
Staphylococcus aureus Induces IFN-β Production via a CARMA3-Independent Mechanism

Abstract
:1. Introduction
2. Results
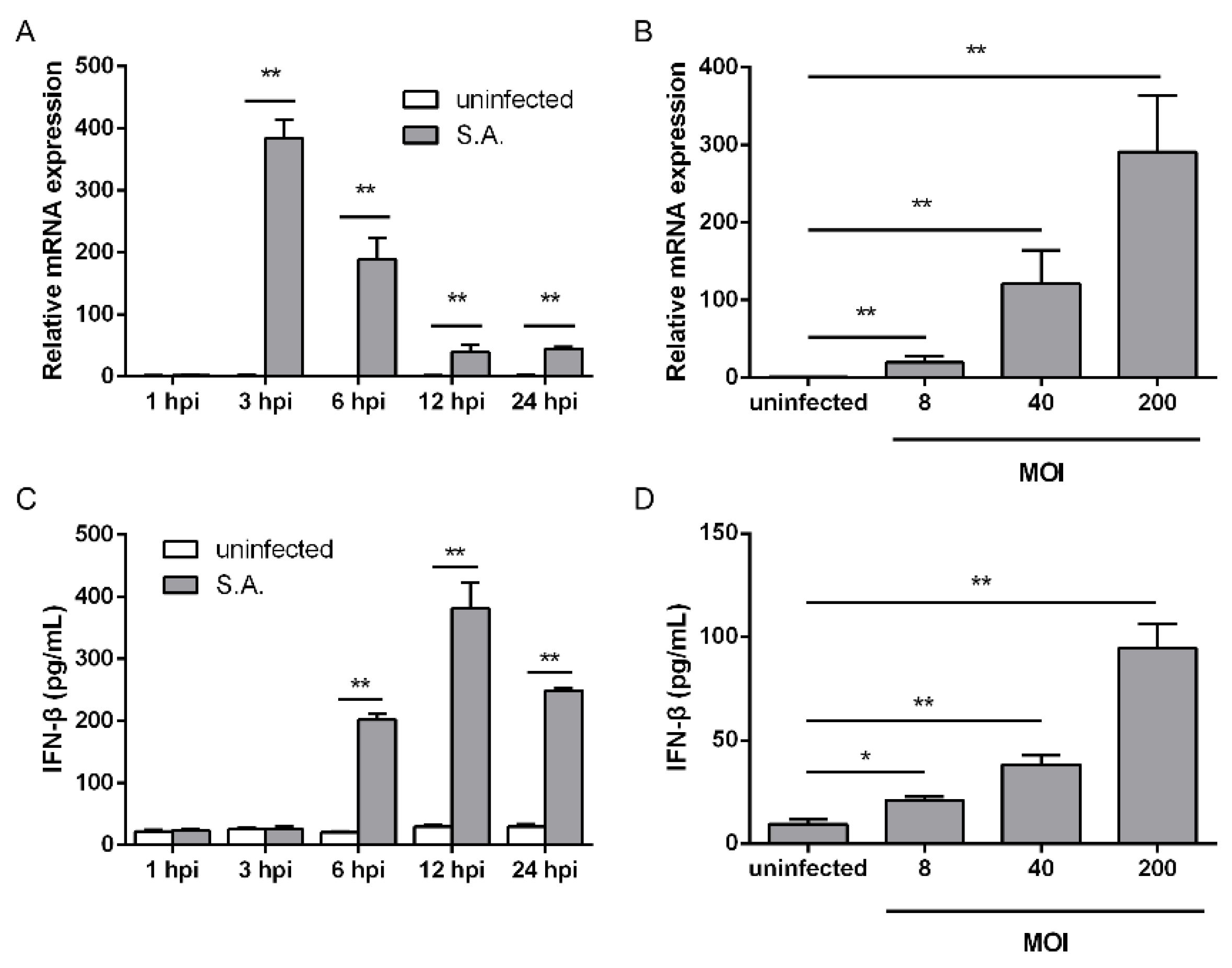
2.1. S. aureus Infection Triggers IFN-β Signaling
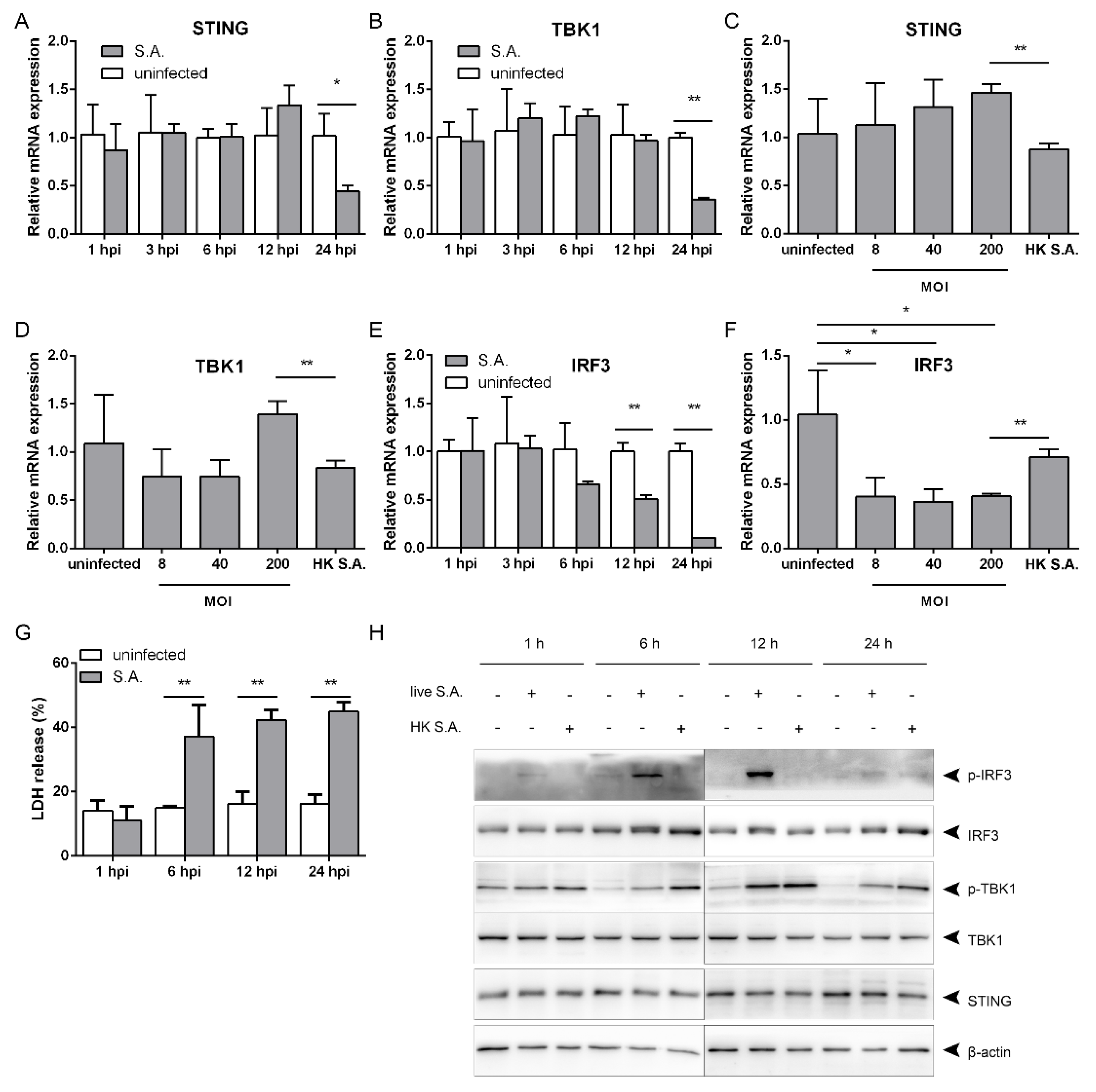
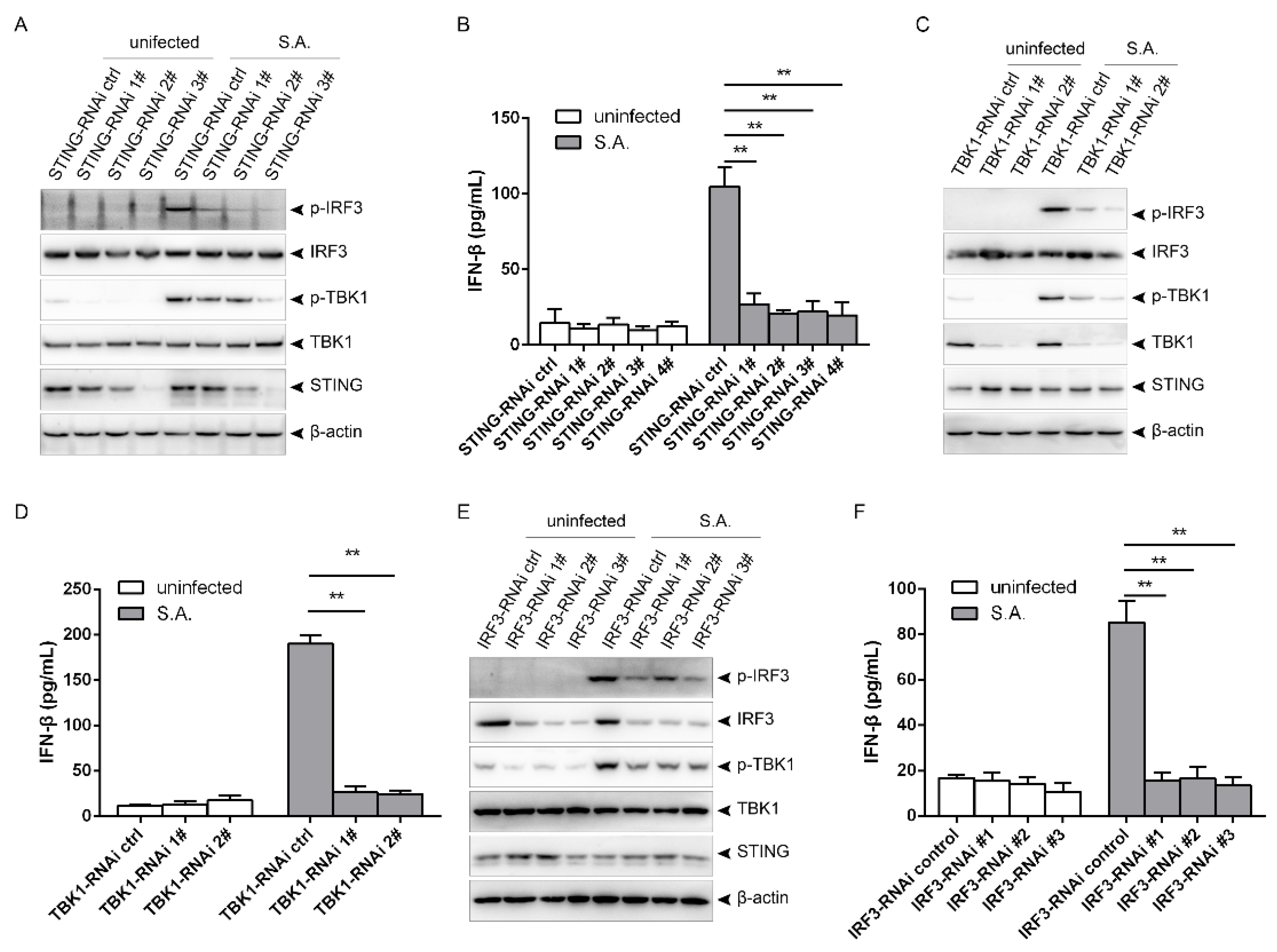
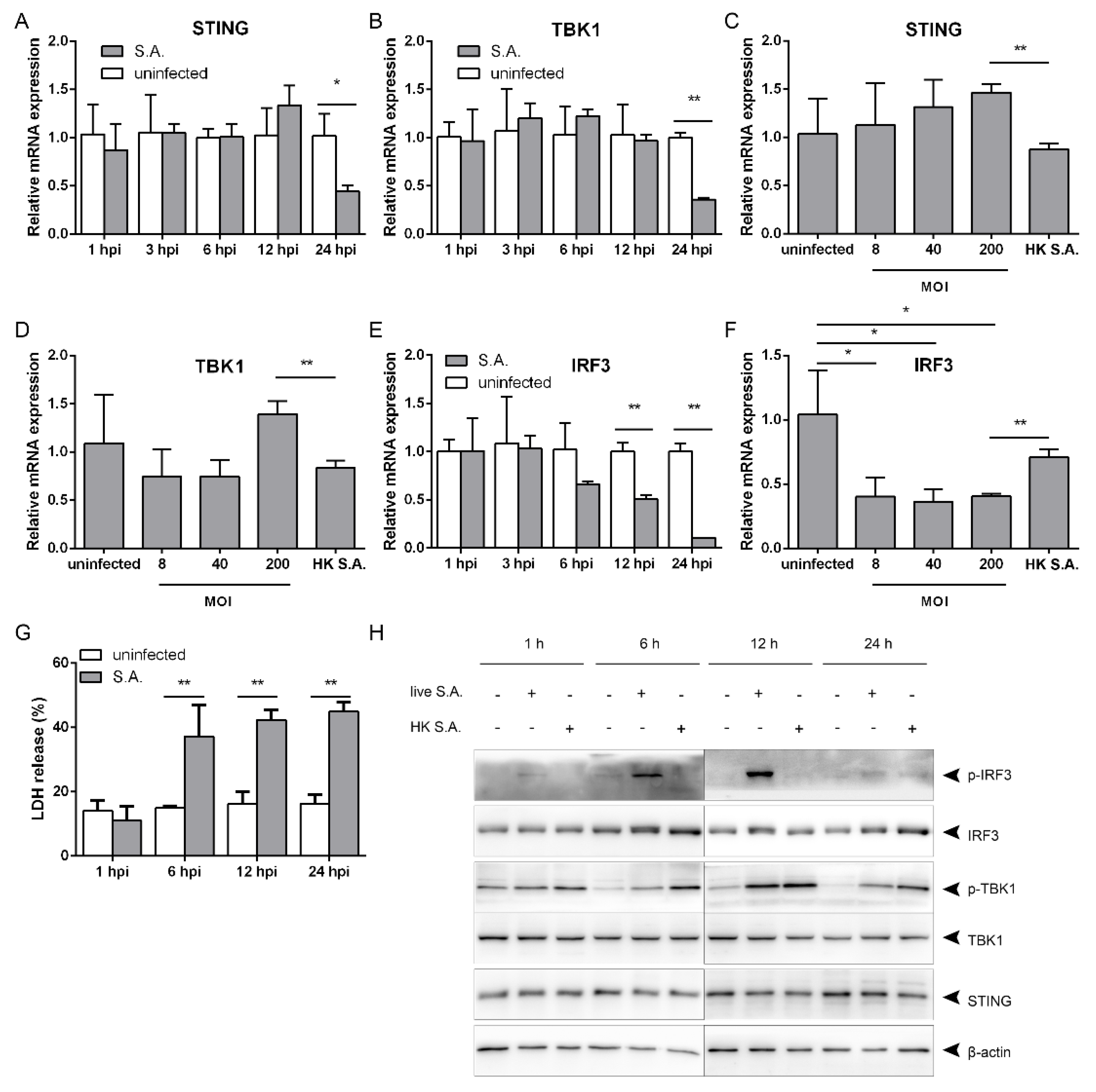
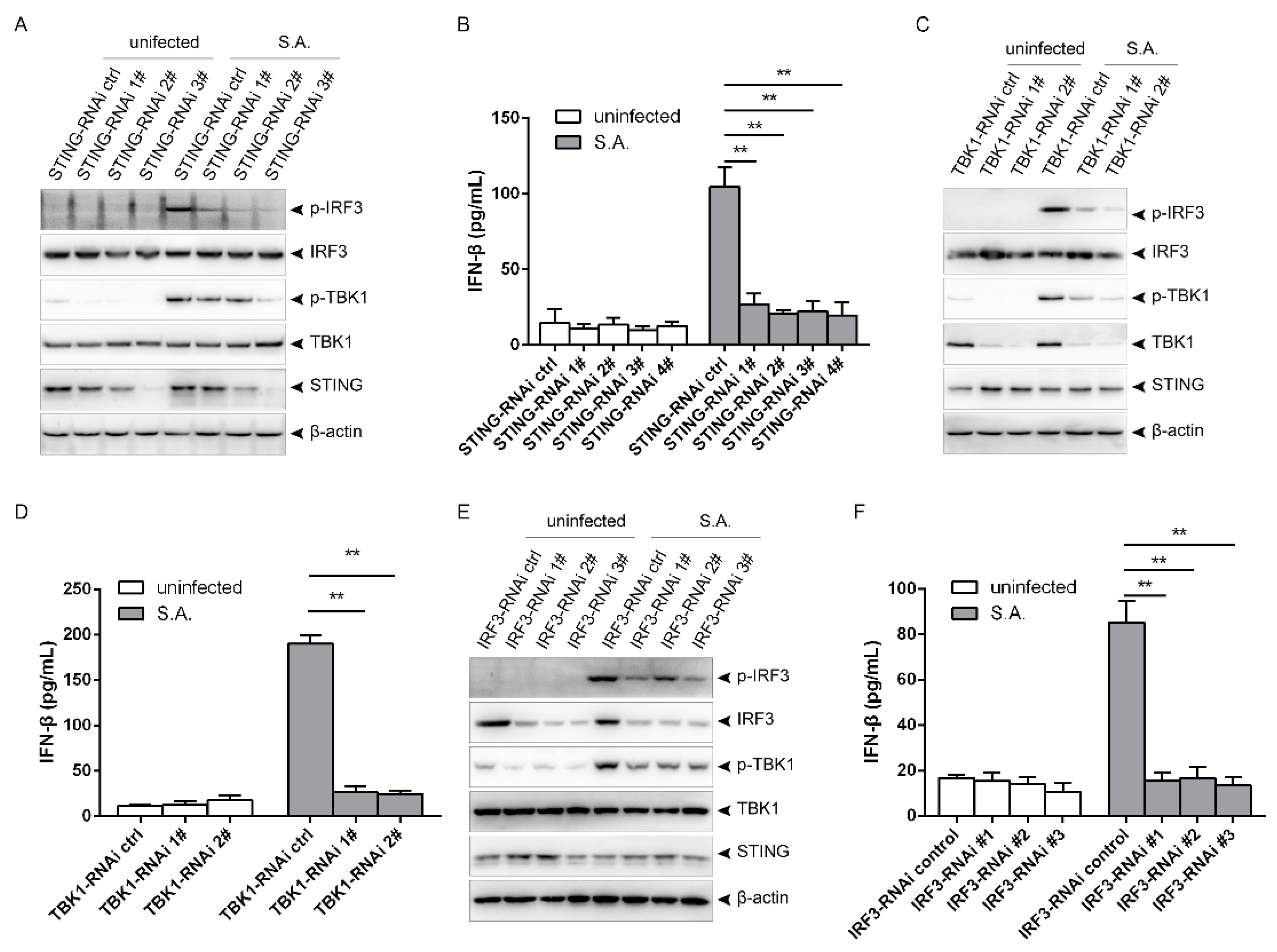
2.2. S. aureus Induces IFN-β Signaling via the STING/TBK1/IRF3 Pathway
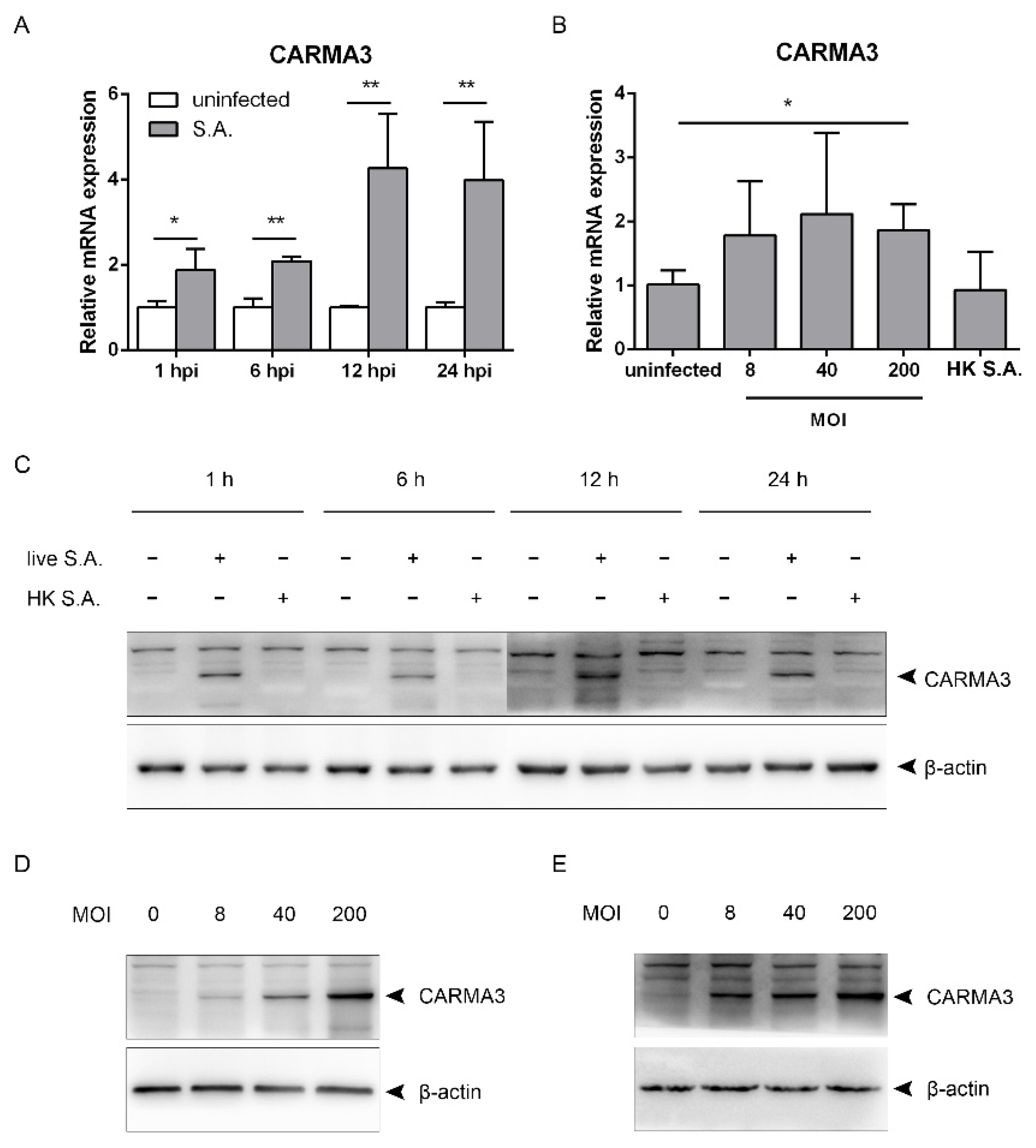
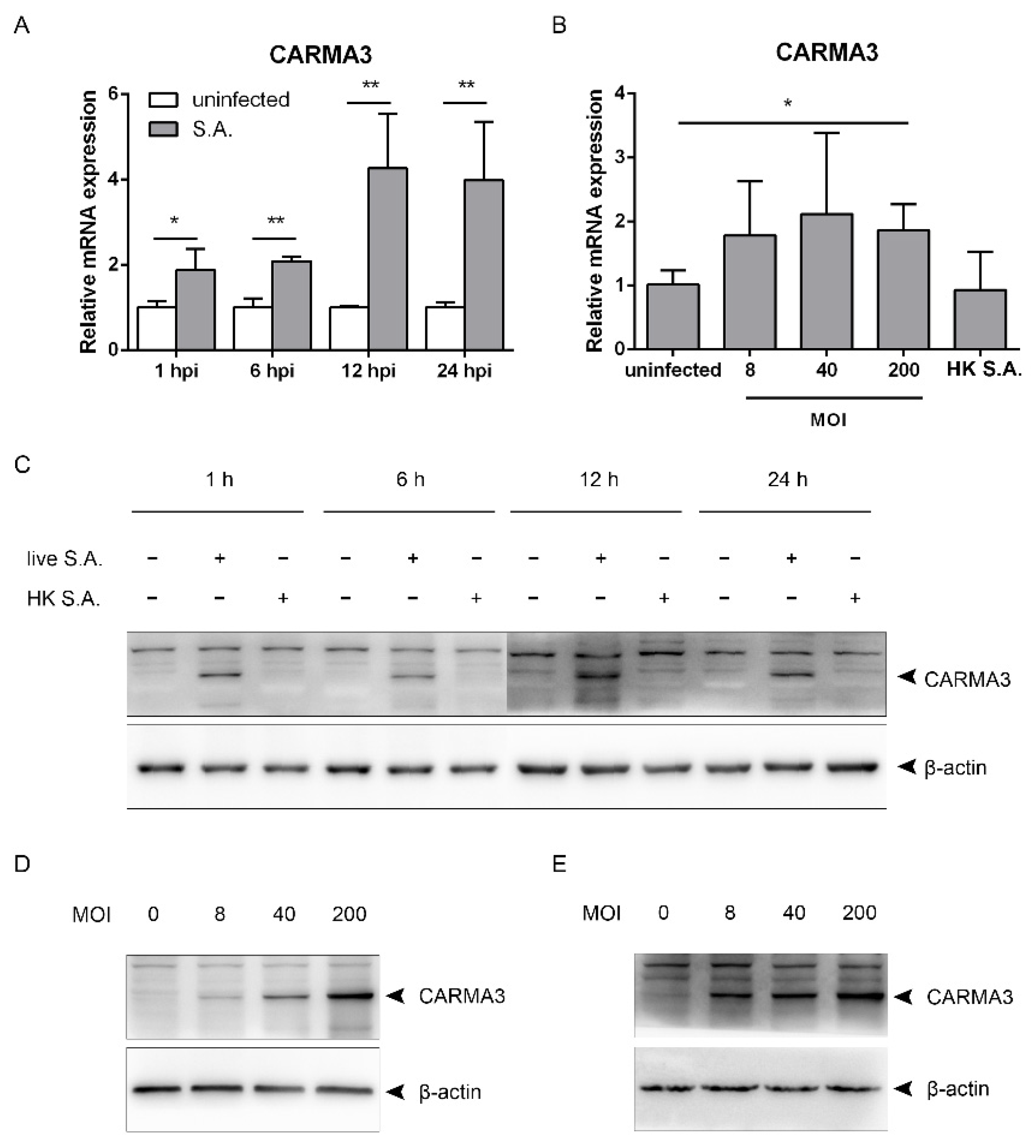
2.3. S. aureus Induces Upregulation of CARMA3
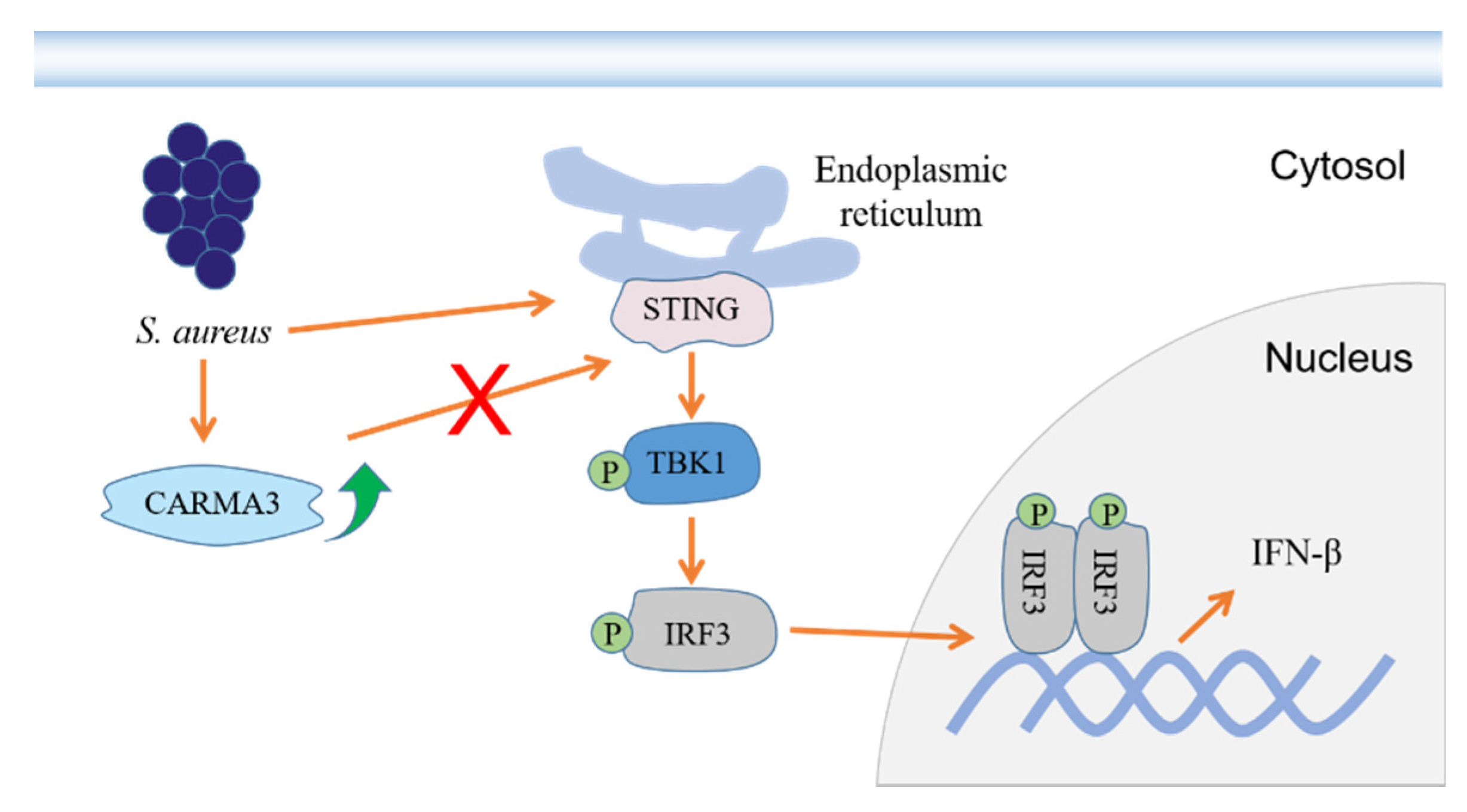
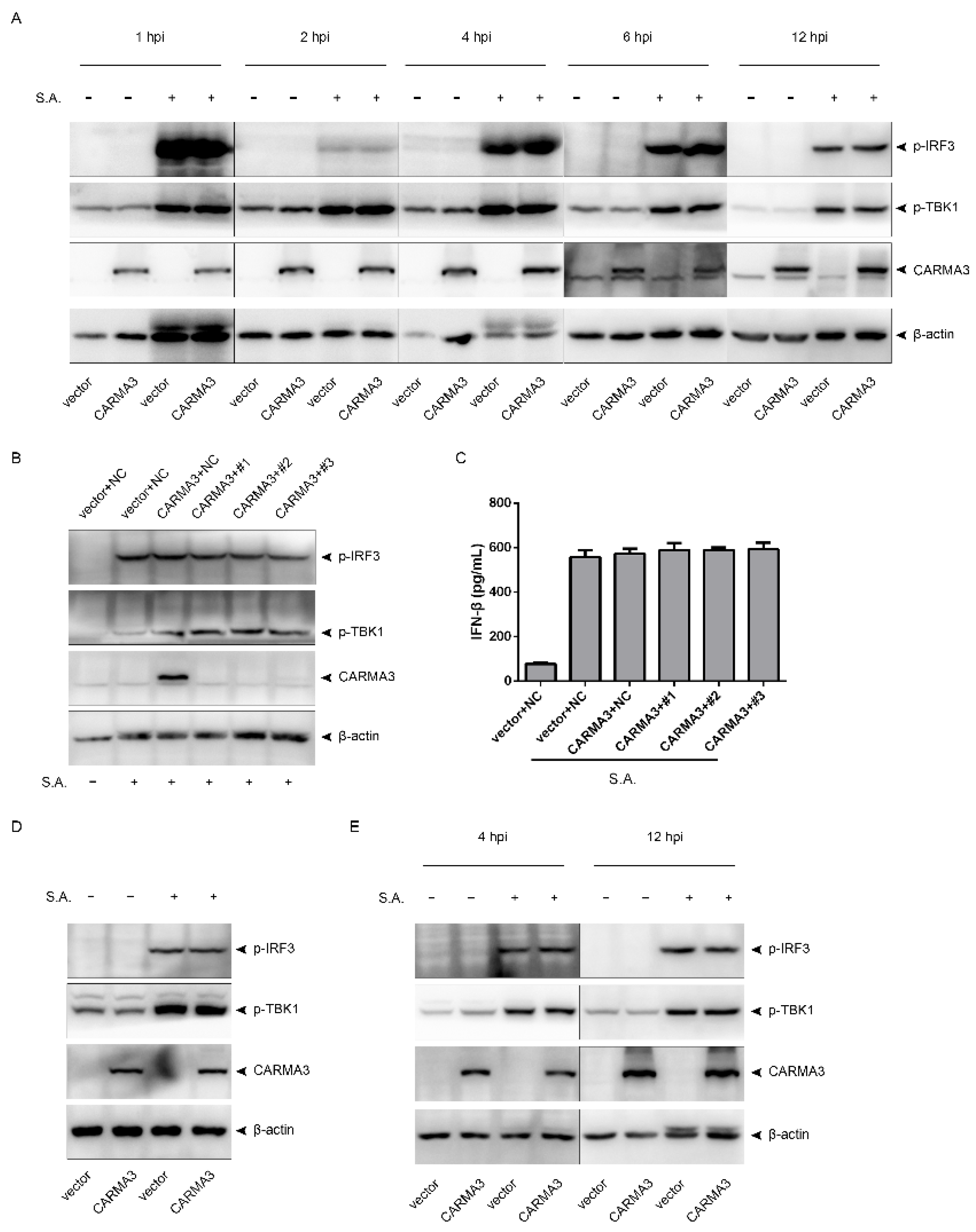
2.4. S. aureus-Induced Activation of the STING/TBK1/IRF3 Pathway Is Independent of CARMA3
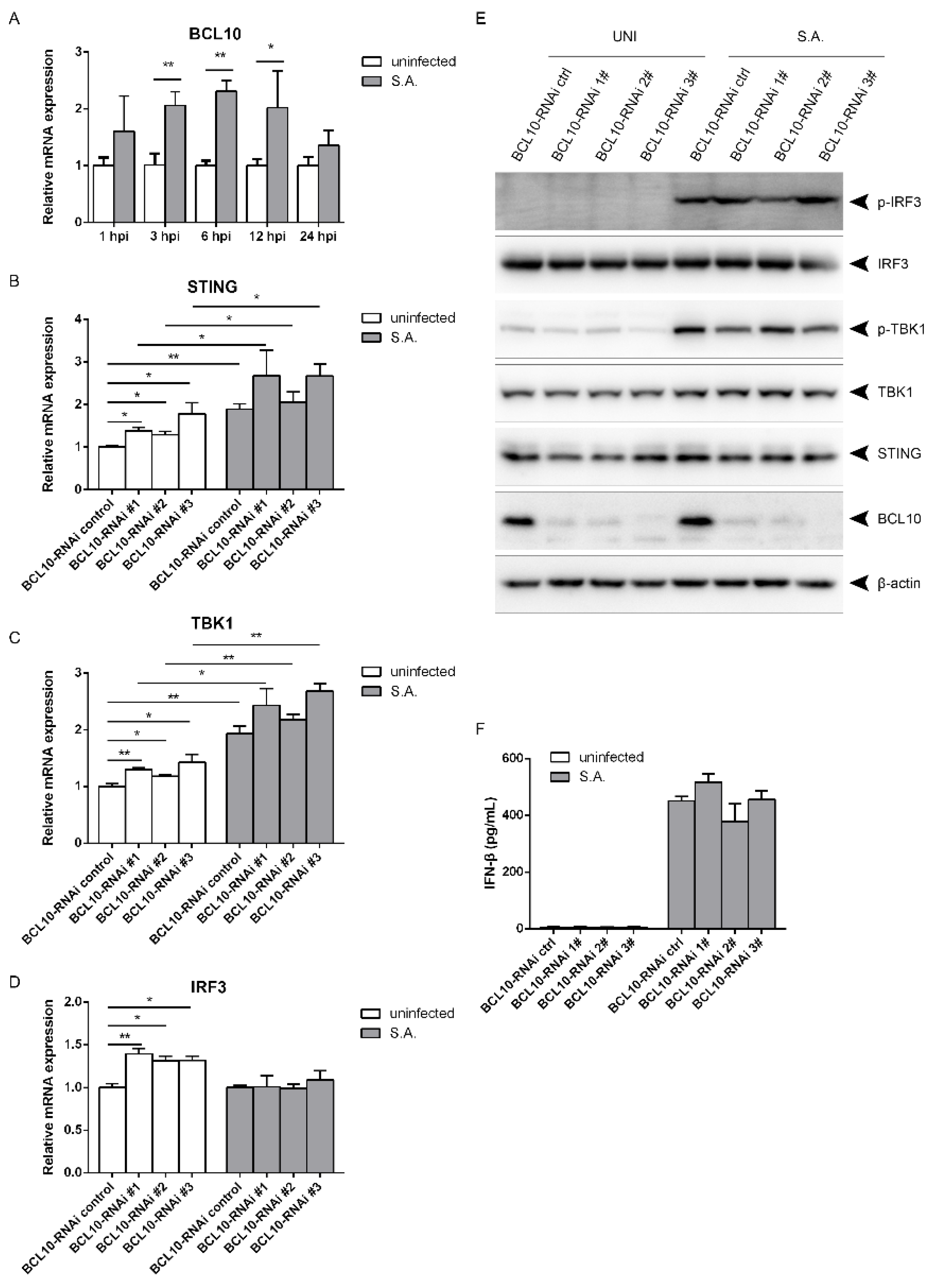
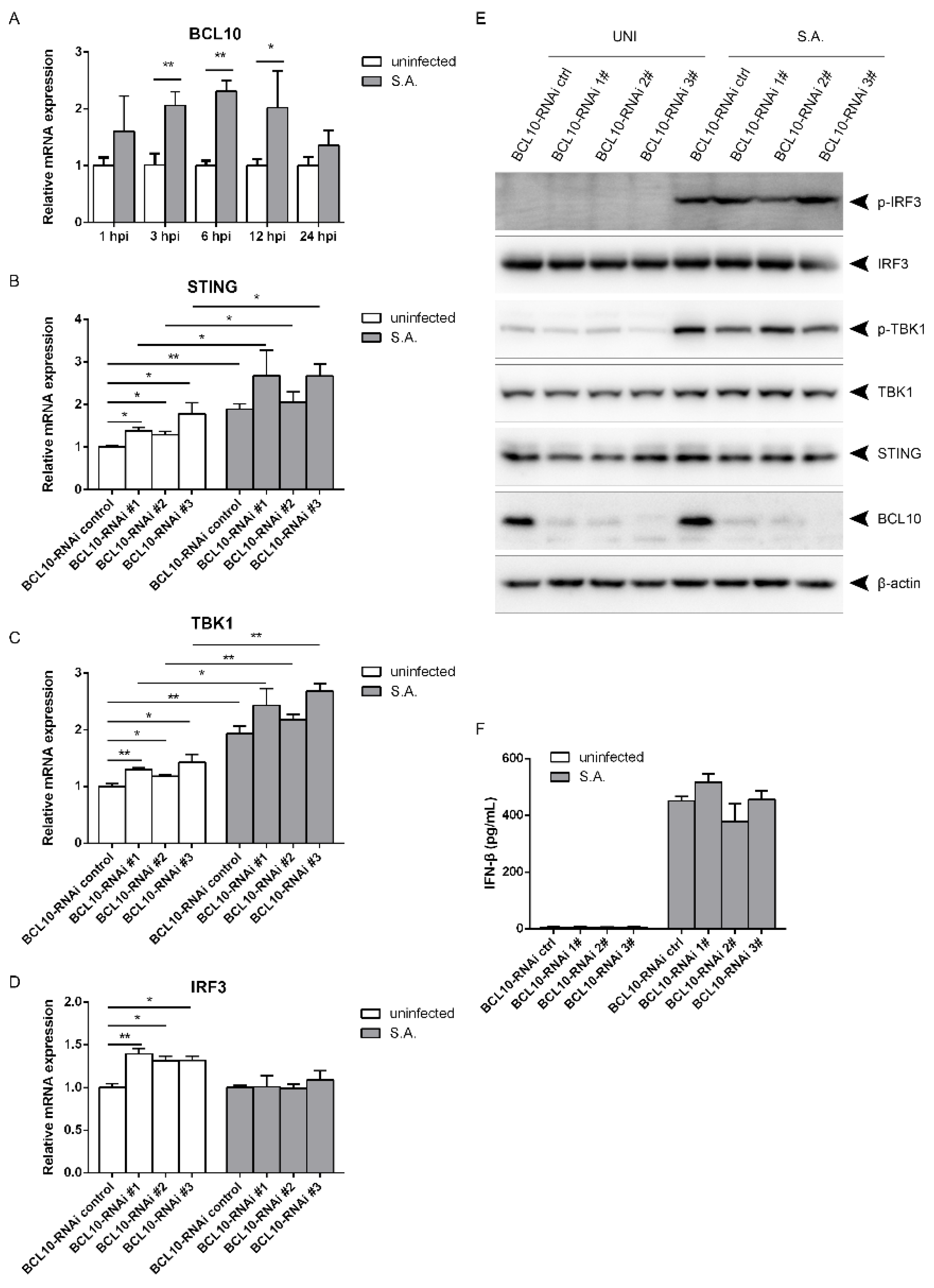
2.5. S. aureus-Induced Activation of the STING/TBK1/IRF3 Pathway Is Independent of B Cell Lymphoma 10 (BCL10)
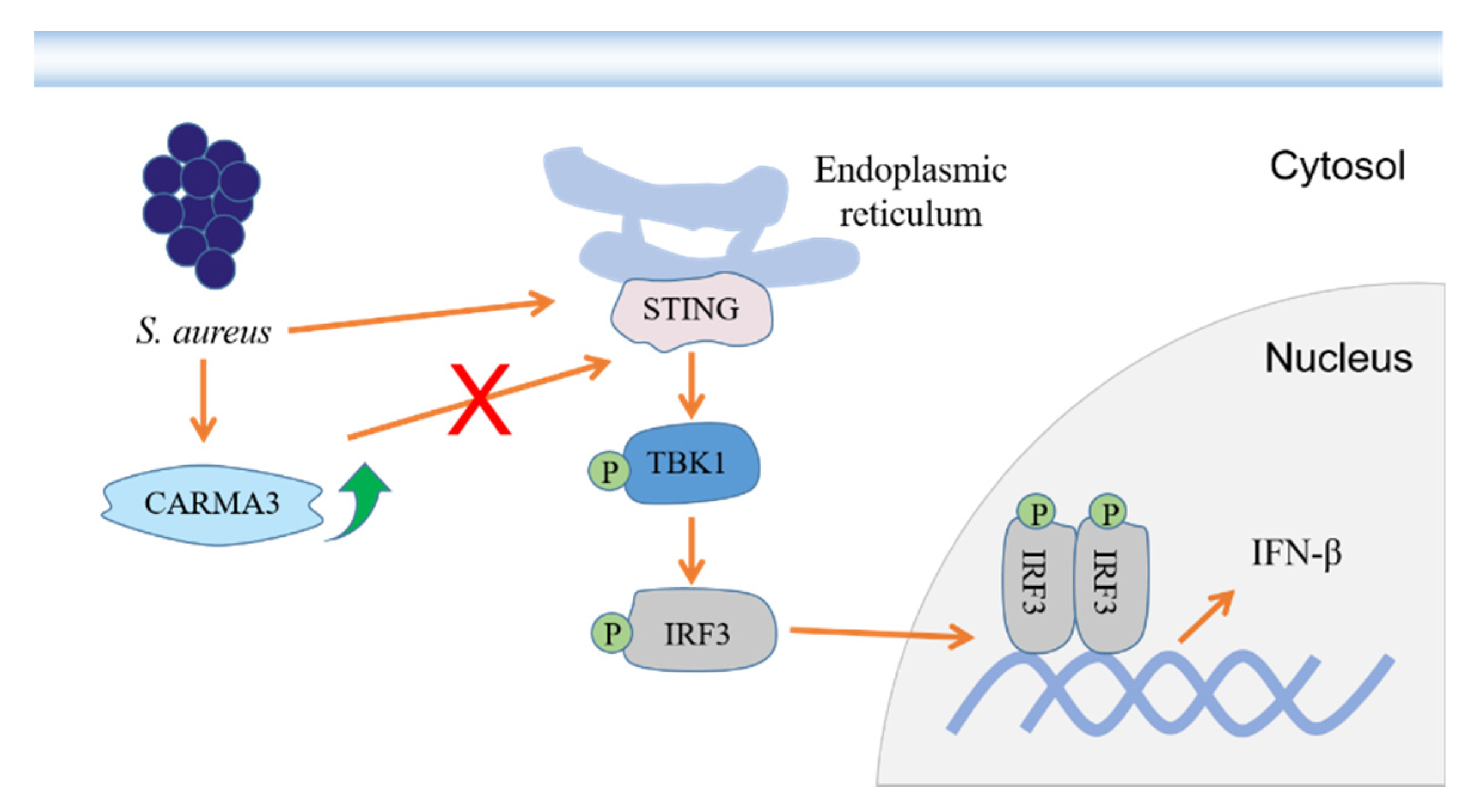
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Bacteria and Cell Culture
4.3. Western Blotting
4.4. Quantitative Real-Time PCR
4.5. IFN-β ELISA
4.6. Lentivirus-Medited RNAi
4.7. Plasmid Transfection
4.8. LDH Release Assay
4.9. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miller, L.S.; Connell, O.R.M.; Gutierrez, M.A.; Pietras, E.M.; Shahangian, A.; Gross, C.E.; Thirumala, A.; Cheung, A.L.; Cheng, G.; Modlin, R.L. MyD88 Mediates Neutrophil Recruitment Initiated by IL-1R but Not TLR2 Activation in Immunity against Staphylococcus aureus. Immunology 2006, 24, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderhaeghen, W.; Hermans, K.; Haesebrouck, F.; Butaye, P. Methicillin-resistantStaphylococcus aureus (MRSA) in food production animals. Epidemiol. Infect. 