
The Dangerous Liaisons in the Oxidative Stress Response to Leishmania Infection


Department of Biochemistry, Faculty of Biology and Medicine, University of Lausanne, 1066 Lausanne, Switzerland
*
Author to whom correspondence should be addressed.
Pathogens 2022, 11(4), 409; https://doi.org/10.3390/pathogens11040409
Submission received: 28 February 2022
/
Revised: 24 March 2022
/
Accepted: 25 March 2022
/
Published: 28 March 2022
(This article belongs to the Special Issue Leishmania & Leishmaniasis)
{kind=link}
{kind=link}
Abstract
:Leishmania parasites preferentially invade macrophages, the professional phagocytic cells, at the site of infection. Macrophages play conflicting roles in Leishmania infection either by the destruction of internalized parasites or by providing a safe shelter for parasite replication. In response to invading pathogens, however, macrophages induce an oxidative burst as a mechanism of defense to promote pathogen removal and contribute to signaling pathways involving inflammation and the immune response. Thus, oxidative stress plays a dual role in infection whereby free radicals protect against invading pathogens but can also cause inflammation resulting in tissue damage. The induced oxidative stress in parasitic infections triggers the activation in the host of the antioxidant response to counteract the damaging oxidative burst. Consequently, macrophages are crucial for disease progression or control. The ultimate outcome depends on dangerous liaisons between the infecting Leishmania spp. and the type and strength of the host immune response.
1. Introduction
During the blood meal of an infected sand fly, Leishmania (L.) parasites are injected into the mammalian host and are internalized by phagocytic cells, principally macrophages. In macrophages, Leishmania promastigotes differentiate into obligate intracellular amastigotes [1]. Amastigotes reside in phagolysosome-like organelles, where they survive and replicate [2]. The phagolysosomes present an increase in temperature and an acidic environment, which trigger the differentiation of promastigotes to amastigotes [3]. They can either host numerous or single amastigotes, depending on the infecting species [4]. Amastigotes replicate until the rupture of the macrophage, the released amastigotes are then internalized by the surrounding phagocytes, leading to the expansion of the infection.
Leishmania spp. principally affect the skin and/or the mucosal tissues depending on the infecting species, which are determinants for the type of cutaneous outcome and clinical pathology, but also for the host inflammatory and anti-inflammatory response [5]. Localized cutaneous leishmaniasis (LCL) remains the most prevalent clinical manifestation of leishmaniasis. It is normally non-life-threatening but can be often associated with a social stigma.
In murine experimental models, protection against cutaneous leishmaniasis is associated with a robust T helper (Th) 1 (Th1) cell immune response and the production of interferon (IFN) gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α), whereas susceptibility is associated with a Th2 response involving IL-10 and IL-4. In humans, the situation is not as clear and there is a large spectrum of immunological responses with different levels in T cell responses and IFN-γ [5,6]. In any case, IFN-γ is central in the defense against Leishmania and boosts parasite control by activating infected macrophages to induce microbicidal effectors to enhance parasite killing [7]. In LCL, the early immune response is mediated by T-cell derived TNF-α and IFN-γ, however, at later stages, Th2 cells producing interleukin (IL)-10 and transforming growth factor-beta (TGF-β) are detected, which could be attributed to a decrease in the proinflammatory cytokine storm once the disease is resolved [5].
In Latin America, infection with L. guyanensis or L. braziliensis mainly leads to self-healing LCL, however, between 5–10% of these infections may disseminate and manifest as metastatic forms such as mucocutaneous leishmaniasis (MCL) [5,8]. The MCL outcome is distinguished by its persistent, dormant, and metastatic behavior, in which parasites disseminate and secondary distant lesions appear, mostly in the oral and nasopharyngeal parts of the face. These clinical features of MCL can be highly disfiguring since parasite dissemination is followed by extensive tissue destruction linked to high immune cell infiltration leading to hyperinflammation [9,10]. Contrary to LCL, MCL lesions are not self-healing and require drug treatment [5,11]. Patients affected by MCL present high levels of TNF-α but decreased levels of IL-10 in comparison to LCL, which results in the hyperinflammatory response characteristic of MCL lesions, whereas the actual detectable parasite load is low [12]. Additionally, the IL-17 inflammation-inducing cytokine and Th17 cells are highly expressed in MCL lesions in comparison to LCL lesions [13].
