layers can significantly influence the heterogeneous nucleation. According to the above analysis, around three liquid Al layers are induced by the (0001) TiB2 substrate, with the first layer (LL1-TiB2) being almost entirely ordered. As the temperature decreases, the LL1-TiB2 becomes a template for more fully ordered liquid Al layers to develop. Such a process can be explained by the epitaxial heterogeneous nucleation model proposed by Fan [60], which reduces to the free growth model [21] when the interfacial mismatch is small enough. However, this nucleation mechanism may not suit the (0001) sapphire–Al system. According to the above discussion, the LL1-sapphire is much less ordered than the LL1-TiB2. Also, even the S/S sapphire–Al interface is in a buckled state. Thus, the LL1-sapphire is hard to become fully ordered, even when the temperature drops below the Al melting point. The low in-plane symmetry and stability of the LL1-sapphire impedes the further liquid Al layering and thus changes the nucleation mechanisms.
5. Conclusions
In this work, liquid Al layering near the (0001) sapphire substrate and the (0001) TiB2 substrate has been simulated using the AIMD method. Structural features and dynamics of the liquid layers induced by these two substrates are compared, respectively. Additionally, the factors that influence the substrate-induced liquid layering are summarized. The main conclusions are as follows:
- For the (0001) TiB2–liquid Al interface, around three liquid Al layers are induced by the substrate. The first liquid layer (nearest to the substrate) is close-packed with almost perfect six-fold symmetry and translational symmetry. The second liquid layer also shows the close-packed order, while the degree of in-plane ordering is much weaker compared with the first liquid layer. The in-plane structure of the third liquid layer is almost disordered, which is similar to the bulk liquid Al.
- For the (0001) sapphire–liquid Al interface, approximately two liquid Al layers are induced near the substrate. Both of the two liquid layers show low 6-fold symmetry and translational symmetry, indicating a poor in-plane ordering. The first liquid layer induced by the (0001) sapphire exhibits a similar degree of ordering to the third liquid layer near the (0001) TiB2, which demonstrates that, compared with the (0001) TiB2, the (0001) sapphire shows a much poorer capability in triggering liquid Al layers.
- Two factors, the adsorption strength (or reduction of interfacial energy) and interfacial lattice mismatch, determine the structural features of the substrate-induced liquid layers. The adsorption strength is the intrinsic driving force for the liquid layering, while the interfacial lattice mismatch between the substrate and the liquid layers is the barrier. The interfacial lattice mismatch may cause compressive or tensile stress in the liquid layers, and the compressive stress makes the liquid more difficult to become ordered than the tensile stress, by impeding the increase of the liquid atomic density.
- The substrate-induced liquid layers play a critical role in the heterogeneous nucleation, and the differences in the structural features and the dynamical properties of the liquid Al layers, induced by the above two substrates, change their heterogeneous nucleation behaviors.
Author Contributions
Conceptualization, S.M. and H.D.; methodology, S.M.; software, S.M.; validation, S.M., R.Y., T.J., and H.D.; formal analysis, S.M.; investigation, S.M. and R.Y.; resources, S.M.; data curation, S.M. and R.Y.; writing (original draft preparation), S.M.; writing (review and editing), R.Y., T.J., and H.D.; visualization, S.M.; supervision, T.J. and H.D.; project administration, H.D.; and funding acquisition, T.J. and H.D.
Funding
This research was funded by the National Natural Science Foundation of China, grant number [51320105003] and [51674153], and the Chinese Scholarship Council (CSC).