2010, 138, 606–625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, L.S.; Pietras, E.M.; Uricchio, L.H.; Hirano, K.; Rao, S.; Lin, H.; Connell, O.R.M.; Iwakura, Y.; Cheung, A.L.; Cheng, G.; et al. Inflammasome-Mediated Production of IL-1β Is Required for Neutrophil Recruitment againstStaphylococcus aureus In Vivo. J. Immunol. 2007, 179, 6933–6942. [Google Scholar] [CrossRef] [Green Version]
- McNab, F.W.; Mayerbarber, K.D.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Siegal, F.P.; Kadowaki, N.; Shodell, M.; Bocarsly, F.P.A.; Shah, K.; Ho, S.; Antonenko, S.; Liu, Y.J. The Nature of the Principal Type 1 Interferon-Producing Cells in Human Blood. Science 1999, 284, 1835–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Chen, Z.J. STING Specifies IRF3 Phosphorylation by TBK1 in the Cytosolic DNA Signaling Pathway. Sci. Signal. 2012, 5, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garzoni, C.; Kelley, W.L. Staphylococcus aureus: New evidence for intracellular persistence. Trends Microbiol. 2009, 17, 59–65. [Google Scholar] [CrossRef]
- Martin, F.J.; Gomez, M.I.; Wetzel, D.M.; Memmi, G.; Seaghdha, O.M.; Soong, G.; Schindler, C.; Prince, A. Staphylococcus aureus activates type I IFN signaling in mice and humans through the Xr repeated sequences of protein A. J. Clin. Investig. 2009, 119, 1931–1939. [Google Scholar] [CrossRef] [Green Version]
- Parker, D.; Planet, P.J.; Soong, G.; Narechania, A.; Prince, A. Induction of Type I Interferon Signaling Determines the Relative Pathogenicity of Staphylococcus aureus Strains. PLoS Pathog. 2014, 10, e1003951. [Google Scholar] [CrossRef] [Green Version]