Another metastatic form developed after LCL is DL (also called disseminated cutaneous leishmaniasis, DCL) and can be caused by L. braziliensis, L. panamensis, L. guyanensis, or L. amazonensis parasites [5]. Parasite dissemination in DL occurs within weeks or days after the initial lesion formation [14,15]. The characteristic clinical picture of DL consists of the formation of multiple nodules, papules, and ulcerated lesions starting at the infection site, which subsequently disseminates preferentially to the limbs. Additionally, MCL nasal mucosal lesions are also found in DL cases [16]. Due to the high number of lesions, DL is difficult to treat. A high pro-inflammatory Th1 response at the lesion site is detected but not in the peripheral blood in DL patients, which suggests that the decreased peripheral Th1 response could allow the spread of the parasite [16].
The presence of a viral endosymbiont, the Leishmania RNA virus (LRV) in the cytoplasm of some Leishmania species has been described already some years ago and may be considered a risk factor for the progression towards exacerbated forms of the disease including MCL and DL [17,18]. LRV belongs to the Totiviridae family, whose members are characterized by icosahedral particles present in a wide range of protozoa including Trichomonas vaginalis, Entamoeba, and Toxoplasma gondi [19,20,21,22]. The viral particles range between 30–40 nm in diameter composed of a non-segmented double-stranded RNA genome encoding a capsid protein and a capsid-RNA-dependent RNA polymerase fusion protein, crucial for the dsRNA virus replication [23]. LRV was first described in L. Viannia subgenus in the L. guyanensis strain [24] and subsequently in the L. braziliensis strain [25]. Additionally, it has also been detected in L. Leishmania subgenus in L. major [26], L. infantum [27], and L. aethiopica [28] strains. The LRV sequence varies between the two L. subgenera, therefore they have been differently categorized as LRV1 and LRV2 in L. Viannia and L. Leishmania, respectively. For example, L. guyanensis parasites are called LgyLRV1+ or LgyLRV1- depending on the presence of the LRV1 particles [29]. LRV1 presence in human L. guyanensis and L. braziliensis infection has been significantly associated with treatment failure and relapse. However, the mechanisms by which LRV1 modulates treatment failure have not as yet been described [30,31]. Taken together, both host and parasite factors will determine the outcome of the disease.
2. The Oxidative Stress Response
Oxidative stress was first reported 30 years ago and describes the imbalance between oxidants and antioxidants in favor of the oxidants, which results in failure of the redox signaling and consequent cell damage [32]. Oxidative stress arises from the excessive release of reactive oxygen species (ROS) and reactive nitrogen species (RNS) [33]. ROS avidly interacts with a broad variety of molecules such as proteins, nucleic acids, lipids, and carbohydrates. Through such interactions, ROS contributes to damage in the biological systems. ROS, in addition, are a key cellular defense against invading pathogens [34]. In response to intracellular pathogens such as Leishmania parasites, macrophages rapidly induce an oxidative stress response as a mechanism of defense to induce pathogen clearance and activate signaling pathways associated with inflammation and immune responses [35,36].
The superoxide, O2−, is the most common oxygen free radical. It can be produced as a byproduct in the mitochondria (Figure 1). The formation of O2− derives from the electrons transfer along the different enzymes of the respiratory chain, which is not totally effective and leads to the leakage of electrons onto molecular oxygen resulting in O2− [37,38]. Superoxide can also be produced from the leakage of electrons through the electron transport chain within the endoplasmic reticulum (ER) [39] or by 5-lipoxygenase [40]. Other oxygen radicals are the hydroxyl (•OH), the peroxyl (RO•2), and the alkoxyl (RO•). The non-radical intermediates that are either oxidizing agents and/or are simply transformed into radicals include hypochlorous acid (HOCl), ozone (O3), singlet oxygen (1O2), and hydrogen peroxide (H2O2). Superoxide is detoxified by the family of enzymes known as superoxide dismutase (SOD), responsible for its transformation into H2O2, which is less reactive than the free radical and which can also participate in signaling pathways [34]. SOD can be cytoplasmic (SOD1) or mitochondrial (SOD2). H2O2 produced by SODs is then detoxified into O2 and H2O by peroxiredoxins. ROS can also be generated by the phagocyte NOX enzymes, whose primary function is ROS production [37]. In the human genome, there are 7 NOX homologs: NOX1 to NOX5 and DUOX1 and DUOX2, which vary in their expression level, organ-specific expression, ROS release, and regulation of their activity [41].