Acknowledgments
This research used the ALICE High Performance Computing Facility at the University of Leicester. The authors acknowledge the support from the Diamond Light Source for the provision of beam time and the National Laboratory for Information Science and Technology at Tsinghua University for access to supercomputing facilities.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kelton, K.; Greer, A.L. Nucleation in Condensed Matter: Applications in Materials and Biology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 15. [Google Scholar]
- Zhu, X.; Jiang, W.; Li, M.; Qiao, H.; Wu, Y.; Qin, J.; Liu, X. The effect of Mg adding order on the liquid structure and solidified microstructure of the Al-Si-Mg-P alloy: An experiment and ab initio study. Metals 2014, 5, 40–51. [Google Scholar] [CrossRef]
- Kurtuldu, G.; Jarry, P.; Rappaz, M. Influence of Cr on the nucleation of primary Al and formation of twinned dendrites in Al–Zn–Cr alloys: Can icosahedral solid clusters play a role? Acta Mater. 2013, 61, 7098–7108. [Google Scholar] [CrossRef] [Green Version]
- Mei, H.; Liu, Q.W.; Liu, L.S.; Lai, X.; She, W.C.; Zhai, P.C. Molecular dynamics simulations of the microstructure of the aluminum/alumina interfacial layer. Appl. Surf. Sci. 2015, 324, 538–546. [Google Scholar] [CrossRef]
- Kaplan, W.D.; Chatain, D.; Wynblatt, P.; Carter, W.C. A review of wetting versus adsorption, complexions, and related phenomena: The rosetta stone of wetting. J. Mater. Sci. 2013, 48, 5681–5717. [Google Scholar] [CrossRef]
- Samyn, P. Wetting and hydrophobic modification of cellulose surfaces for paper applications. J. Mater. Sci. 2013, 48, 6455–6498. [Google Scholar] [CrossRef]
- Schulli, T.U.; Daudin, R.; Renaud, G.; Vaysset, A.; Geaymond, O.; Pasturel, A. Substrate-enhanced supercooling in AuSi eutectic droplets. Nature 2010, 464, 1174–1177. [Google Scholar] [CrossRef] [PubMed]
- Comtet, J.; Nigues, A.; Kaiser, V.; Coasne, B.; Bocquet, L.; Siria, A. Nanoscale capillary freezing of ionic liquids confined between metallic interfaces and the role of electronic screening. Nat. Mater. 2017, 16, 634–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandman, M.; Kauffmann, Y.; Koch, C.T.; Kaplan, W.D. Direct Quantification of Ordering at a Solid-Liquid Interface Using Aberration Corrected Transmission Electron Microscopy. Phys. Rev. Lett. 2013, 110, 086106. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.H.; Kauffmann, Y.; Scheu, C.; Kaplan, W.D.; Ruhle, M. Ordered liquid aluminum at the interface with sapphire. Science 2005, 310, 661–663. [Google Scholar] [CrossRef] [PubMed]
- Haghayeghi, R.; Qian, M. Initial crystallisation or nucleation in a liquid aluminium alloy containing spinel seeds. Mater. Lett. 2017, 196, 358–360. [Google Scholar] [CrossRef]
- Wang, J.; Horsfield, A.; Schwingenschlögl, U.; Lee, P.D. Heterogeneous nucleation of solid Al from the melt by TiB 2 and Al 3 Ti: An ab initio molecular dynamics study. Phys. Rev. B 2010, 82, 184203. [Google Scholar] [CrossRef]
- Zhang, H.L.; Han, Y.F.; Zhou, W.; Dai, Y.B.; Wang, J.; Sun, B.D. Atomic study on the ordered structure in Al melts induced by liquid/substrate interface with Ti solute. Appl. Phys. Lett. 2015, 106, 041606. [Google Scholar]
- Kang, J.; Zhu, J.; Curtis, C.; Blake, D.; Glatzmaier, G.; Kim, Y.-H.; Wei, S.-H. Atomically Abrupt Liquid-Oxide Interface Stabilized by Self-Regulated Interfacial Defects: The Case of Al/Al2O3 Interfaces. Phys. Rev. Lett. 2012, 108, 226105. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yang, L.; Zhang, D.; Xia, M.; Wang, Y.; Li, J.G. The Role of Lattice Misfit on Heterogeneous Nucleation of Pure Aluminum. Metall. Mater. Trans. A 2016, 47, 5012–5022. [Google Scholar] [CrossRef]
- Men, H.; Fan, Z. Prenucleation Induced by Crystalline Substrates. Metall. Mater. Trans. A 2018, 49, 2766–2777. [Google Scholar] [CrossRef]
- Greer, A.L. Liquid metals: Supercool order. Nat. Mater. 2006, 5, 13–14. [Google Scholar] [CrossRef]
- Easton, M.A.; Qian, M.; Prasad, A.; StJohn, D.H. Recent advances in grain refinement of light metals and alloys. Curr. Opin. Solid State Mater. Sci. 2016, 20, 13–24. [Google Scholar] [CrossRef] [Green Version]