- Parker, D.; Prince, A. Staphylococcus aureus Induces Type I IFN Signaling in Dendritic Cells Via TLR. J. Immunol. 2012, 189, 4040–4046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, C.; Zhou, Z.; Quan, Y.; Zhang, S.; Wang, T.; Zhao, X.; Morrison, C.; Heise, M.T.; He, W.; Miller, M.S.; et al. CARMA3 Is a Host Factor Regulating the Balance of Inflammatory and Antiviral Responses against Viral Infection. Cell Rep. 2016, 14, 2389–2401. [Google Scholar] [CrossRef] [Green Version]
- Bowie, A. The STING in the Tail for Cytosolic DNA-Dependent Activation of IRF. Sci. Signal. 2012, 5, 9. [Google Scholar] [CrossRef]
- Missiakas, D.; Winstel, V. Selective Host Cell Death by Staphylococcus aureus: A Strategy for Bacterial Persistence. Front. Immunol. 2021, 11, 1733. [Google Scholar] [CrossRef] [PubMed]
- Hebert, A.; Sayasith, K.; Senechal, S.; Dubreuil, P.; Lagace, J. Demonstration of intracellularStaphylococcus aureusin bovine mastitis alveolar cells and macrophages isolated from naturally infected cow milk. FEMS Microbiol. Lett. 2000, 193, 57–62. [Google Scholar] [CrossRef]
- Kubica, M.; Guzik, K.; Koziel, J.; Zarebski, M.; Richter, W.; Gajkowska, B.; Golda, A.; Gudowska, M.A.; Brix, K.; Shaw, L.; et al. A Potential New Pathway for Staphylococcus aureus Dissemination: The Silent Survival of S. aureus Phagocytosed by Human Monocyte-Derived Macrophages. PLoS ONE 2008, 3, e1409. [Google Scholar] [CrossRef] [Green Version]
- Sendi, P.; Proctor, R.A. Staphylococcus aureus as an intracellular pathogen: The role of small colony variants. Trends Microbiol. 2009, 17, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Keeffe, O.K.M.; Wilk, M.M.; Leech, J.M.; Murphy, A.G.; Laabei, M.; Monk, I.R.; Massey, R.C.; Lindsay, J.A.; Foster, T.J.; Geoghegan, J.A.; et al. Manipulation of Autophagy in Phagocytes Facilitates Staphylococcus aureus Bloodstream Infection. Infect. Immun. 2015, 83, 3445–3457. [Google Scholar] [CrossRef] [Green Version]