NOX2 (also known as gp91phox) is the prototype of NADPH oxidases and thus the best-characterized isoform [42]. A complex series of protein/protein interactions are responsible for the activation of NOX2. In macrophages, NOX2 comprises the principal source of ROS. NOX2 is composed of six hetero-subunits, which connect in response to a stimulus to activate the enzyme complex and consequently produce superoxide (Figure 1) [43]. The two NOX2 subunits gp91phox and p22phox are integral membrane proteins that together comprise the large heterodimeric subunit called flavocytochrome b558 (cyt b558). The multidomain regulatory subunits, p40phox, p47phox, and p67phox exist in the cytosol as a complex under basal conditions. Upon stimulation, p47phox is phosphorylated and the whole complex is translocated to the membrane where it interacts with cyt b558 to compose the activate oxidase enzyme. To be active, the complex needs two low-molecular-weight guanine nucleotide-binding proteins, called Rac2 and Rap1A. Rac2 binds guanosine triphosphate (GTP) and translocates to the membrane along with the p40phox, p47phox, and p67phox complex. During the process of phagocytosis, the plasma membrane is internalized and becomes the interior membrane of the phagocytic vesicle leading to the release of O2•− through the enzyme complex [34].
RNS comprises nitrogen-containing oxidants, such as NO• and its by-products including nitrate (NO3−), nitrite (NO2−), and peroxynitrite (ONOO•) [44]. In the organism, NO• is synthesized from L-arginine and molecular oxygen using NADPH as an electron donor. Overall, the reaction involves a two-step oxidative conversion of L-arginine to NO and L-citrulline via N-hydroxy-L-arginine as an intermediate (Figure 1) [45]. The enzymes responsible for NO• generation are nitric oxide synthases (NOSs). There are three different subtypes of NOS enzymes depending on the tissue type: eNOS (endothelial NOS), iNOS (inducible NOS), and nNOS (neuronal NOS). The iNOS isoform is the most relevant to phagocyte-pathogen interactions [46,47]. Stimulation of pattern recognition receptors (PRRs) together with signaling from pro-inflammatory cytokines can lead to iNOS transcription. In addition, the L-arginine substrate used by iNOS for generating NO• is also used by Leishmania parasites for the production of their essential nutrients L-ornithine and urea, resulting in the decrease of NO• and therefore favoring parasite survival [48]. Arginase 1 is the cytosolic enzyme responsible to convert L-arginine into urea and ornithine. Ornithine is a precursor of polyamines, which induce the synthesis of trypanothione and the proliferation of parasites. Trypanothione is an analog of glutathione essential for parasite protection against oxidants [49,50].
3. Oxidative Stress as Host Defense against Leishmania
In infection, the role of oxidative stress is dual: free radicals serve as protection against invading pathogens but consequently can also lead to inflammation resulting in tissue damage [51]. Phagocytic cells, such as macrophages and neutrophils, induce the antimicrobial response when infected by pathogens. The free radicals from oxygen, nitrogen, and chlorine derived from the macrophage respiratory burst are toxic to Leishmania. The first and foremost player among the prooxidants is the superoxide anion produced by the membrane-bound NOX2. This is the initial molecule of a storm of free radicals resulting in an oxidant milieu aiming at parasite killing. The use of mouse strains deficient in the gp91phox (gp91phox−/−) subunit of NOX2 complex revealed that ROS are crucial for parasite killing. For instance, gp91phox−/− mice infected with L. amazonensis presented severe pathology at the later stage of infection [52]
Nonetheless, Leishmania parasites have evolved strategies to antagonize the host immune system, therefore, contributing to their persistence and proliferation within macrophages [35]. Lipophosphoglycans (LPGs) molecules on the Leishmania surface have been described to inhibit the phosphorylation of the p47phox subunit and therefore block superoxide generation by NOX2 [53]. The induced oxidative stress in parasitic infections triggers the activation in the host of the antioxidant response to counteract the damaging oxidative burst. Such a response is principally mounted by the nuclear factor-erythroid 2-related factor 2 (NRF2) transcription factor leading to the decrease of oxidative free radicals, that consequently favor parasite persistence.