- Cantor, B. Heterogeneous nucleation and adsorption. Philos. Trans. R. Soc. Lond. A Math. Phys. Eng. Sci. 2003, 361, 409–417. [Google Scholar] [CrossRef]
- Kim, W.T.; Cantor, B. An adsorption model of the heterogeneous nucleation of solidification. Acta Metall. Mater. 1994, 42, 3115–3127. [Google Scholar] [CrossRef]
- Greer, A.; Bunn, A.; Tronche, A.; Evans, P.; Bristow, D. Modelling of inoculation of metallic melts: Application to grain refinement of aluminium by Al–Ti–B. Acta Mater. 2000, 48, 2823–2835. [Google Scholar] [CrossRef]
- Fan, Z.; Wang, Y.; Zhang, Y.; Qin, T.; Zhou, X.; Thompson, G.; Pennycook, T.; Hashimoto, T. Grain refining mechanism in the Al/Al–Ti–B system. Acta Mater. 2015, 84, 292–304. [Google Scholar] [CrossRef]
- Murty, B.S.; Kori, S.A.; Chakraborty, M. Grain refinement of aluminium and its alloys by heterogeneous nucleation and alloying. Int. Mater. Rev. 2002, 47, 3–29. [Google Scholar] [CrossRef]
- Quested, T.; Greer, A. The effect of the size distribution of inoculant particles on as-cast grain size in aluminium alloys. Acta Mater. 2004, 52, 3859–3868. [Google Scholar] [CrossRef]
- Liotti, E.; Arteta, C.; Zisserman, A.; Lui, A.; Lempitsky, V.; Grant, P.S. Crystal nucleation in metallic alloys using x-ray radiography and machine learning. Sci. Adv. 2018, 4, eaar4004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Lu, W.Q.; Hu, Q.D.; Xia, M.X.; Wang, Y.; Li, J.G. Interfacial tuning for the nucleation of liquid AlCu alloy. Acta Mater. 2017, 139, 75–85. [Google Scholar] [CrossRef]
- Brown, A.J.; Dong, H.B.; Howes, P.B.; Nicklin, C.L. In situ observation of the orientation relationship at the interface plane between substrate and nucleus using X-ray scattering techniques. Scr. Mater. 2014, 77, 60–63. [Google Scholar] [CrossRef]
- Han, Y.; Dai, Y.; Shu, D.; Wang, J.; Sun, B. First-principles calculations on the stability of Al/TiB2 interface. Appl. Phys. Lett. 2006, 89, 144107. [Google Scholar] [CrossRef]
- Zhang, H.L.; Han, Y.F.; Dai, Y.B.; Wang, J.; Sun, B.D. An ab initio molecular dynamics study: Liquid-Al/solid-TiB2 interfacial structure during heterogeneous nucleation. J. Phys. D Appl. Phys. 2012, 45, 455307. [Google Scholar] [CrossRef]
- Liang, H.T.; Laird, B.B.; Asta, M.; Yang, Y. In-plane characterization of structural and thermodynamic properties for steps at faceted chemically heterogeneous solid/liquid interfaces. Acta Mater. 2018, 143, 329–337. [Google Scholar] [CrossRef]
- Yang, Y.; Olmsted, D.L.; Asta, M.; Laird, B.B. Atomistic characterization of the chemically heterogeneous Al–Pb solid–liquid interface. Acta Mater. 2012, 60, 4960–4971. [Google Scholar] [CrossRef]
- Guo, C.; Wang, J.; Li, J.; Wang, Z.; Tang, S. Kinetic Pathways and Mechanisms of Two-Step Nucleation in Crystallization. J. Phys. Chem. Lett. 2016, 7, 5008–5014. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, C.; Zhang, Q.; Tang, S.; Li, J.; Wang, Z. Recent Progresses in Modeling of Nucleation During Solidification on the Atomic Scale. Acta Metall. Sin. 2017, 54, 204–216. [Google Scholar]
- Qiao, J.; Zhang, L.; Hui, X.; Lin, J. Kinetic and thermodynamic properties of liquid zinc: An ab initio molecular dynamics study. Comput. Mater. Sci. 2018, 141, 180–184. [Google Scholar] [CrossRef]
- Debela, T.T.; Wang, X.D.; Cao, Q.P.; Lu, Y.H.; Zhang, D.X.; Fecht, H.J.; Tanaka, H.; Jiang, J.Z. Nucleation driven by orientational order in supercooled niobium as seen via ab initio molecular dynamics. Phys. Rev. B 2014, 89, 104205. [Google Scholar] [CrossRef]
- Wang, W.Y.; Han, J.J.; Fang, H.Z.; Wang, J.; Liang, Y.F.; Shang, S.L.; Wang, Y.; Liu, X.J.; Kecskes, L.J.; Mathaudhu, S.N.; et al. Anomalous structural dynamics in liquid Al80Cu20: An ab initio molecular dynamics study. Acta Mater. 2015, 97, 75–85. [Google Scholar] [CrossRef]