- Koziel, J.; Gudowska, M.A.; Mikolajczyk, T.; Bzowska, M.; Sturdevant, D.E.; Whitney, A.R.; Shaw, L.N.; DeLeo, F.R.; Potempa, J. Phagocytosis of Staphylococcus aureus by Macrophages Exerts Cytoprotective Effects Manifested by the Upregulation of Antiapoptotic Factors. PLoS ONE 2009, 4, e5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musilova, J.; Mulcahy, M.E.; Kuijk, M.M.; McLoughlin, R.M.; Bowie, A.G. Toll-like receptor 2–dependent endosomal signaling by Staphylococcus aureus in monocytes induces type I interferon and promotes intracellular survival. J. Biol. Chem. 2019, 294, 17031–17042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, M.; Liu, J.; Chen, X.; Xie, Y.; Yuan, C.; Xiang, Y.; Sun, B.; Lan, K.; Chen, M.; James, S.J.; et al. Casein Kinase II Controls TBK1/IRF3 Activation in IFN Response against Viral Infection. J. Immunol. 2015, 194, 4477–4488. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Q.; Hou, J.; Zhou, Y.; Yang, Y.; Xie, B.; Cao, X. Siglec1 suppresses antiviral innate immune response by inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell Res. 2015, 25, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Bucktrout, B.S.; Xi, Y.; Xu, D.; Du, D.; Zhang, Q.; Xiang, W.; Liu, J.; Melton, A.; Sheppard, D.; et al. Innate Antiviral Host Defense Attenuates TGF-β Function through IRF3-Mediated Suppression of Smad Signaling. Mol. Cell 2014, 56, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Zhao, Z.; Yang, Z.; Meng, Q.; Tan, P.; Xie, W.; Qingcai, M.; Wang, R.F.; Cui, J. USP38 Inhibits Type I Interferon Signaling by Editing TBK1 Ubiquitination through NLRP4 Signalosome. Mol. Cell 2016, 64, 267–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakshi, S.; Taylor, J.; Strickson, S.; McCartney, T.; Cohen, P. Identification of TBK1 complexes