4. The Antioxidant Stress Response: The NRF2 Transcription Factor
Infection induces oxidative stress and inflammation, whereas the host immune system induces the antioxidant and anti-inflammatory response as a mechanism of defense to limit infection and favor pathogen clearance. In this regard, the cytoprotective role of NRF2 has been widely studied as a therapeutic strategy for protection against viruses such as influenza virus [54] or Leishmania parasites [55].
NRF2 was first described in the early 90s as a protein that recognizes the NF-E2 binding site of human β-globin genes (Figure 2) [56]. Some years later the role of NRF2 was described in the antioxidant response evidenced by the transcriptional modulation of the Nqo1 gene [57]. However, the detailed function of NRF2 in the antioxidant response was reported later by Itoh et al. [58]. This opened the door to thousands of studies involving NRF2 in the past decades. Indeed, from birth, animals must fight against multiple stressors that interfere with their homeostasis. Consequently, animals have been forced to evolve detoxifying systems like the NRF2 system that protects against a broad spectrum of stressors, including oxidative stress [59].
NRF2 is part of the cap’n’collar family of transcription factors, a family of basic leucine zipper transcription factors broadly conserved from worms to humans, but not present in plants or fungi [60]. Currently, NRF2 is related to a wide range of diseases in the field of inflammation, cancer, and metabolism [61,62,63]. Accordingly, NRF2 deficient (Nrf2−/−) mice have been described to be more vulnerable to chemical and radiation-induced tumorigenesis [62], exhibit more severe lung inflammation and damage upon exposure to cigarette smoke [64], and hyperoxia [65] in comparison to wild-type (WT) mice. The NRF2 stress response pathway is defined as the principal inducible defense against oxidative and electrophilic stresses. The antioxidant response controlled by NRF2 comprises the regulation of phase II enzymes which include the glutathione (GSH) thioredoxin, thioredoxin reductase 1, sulfiredoxin, and peroxiredoxin, which play an essential role in the reduction of oxidized protein thiols, as well as enzymes involved in NADPH generation, drug efflux, xenobiotic detoxification, and heme metabolism [66,67,68].
Years later after NRF2 description, KEAP1, an inhibitor of NRF2, was discovered [69]. KEAP1 induces NRF2 degradation under non-stressed conditions, whereas oxidative insults directly modify KEAP1 thiols groups, resulting in the inactivation of KEAP1 function, subsequent stabilization of NRF2, and induction of cytoprotective genes. Under normal conditions, NRF2 is constantly degraded via the ubiquitin-proteasome pathway in a KEAP1- dependent manner. Upon oxidative or electrophilic stress, the KEAP1 homodimer is inactivated leading to NRF2 stabilization and translocation to the nucleus. Nuclear NRF2 forms heterodimers with small Maf proteins and induces the expression of its target genes through binding to specific regions of the DNA named antioxidant response elements or electrophile response elements. NRF2 activates many cytoprotective genes [66]. Alternatively, KEAP1 is also regulated by the autophagy receptor, SQSTM1/p62, which acts both as a target and positive regulator of NRF2 [70,71]. SQSTM1/p62 can directly bind to KEAP1 [72]. On disruption of autophagy, there is the accumulation of SQSTM1/p62, which activates NRF2 by competing for the binding to KEAP1 [71]. Importantly, SQSTM1/p62 regulates NRF2 independently of the cellular redox state, which may connect NRF2 activity to the autophagy response pathway [61]. Overall, several mechanisms may lead to NRF2 activation and NRF2 is a key host factor in the host’s antioxidant response during an infection to limit over exacerbated tissue damage, however, this comes with a cost since its activation can favor pathogen persistence.
5. Interplay between Inflammation, NF-κB, and NRF2 Transcription Factors
Macrophages are myeloid innate immune cells that reside in many organs throughout the body and present distinguished tissue-specific cellular functions. They are specialized in the detection, phagocytosis, and destruction of harmful invading organisms [73]. Inflammation is triggered when host cells recognize conserved structures on pathogens, called microbial-associated molecular patterns (MAMPs), or endogenous stress signals such as ROS, called danger-associated molecular patterns through pattern recognition receptors (PRRs). These receptors are expressed by myeloid cells, such as monocytes, macrophages, neutrophils, and dendritic cells [74] as well as by several non-immune cells including epithelial cells and fibroblasts [75,76]. Classes of PRRs include membrane-bound Toll-like receptors (TLRs), C-type lectin receptors, cytosolic RIG-I-like receptors (RLRs), and Nod-like receptors (NLRs). Their activation leads to the induction of pro-inflammatory pathways [77]. In the context of Leishmania, most studies have focused on the role of TLRs.