- Weber, H.; Schumacher, M.; Jovari, P.; Tsuchiya, Y.; Skrotzki, W.; Mazzarello, R.; Kaban, I. Experimental and ab initio molecular dynamics study of the structure and physical properties of liquid GeTe. Phys. Rev. B 2017, 96, 054204. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmuller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular-Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Mishin, Y.; Farkas, D.; Mehl, M.J.; Papaconstantopoulos, D.A. Interatomic potentials for monoatomic metals from experimental data and ab initio calculations. Phys. Rev. B 1999, 59, 3393–3407. [Google Scholar] [CrossRef]
- Alfe, D. First-principles simulations of direct coexistence of solid and liquid aluminum. Phys. Rev. B 2003, 68, 064423. [Google Scholar] [CrossRef]
- Bouchet, J.; Bottin, F.; Jomard, G.; Zerah, G. Melting curve of aluminum up to 300 GPa obtained through ab initio molecular dynamics simulations. Phys. Rev. B 2009, 80, 094102. [Google Scholar] [CrossRef]
- Davidchack, R.L.; Laird, B.B. Simulation of the hard-sphere crystal–melt interface. J. Chem. Phys. 1998, 108, 9452–9462. [Google Scholar] [CrossRef] [Green Version]
- Palafox-Hernandez, J.P.; Laird, B.B. Orientation dependence of heterogeneous nucleation at the Cu–Pb solid-liquid interface. J. Chem. Phys. 2016, 145, 211914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buta, D.; Asta, M.; Hoyt, J.J. Atomistic simulation study of the structure and dynamics of a faceted crystal-melt interface. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2008, 78, 031605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K. On the Concept of Static Structure Factor. arXiv, 2016; arXiv:1606.03610. [Google Scholar]
- Ma, S.; Brown, A.J.; Yan, R.; Davidchack, R.L.; Howes, P.B.; Nicklin, C.; Zhai, Q.; Jing, T.; Dong, H. Atomistics of adsorption-induced pre-nucleation layering of liquid metals at the interface with poor nucleant substrates. Unpublished work. 2018. [Google Scholar]
- Russo, J.; Tanaka, H. Crystal nucleation as the ordering of multiple order parameters. J. Chem. Phys. 2016, 145, 211801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.H.; Chang, B.; Oyola-Reynoso, S.; Wang, Z.J.; Thuo, M. Quantifying Gauche Defects and Phase Evolution in Self-Assembled Monolayers through Sessile Drops. ACS Omega 2017, 2, 2072–2084. [Google Scholar] [CrossRef]
- Chen, J.; Wang, Z.; Oyola-Reynoso, S.; Thuo, M.M. Properties of Self-Assembled Monolayers Revealed via Inverse Tensiometry. Langmuir 2017, 33, 13451–13467. [Google Scholar] [CrossRef] [PubMed]
- Hinnemann, B.; Carter, E.A. Adsorption of Al, O, Hf, Y, Pt, and S atoms on α-Al2O3 (0001). J. Phys. Chem. C 2007, 111, 7105–7126. [Google Scholar] [CrossRef]
- Siegel, D.J.; Hector Jr, L.G.; Adams, J.B. Adhesion, atomic structure, and bonding at the Al (111)/α−Al2O3 (0001) interface: A first principles study. Phys. Rev. B 2002, 65, 085415. [Google Scholar] [CrossRef]
- Xiao, L.; Schneider, W.F. Surface termination effects on metal atom adsorption on α-alumina. Surf. Sci. 2008, 602, 3445–3453. [Google Scholar] [CrossRef]
- Duan, Y.; Li, J.; Li, T.; Zhang, X.; Wang, Z.; Li, H. Density dependent structural phase transition for confined copper: Origin of the layering. Phys. Chem. Chem. Phys. 2018, 20, 9337–9342. [Google Scholar] [CrossRef] [PubMed]
- Alert, R.; Casademunt, J.; Tierno, P. Landscape-inversion phase transition in dipolar colloids: Tuning the structure and dynamics of 2D crystals. Phys. Rev. Lett. 2014, 113, 198301. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.Y. An Epitaxial Model for Heterogeneous Nucleation on Potent Substrates. Metall. Mater. Trans. A 2013, 44a, 1409–1418. [Google Scholar] [CrossRef]
Figure 1.
Initial (the left column) and evolved (the right column) solid/liquid (S/L) interfacial structure of (a) (0001) sapphire–liquid Al and (b) (0001) TiB2–liquid Al systems. (Al: gray spheres; O: red spheres; Ti: green spheres; B: blue spheres).
Figure 1.
Initial (the left column) and evolved (the right column) solid/liquid (S/L) interfacial structure of (a) (0001) sapphire–liquid Al and (b) (0001) TiB2–liquid Al systems. (Al: gray spheres; O: red spheres; Ti: green spheres; B: blue spheres).

Figure 2.
Atomic density profiles (averaged from 3–6 ps) along the z-direction of the (0001) sapphire–liquid Al and the (0001) TiB2–liquid Al interfaces. The first liquid layers (LL1) of these two systems are aligned to z = 0.