required for the phosphorylation of IRF3 and the production of interferon β. Biochem. J. 2017, 474, 1163–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Li, C.; Xue, P.; Zhong, B.; Mao, A.P.; Ran, Y.; Chen, H.; Wang, Y.Y.; Yang, F.; Shu, H.B. ISG56 is a negative-feedback regulator of virus-triggered signaling and cellular antiviral response. Proc. Natl. Acad. Sci. USA 2009, 106, 7945–7950. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Tian, Y.; Wang, R.P.; Gao, D.; Zhang, Y.; Diao, F.C.; Chen, D.Y.; Zhai, Z.H.; Shu, H.B. Negative feedback regulation of cellular antiviral signaling by RBCK1-mediated degradation of IRF. Cell Res. 2008, 18, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.Q.; Xia, T.; Hu, Y.H.; Sun, M.S.; Yan, S.; Lei, C.Q.; Shu, H.B.; Guo, J.H.; Liu, Y. IFITM3 inhibits virus-triggered induction of type I interferon by mediating autophagosome-dependent degradation of IRF. Cell. Mol. Immunol. 2018, 15, 858–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, C.Q.; Zhang, Y.; Xia, T.; Jiang, L.Q.; Zhong, B.; Shu, H.B. FoxO1 Negatively Regulates Cellular Antiviral Response by Promoting Degradation of IRF3*. J. Biol. Chem. 2013, 288, 12596–12604. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early transmission dynamics in Wuhan, China, of Novel Coronavirus–infected pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Jamrozy, D.M.; Coldham, N.G.; Butaye, P.; Fielder, M.D. Identification of a novel plasmid-associated spectinomycin adenyltransferase gene spd in methicillin-resistant Staphylococcus aureus ST398 isolated from animal and human sources. J. Antimicrob. Chemother. 2014, 69, 1193–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wang, Y.; Wu, C.; Shen, Z.; Schwarz, S.; Du, X.D.; Dai, L.; Zhang, W.; Zhang, Q.; Shen, J. First Report of the Multidrug Resistance Genecfrin Enterococcus faecalis of Animal Origin. Antimicrob. Agents Chemother. 2011, 56, 1650–1654. [Google Scholar] [CrossRef] [Green Version]