Macrophages activated by intracellular pathogens have been classically designated as pro-inflammatory M1 macrophages. M1 macrophages are found in an inflammatory scenario that is controlled by the signaling of TLR and IFN pathways, which guide acute inflammatory responses with the release of pro-inflammatory cytokines such as IL-1β, IL-6, IL-12, IL-18, IL-23, and TNF-α. This pro-inflammatory signaling cascade induces Th1 response activation and facilitates complement-mediated phagocytosis [78,79]. On the contrary, M2 macrophages are responsible for the induction of the anti-inflammatory response with the production of cytokines including IL-10 and IL-13, or chemokines such as the C-C motif ligand 22. M2 macrophages have been described to particularly participate during parasitic, helminthic, and fungal infections [78].
The binding of MAMPs to TLRs initiates signaling cascades that induce the nuclear translocation of nuclear factor-kappa B (NF-κB) transcription factors leading to transcription of IL-6 and TNF-α cytokines, and type I IFN (IFN-I) [80]. The NF-κB is a family of transcription factors that consists of five members: p50, p52, p65 (RelA), c-Rel, and RelB. Dimerization of the NF-κB family is necessary for their DNA-binding properties. In unstimulated cells, NF-κB dimers are mainly cytoplasmic due to the binding of a set of inhibitory proteins known as the inhibitor of the NF-κB (IκB or IKK) family [81]. In contrast, in stimulatory conditions, such as infection, exposure to pro-inflammatory cytokines and oxidative species [82,83] will lead to the first step of NF-κB activation requiring post-translation modification of IκB inhibitors. Two defined mechanisms called the canonical pathway and, the alternative or non-canonical pathway, have been described for the induction of the NF-κB complex [84,85]. The pathways differ in the receptor inducing the signaling cascade, the NF-κB dimers, and the IKK components involved.
Contrarily to its activation, the mechanisms responsible for terminating the NF-κB pathway remain still poorly understood. Most of the studies have been centered on mechanisms involving IκB proteins and upstream signaling intermediates [86,87]. However, NF-κB signaling is also regulated by negative feedback mechanisms such as by A20 deubiquitinase (also known as TNFAIP3). A20 is essential for maintaining immune homeostasis and downregulating inflammation as confirmed by A20 deficient mice that prematurely die due to spontaneous multi-organ inflammation [88]. The inflammatory NF-κB signaling can also be controlled by NRF2 [89]. Indeed, numerous studies over the past years demonstrate the connection between the NRF2 and NF-κB pathways to regulate the transcription or function of downstream pro-inflammatory proteins [90,91,92].
Several mechanisms have been described on how NRF2 negatively regulates inflammation. The NRF2-Heme Oxygenase-1 (HO-1) axis not only helps the antioxidant response but also plays an anti-inflammatory role. NRF2 indirectly inhibits inflammation by HO-1 induction that inhibits NF-κB (i.e., RelA) phosphorylation at S276, a critical site for sustaining TNF-dependent NF-κB activation [93]. Interestingly, NF-κB activation is upregulated in Nrf2−/− mice leading to acute inflammation [91]. Additionally, NRF2 regulates NF-κB activation by modulating the degradation of IκBα as described by using mouse embryonic fibroblasts deficient in NRF2 [91]. Accordingly, the NRF2 activator, sulforaphane (SFN), present in cruciferous vegetables like broccoli and cabbage, has been described as a negative regulator of inflammation by decreasing the expression of NF-κB-induced pro-inflammatory cytokines, such as IL-1β and TNF-α, and the inflammatory mediators cyclooxygenase 2 and iNOS [90]. Similarly, NQO1 activation downregulates the LPS-induced expression of proinflammatory cytokines [94]. The NRF2 activator and target protein p62/SQSTM1 has also been described as a negative modulator of the inflammatory pathway [95]. p62/SQSTM1 deficiency resulted in higher levels of the proinflammatory cytokine IL-1β. NRF2 and NF-κB pathways synergize to induce p62/SQSTM1 to counteract uncontrolled inflammation and prevent NLRP3-inflammasome activation [70]. The pro-inflammatory cytokine IL-17 is repressed by NRF2 in the case of autoimmune encephalomyelitis [96]. Furthermore, the induced levels of GSH by NRF2 have been reported to affect TNF-α levels [97].