Figure 2.
Atomic density profiles (averaged from 3–6 ps) along the z-direction of the (0001) sapphire–liquid Al and the (0001) TiB2–liquid Al interfaces. The first liquid layers (LL1) of these two systems are aligned to z = 0.
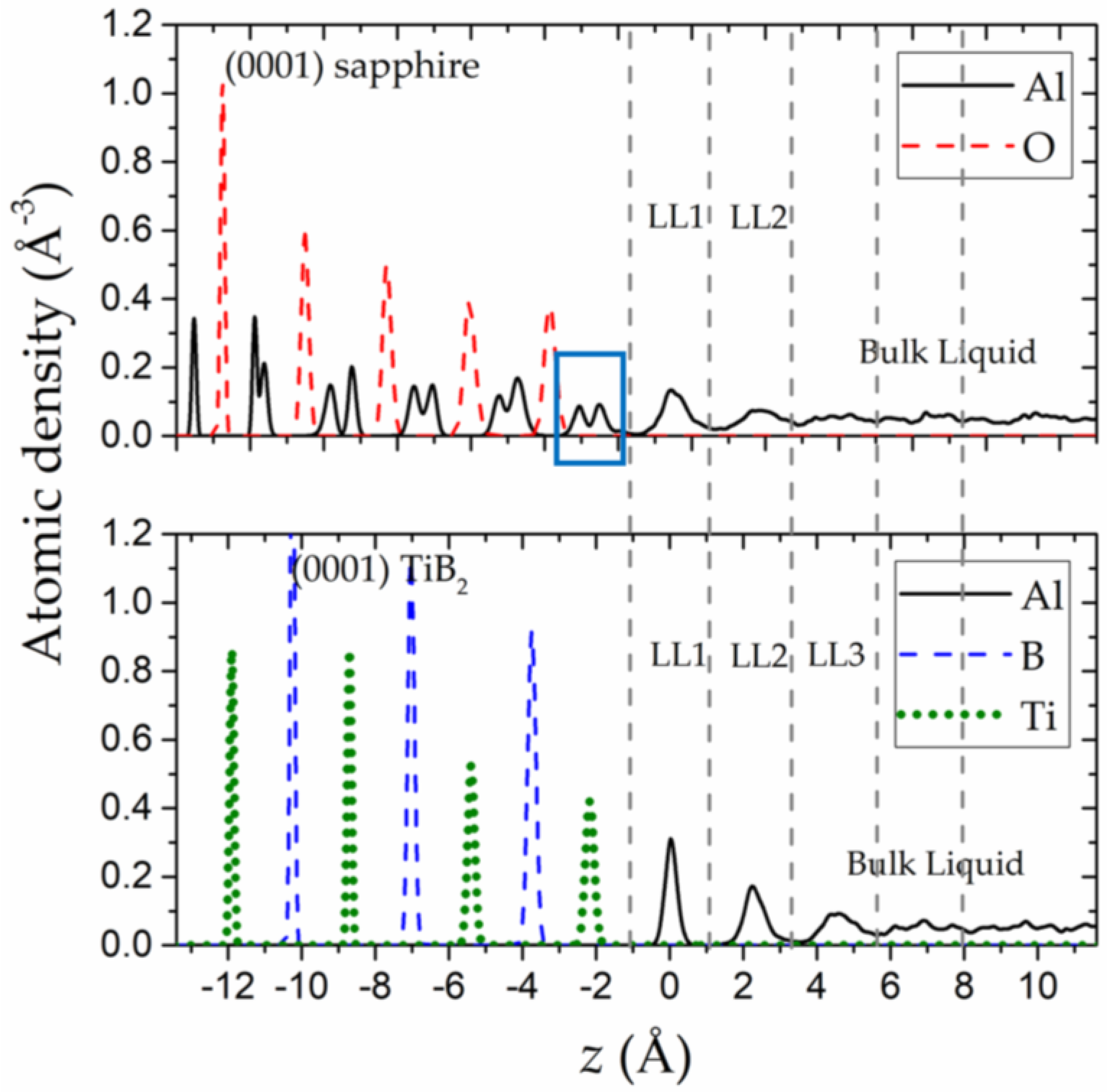
Figure 3.
In-plane atomic distribution (the left one in each picture) and two-dimensional (2D) structural factor (the right panel in each image) of (a) LL1-TiB2, (b) LL2-TiB2, (c) LL3-TiB2, (d) LL1-sapphire, and (e) LL2-sapphire.
Figure 3.