- Cai, X.; Chiu, Y.H.; Chen, Z.J. The cGAS-cGAMP-STING Pathway of Cytosolic DNA Sensing and Signaling. Mol. Cell 2014, 54, 289–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, J.; Liu, F. The cGAS-cGAMP-STING Pathway: A Molecular Link Between Immunity and Metabolism. Diabetes 2019, 68, 1099–1108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, F.; Li, B.; Liu, S.Y.; Iyer, S.S.; Yu, Y.; Wu, A.; Cheng, G. Positive Feedback Regulation of Type I IFN Production by the IFN-Inducible DNA Sensor cGAS. J. Immunol. 2015, 194, 1545–1554. [Google Scholar] [CrossRef]
- Zhang, S.; Pan, D.; Jia, X.M.; Lin, X.; Zhao, X. The CARMA3-BCL10-MALT1 (CBM) complex contributes to DNA damage-induced NF-κB activation and cell survival. Protein Cell 2017, 8, 856–860. [Google Scholar] [CrossRef] [Green Version]
- Qu, J.; Hou, Z.; Han, Q.; Jiang, W.; Zhang, C.; Tian, Z.; Zhang, J. Intracellular Poly(I:C) Initiated Gastric Adenocarcinoma Cell Apoptosis and Subsequently Ameliorated NK Cell Functions. J. Interferon Cytokine Res. 2014, 34, 52–59. [Google Scholar] [CrossRef] [Green Version]
- Koarai, A.; Sugiura, H.; Yanagisawa, S.; Ichikawa, T.; Minakata, Y.; Matsunaga, K.; Hirano, T.; Akamatsu, K.; Ichinose, M. Oxidative Stress Enhances Toll-Like Receptor 3 Response to Double-Stranded RNA in Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2010, 42, 651–660. [Google Scholar] [CrossRef] [Green Version]
- Fang, R.; Jiang, Q.; Zhou, X.; Wang, C.; Guan, Y.; Tao, J.; Xi, J.; Feng, J.M.; Jiang, Z. MAVS activates TBK1 and IKKε through TRAFs in NEMO dependent and independent manner. PLoS Pathog. 2017, 13, e1006720. [Google Scholar] [CrossRef]