NRF2 has also been connected with the activation of T cells. Activation of NRF2 in CD4+ T cells has been associated with decreased expression of activation markers such as CD25 and CD69 as well as reduced activation of NF-κB [98]. The maintenance of ROS and antioxidant protein balance within the cell is critical for keeping the integrity of T-cell mediated immunity, therefore NRF2 plays a crucial role in limiting T cell activation. Accordingly, NRF2 activation reduces IFN-γ production and raises IL-4 and IL-13 cytokines in CD4+ T cells, and skews them into a Th2 differentiation promoting the anti-inflammatory response [99,100]. Thus, NRF2 aside from having a clear antioxidant role could represent a key player in attenuating inflammation to limit pro-inflammatory responses and tissue damage in Leishmania infection.
6. Leishmania Parasites and Inflammation
Macrophages are crucial for disease progression and the favorable/unfavorable outcome depends on the interplay between the infecting Leishmania spp. and the type and magnitude of the host immune response [49,101]. During Leishmania infection, both pro-inflammatory M1 and anti-inflammatory M2 macrophages are induced. However, macrophage polarization phenotypes are not mutually exclusive which complicates the immune scenario during leishmaniasis [49]. Additionally, Leishmania parasites have developed strategies to subvert the pro-inflammatory response to promote parasite survival [101,102].
The immune response led by TLRs plays a crucial role in determining the response of the host during Leishmania infection [103]. Parasite surface molecules are recognized by TLRs. For example, LPGs are recognized by TLR-2 resulting in the activation of NF-κB [104,105]. Similarly, the dsRNA of LRV1 within L. guyanensis (Lgy) parasites induces the cascade of pro-inflammatory cytokines via TLR-3 signaling [18]. Bone marrow-derived macrophages (BMDMs) infected with Lgy parasites bearing the LRV1 endosymbiont demonstrate that LRV1 is recognized by TLR-3 promoting hyper-inflammation and IFN-I production resulting in tissue destruction and parasite persistence as observed in MCL patients [18]. These findings are supported by in vivo infection of mice deficient in TLR-3 (Tlr3−/−), which present a significant decrease in footpad swelling at the peak of infection in comparison to the control C57Bl/6 WT mice (Figure 2) [18]. Moreover, LRV1 presence is associated with IL-17 secretion, which contributes to LRV1-mediated disease severity (Figure 2). The IL-17 cytokine is produced by Th17 in hyperinflammatory situations, such as LCL. Interestingly, in vivo infection of IL-17 deficient (Il17−/−) mice with LgyLRV1+ showed decreased LRV1-mediated pathology [17]. Furthermore, LRV1-induced TLR-3 activation has been associated with parasite persistence by promoting macrophage survival through AKT signaling partially relying on the microRNA miR-155. miR-155 has been described to be the only microRNA upregulated by the presence of LRV1. Infection of miR155 deficient (Mir155−/−) mice with LgyLRV1+ results in decreased disease pathology, which indicates that LRV1 uses miR1-55 as another survival strategy [106]. The relationship between LRV1 and IFN-I (ie IFN-alpha and IFN-beta) production has been further investigated by Rossi et al., who demonstrate that the IFN-I response is responsible for worsening the outcome of leishmaniasis. Injection of recombinant IFN-I to mice infected with LgyLRV1- or coinfection with lymphocytic choriomeningitis virus (LCMV) or Toscana virus (TOSV) resulted in higher lesion size and parasite load as well as downregulation of the IFN-γ receptor (IFN-γR), which is responsible for mediating the antileishmanial response induced by IFN-γ [7,107]. Similarly, L. braziliensis parasites bearing the LRV1 endosymbiont have been reported to promote aggressive pathogenesis in the context of HIV coinfection. The immunosuppressed immune status of HIV patients represents a favorable environment for disease exacerbation exerted by LRV1 [108]. The exact mechanism as to how LRV1 and HIV may synergize to worsen leishmaniasis outcomes has not, as yet, been described.