In-plane atomic distribution (the left one in each picture) and two-dimensional (2D) structural factor (the right panel in each image) of (a) LL1-TiB2, (b) LL2-TiB2, (c) LL3-TiB2, (d) LL1-sapphire, and (e) LL2-sapphire.

Figure 4.
(a) Orientational order parameter and (b) relative in-plane CN calculated for the (0001) sapphire–liquid Al and the (0001) TiB2–liquid Al systems. (c–f) Bond angle distributions and in-plane coordination number (CN) distributions of the substrate-induced liquid layers.
Figure 4.
(a) Orientational order parameter and (b) relative in-plane CN calculated for the (0001) sapphire–liquid Al and the (0001) TiB2–liquid Al systems. (c–f) Bond angle distributions and in-plane coordination number (CN) distributions of the substrate-induced liquid layers.
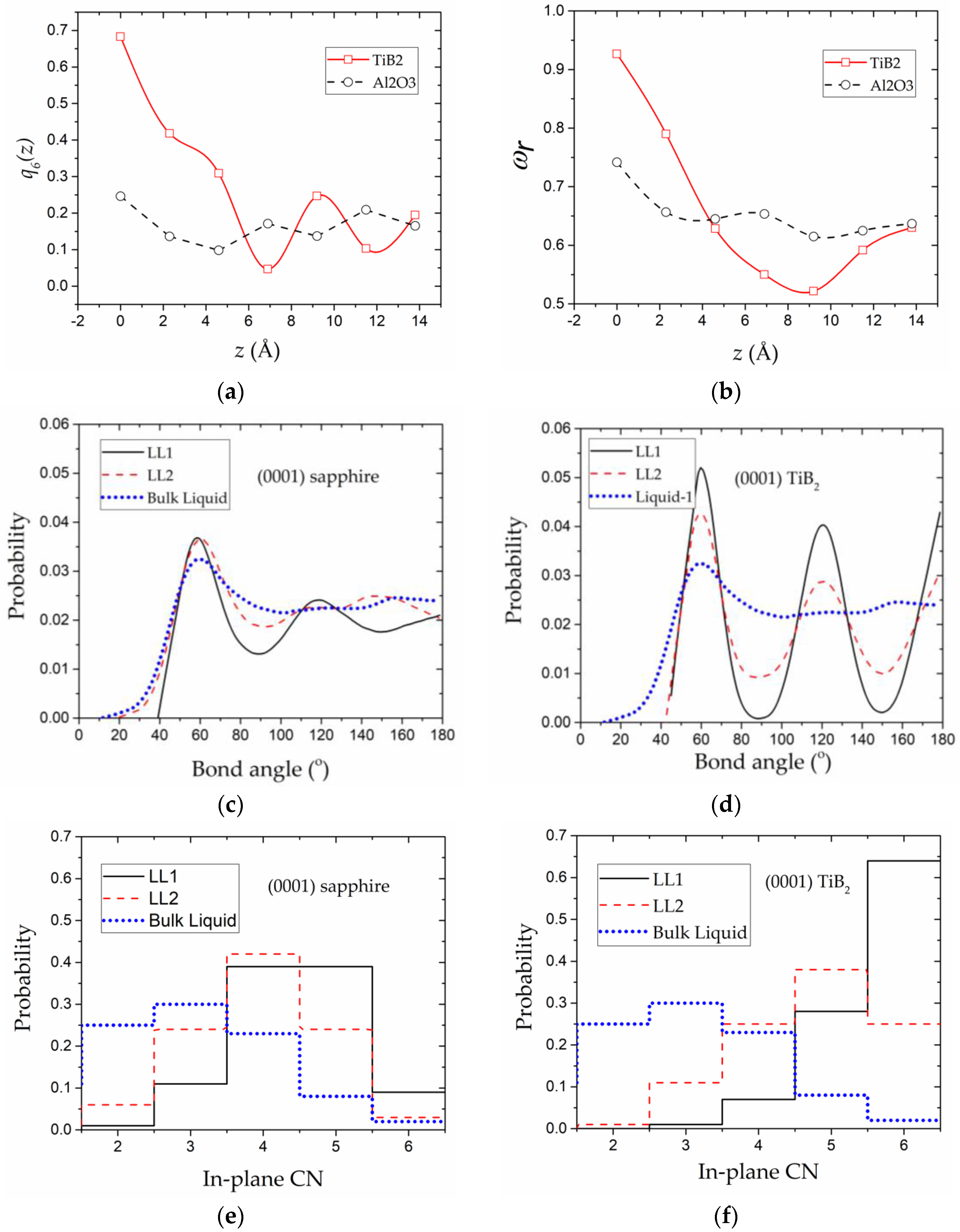
Figure 5.