- Franz, K.M.; Neidermyer, W.J.; Tan, Y.J.; Whelan, S.P.J.; Kagan, J.C. STING-dependent translation inhibition restricts RNA virus replication. In Proceedings of the National Academy of Sciences, Yale University School of Medicine, New Haven, CT, USA, 23 October 2018; Volume 115, pp. E2058–E2067. [Google Scholar]
- Zhou, Y.; Shah, S.Z.A.; Yang, L.; Zhang, Z.; Zhou, X.; Zhao, D. Virulent Mycobacterium bovis Beijing Strain Activates the NLRP7 Inflammasome in THP-1 Macrophages. PLoS ONE 2016, 11, e0152853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, C.Q.; Wu, X.; Zhong, X.; Jiang, L.; Zhong, B.; Shu, H.B. USP19 Inhibits TNF-α– and IL-1β–Triggered NF-κB Activation by Deubiquitinating TAK. J. Immunol. 2019, 203, 259–268. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer (5′–3′) | Reverse Primer (5′–3′) |
|---|---|---|
| IFN-β | GCACTGGGTGGAATGAGACT | AGTGGAGAGCAGTTGAGGACA |
| CARMA3 | AGGCAGGAGTGGTTCTGTACT | TCTTCAGGTTGCTTCGAGGAC |
| STING | TGGCTGCTGATGCCATACTC | CACAGCTCTTCAGCCAGACA |
| TBK1 | ACACATGACGGCGCATAAGA | CGGCTCGTGACAAAGATAGGA |
| IRF3 | ACAGATGGCTGACTTTGGCA | GCAGCTAACCGCAACACTTC |
| BCL10 | GAGCATCCACTGTCATGTACCA | AGGAAGAGTGGCTGAAGAGAA |
| β-actin | CAGAGCAAGAGAGGTATCCTGAC | AAGGTCTCAAACATGATCTGGGT |
| Name | Sequence (Sense, Antisense) |
|---|---|
| STING-targeting sequences | |
| Target sequence 1: | CACCGTTAGAGGAATTCGGAGTGCG |
| AAACCGCACTCCGAATTCCTCTAAC | |
| Target sequence 2: | CACCGCGCACTCCGAATTCCTCTAA |
| AAACTTAGAGGAATTCGGAGTGCGC | |
| Target sequence 3: | CACCGAGCGGTGACCTCTGGGCCGT |
| AAACACGGCCCAGAGGTCACCGCTC | |
| Target sequence 4: | CACCGTGTTGGGGTCAACTACACTC |
| AAACGAGTGTAGTTGACCCCAACAC | |
| TBK1-targeting sequences | |
| Target sequence 1: | CACCGCATAAGCTTCCTTCGCCCAG |