Out-of-plane and in-plane diffusion coefficients, and , of the substrate-induced liquid layers.
Figure 5.
Out-of-plane and in-plane diffusion coefficients, and , of the substrate-induced liquid layers.
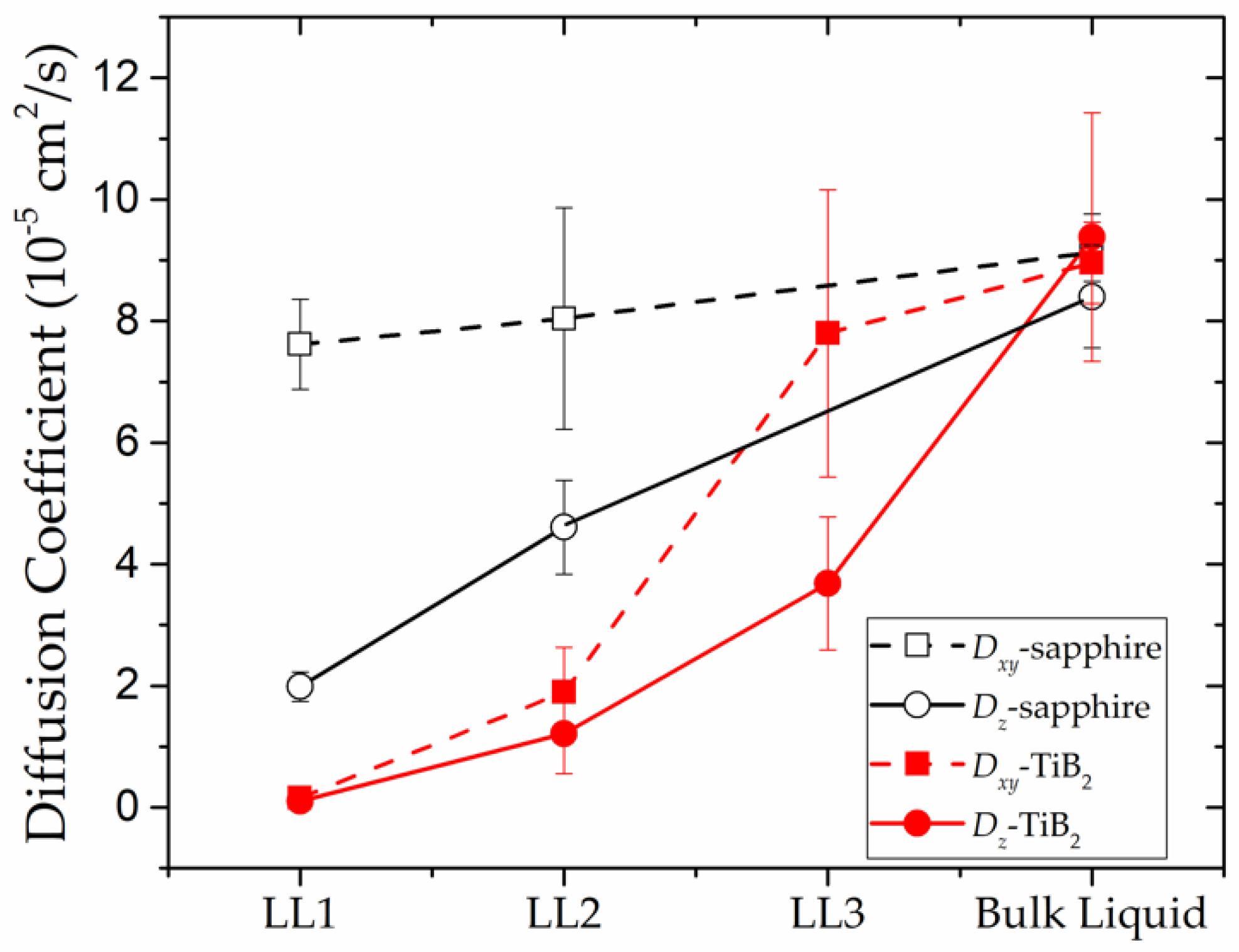
Figure 6.
(a,b) Top view of the (0001) sapphire surface and the (0001) TiB2 surface relaxed at 0 K. Two interatomic distances, and , exist along the direction at the (0001) sapphire surface, resulting in ~−10.75% and ~1.4% mismatches with the (111) Al (d<110> ≈ 2.86 Å). The large negative sapphire–Al interfacial mismatch is the main resistance for the liquid Al layering. is the interatomic distance along the <> direction at the (0001) TiB2 surface. The TiB2–Al interfacial mismatch is ~5.43%. (c,d) The relaxed structures of the (0001) sapphire– (111) Al and the (0001) TiB2–(111) Al S/S interfaces. (Al: gray spheres; O: red spheres; Ti: green spheres; B: blue spheres).
Figure 6.
(a,b) Top view of the (0001) sapphire surface and the (0001) TiB2 surface relaxed at 0 K. Two interatomic distances, and , exist along the direction at the (0001) sapphire surface, resulting in ~−10.75% and ~1.4% mismatches with the (111) Al (d<110> ≈ 2.86 Å). The large negative sapphire–Al interfacial mismatch is the main resistance for the liquid Al layering. is the interatomic distance along the <> direction at the (0001) TiB2 surface. The TiB2–Al interfacial mismatch is ~5.43%. (c,d) The relaxed structures of the (0001) sapphire– (111) Al and the (0001) TiB2–(111) Al S/S interfaces. (Al: gray spheres; O: red spheres; Ti: green spheres; B: blue spheres).

Figure 7.
Temperature influence on the mismatches of the (0001) sapphire– (111) Al and the (0001) TiB2– (111) Al interfaces.
Figure 7.
Temperature influence on the mismatches of the (0001) sapphire– (111) Al and the (0001) TiB2– (111) Al interfaces.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Main results showing the out-of-plane and in-plane structural features of the substrate-induced liquid layers.
Table 1.
Main results showing the out-of-plane and in-plane structural features of the substrate-induced liquid layers.
| Interfacial Systems | Out-of-Plane Structural Features | In-Plane Structural Features | ||||
|---|---|---|---|---|---|---|
| 1 (Å) | (Å) | (Å−3) | In-Plane CN | |||
| Sapphire–liquid Al | LL1 | 2.19 | 0.47 | 0.14 | 4.45 | 0.25 |
| LL2 | 2.30 | 0.62 | 0.08 | 3.94 | 0.14 | |
| TiB2–liquid Al | LL1 | 2.22 | 0.15 | 0.34 | 5.56 | 0.68 |
| LL2 | 2.33 | 0.31 | 0.18 | 4.74 | 0.42 | |
| LL3 | 2.40 | 0.49 | 0.09 | 3.77 | 0.31 | |
1 For the first liquid layers (LL1), d is the distance from S0 to LL1; for the second liquid layers (LL2), d is the distance from LL1 to LL2; for the third liquid layers (LL3), d is the distance from LL2 to LL3. CN—coordination number.
Table 2.
Adsorption energy and the vibrational frequencies of the single Al adsorbed (0001) sapphire and (0001) TiB2 surfaces.
Table 2.
Adsorption energy and the vibrational frequencies of the single Al adsorbed (0001) sapphire and (0001) TiB2 surfaces.
| Substrates | W (eV/atom) | Vibrational Frequencies (cm−1) Involving Significant Adsorbate Motion | |
|---|---|---|---|
| In-Plane | Out-Of-Plane | ||
| Sapphire | −2.05 | 171.69; 204.20 | 219.28 |
| TiB2 | −3.36 | 113.98; 115.41 | 185.95 |
Table 3.
Temperature influence on the lattice parameters of sapphire, TiB2, and Al, based on the ab initio molecular dynamics (AIMD) calculations.
Table 3.
Temperature influence on the lattice parameters of sapphire, TiB2, and Al, based on the ab initio molecular dynamics (AIMD) calculations.
| Temperature (K) | Sapphire (Å) 1 | TiB2 (Å) 2 | Al (Å) |
|---|---|---|---|
| 100 | 4.813 | 3.041 | 4.057 |
| 200 | 4.817 | 3.043 | 4.064 |
| 300 | 4.820 | 3.045 | 4.075 |
| 400 | 4.826 | 3.047 | 4.082 |
| 500 | 4.831 | 3.049 | 4.091 |
1 lattice parameter a of sapphire; 2 lattice parameter a of TiB2.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ma, S.; Yan, R.; Jing, T.; Dong, H. Substrate-Induced Liquid Layering: A New Insight into the Heterogeneous Nucleation of Liquid Metals. Metals 2018, 8, 521. https://doi.org/10.3390/met8070521
AMA Style
Ma S, Yan R, Jing T, Dong H. Substrate-Induced Liquid Layering: A New Insight into the Heterogeneous Nucleation of Liquid Metals. Metals. 2018; 8(7):521. https://doi.org/10.3390/met8070521
Chicago/Turabian StyleMa, Sida, Rui Yan, Tao Jing, and Hongbiao Dong. 2018. "Substrate-Induced Liquid Layering: A New Insight into the Heterogeneous Nucleation of Liquid Metals" Metals 8, no. 7: 521. https://doi.org/10.3390/met8070521
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.