| AAACCTGGGCGAAGGAAGCTTATGC | |
| Target sequence 2: | CACCGGAGGAGCCGTCCAATGCGTA |
| AAACTACGCATTGGACGGCTCCTCC | |
| IRF3-targeting sequences | |
| Target sequence 1: | CACCGACCAGCCAGGGCAAAATCCG |
| AAACCGGATTTTGCCCTGGCTGGTC | |
| Target sequence 2: | CACCGGAACGAGGTTCAGGATCCCG |
| AAACCGGGATCCTGAACCTCGTTCC | |
| Target sequence 3: | CACCGCCAGTGGTGCCTACACCCCG |
| AAACCGGGGTGTAGGCACCACTGGC | |
| BCL10-targeting sequences | |
| Target sequence 1: | CACCGCACTGTCATGTACCACCCGG |
| AAACCCGGGTGGTACATGACAGTGC | |
| Target sequence 2: | CACCGCCGAACTTCAAGTAGAAAAC |
| AAACGTTTTCTACTTGAAGTTCGGC | |
| Target sequence 3: | CACCGCGCACCGTCCCTCACGGAGG |
| AAACCCTCCGTGAGGGACGGTGCGC |
| Name | Sequence (Sense, Antisense) |
|---|---|
| Target sequence 1: | CACCGACGGCTCCGGAGGCGCGACG |
| AAACCGTCGCGCCTCCGGAGCCGTC | |
| Target sequence 2: | CACCGCGGTACGGTTAGCGCGGCAC |
| AAACGTGCCGCGCTAACCGTACCGC | |
| Target sequence 3: | CACCGTGGCGGCGGGCTCACCGGTA |
| AAACTACCGGTGAGCCCGCCGCCAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Zhao, S.; Gao, X.; Jiang, S.; Ma, J.; Wang, R.; Li, Q.; Qin, L.; Tong, Z.; Wu, J.; et al. Staphylococcus aureus Induces IFN-β Production via a CARMA3-Independent Mechanism. Pathogens 2021, 10, 300. https://doi.org/10.3390/pathogens10030300
Zhou Y, Zhao S, Gao X, Jiang S, Ma J, Wang R, Li Q, Qin L, Tong Z, Wu J, et al. Staphylococcus aureus Induces IFN-β Production via a CARMA3-Independent Mechanism. Pathogens. 2021; 10(3):300. https://doi.org/10.3390/pathogens10030300
Chicago/Turabian StyleZhou, Yang, Shasha Zhao, Xiao Gao, Songhong Jiang, Jialu Ma, Rui Wang, Qing Li, Leiying Qin, Zhizi Tong, Junwei Wu, and et al. 2021. "Staphylococcus aureus Induces IFN-β Production via a CARMA3-Independent Mechanism" Pathogens 10, no. 3: 300. https://doi.org/10.3390/pathogens10030300