LRV1 signaling has also been studied in the context of other PRRs, such as the cytoplasmic dsRNA sensor RLRs and the inflammasome-independent NLRs [109]. RLRs are cytosolic receptors and promote the IFN-I response upon recognition of viral RNA [110]. No role for RLR-signaling in response to LRV1 was detected, which indicated that LRV1 was not likely to exit the phagolysosome to engage cytoplasmic receptors [109]. In contrast, LRV1 has been reported to induce the expression of inflammasome-independent NLRs, such as NLRC2 and NLRC5, which are known to induce anti-viral responses. NLRC2 has been described to direct monocytes to the infection site, which correlated to the phenotype observed in mice deficient in NLRC2 (Nlrc2−/−) infected with LgyLRV1+ presenting decreased lesion size and parasite load [109]. NLRC5, on the other hand, regulates MHC-I expression. MHC-I peptide complexes bind to CD8 cytotoxic (CD8+) T cells. The role of CD8+ T cells during Leishmania infection has been controversial since discrepancies among infection with different strains have been debated [111]. In the case of L. braziliensis LCL, CD8+ T cells are described to mediate tissue injury [112]. However, NLRC5 deficiency in mice had no consequence on LgyLRV1+ pathogenicity, which indicated that CD8+ T cells may not play a role in LRV1-mediated pathology in L. guyanensis infection. Additionally, no inflammasome activation was detected in vivo and in vitro in LgyLRV1+ infection [109]. These findings confirmed that LRV1 induces the pro-inflammatory signaling pathway via TLR-3 in L. guyanensis LCL. Recently, it has been described that LRV1 subverts the NLRP3 inflammasome activation via TLR-3-induced autophagy in L. guyanensis and L. braziliensis infections [113].
Several mechanisms used by LRV1 to promote aggressive MCL pathology have been identified. However, the way how this viral endosymbiont is exposed to the host is still under debate. Recent studies have demonstrated that exosomes are secreted by Leishmania within the sand fly gut, and are transmitted along with the parasites during the sand fly blood meal. Additionally, LRV1 viral particles have been detected in Leishmania exosomes, indicating that LRV1, as many other viruses infecting higher eukaryotes [114] could take advantage of the exosomal pathway to be externally released and become better suited to interact with the macrophage dsRNA sensing machinery [115,116]. This more direct interaction with the host could be used by LRV1 to promote its propagation and infectivity towards its hosts, ultimately, resulting in severe leishmaniasis outcomes. Taken together, infection with Leishmania parasites can lead to the simultaneous activation of multiple PRRs leading to the induction of pro-inflammatory pathways to restrain infection, however, often leading to increased pathology.
7. NRF2 Role in Leishmania Infection
The antioxidant and anti-inflammatory pathways induced by NRF2 have been associated with resistance to infection with multiple pathogens including viruses, bacteria, and protozoan microorganisms such as Entamoeba histolytica, Plasmodium spp., Toxoplasma gondii, Cryptosporidium parvum, and Leishmania spp. [117,118,119,120]. The role of the NRF2 pathway in Leishmania infection has not been widely explored. Infection with L. amazonensis has been reported to induce NRF2 activation through dsRNA-dependent protein kinase (PKR). This kinase activates Phosphoinositide-3-kinase (PI3K)/AKT signaling triggering NRF2 release from KEAP1 as well inducing NRF2 activator and target protein p62/SQSTM1. Activation of NRF2 in L. amazonensis decreases oxidative stress levels and favors parasite survival as observed by infecting NRF2-knockdown macrophages [121]. Our own findings revealed that the KEAP1-NRF2 pathway is activated in different Leishmania spp. suggesting that the activation of the antioxidant response is conserved among species and the difference in oxidative levels is not crucial to mount the antioxidant response in Leishmania infection (Figure 2). Consequently, the induced antioxidant response favors parasite survival. At late phases of macrophage infection in L. guyanensis infection, Nrf2−/− cells presented reduced parasite burden independently of LRV1. These data correlated with the increased expression of the antioxidant genes Hmox1 and Nqo1 regulated by NRF2 in L. guyanensis infection [122].
In Lgy infection, the ROS produced by NOX2 initiates the signaling responsible for NRF2 activation (Figure 2) [122]. Additionally, NOX2 regulation of NRF2 pathway is preserved among different Leishmania spp., confirming that Leishmania parasites have evolutionarily managed to exploit the NRF2 signaling pathway in the host cell in a similar way for their own benefit. Contrary to L. amazonensis infection, the PI3K/AKT axis does not participate in NRF2 pathway activation in Lgy infection as tested by pharmacological inhibition of either PI3K or AKT. Furthermore, PKR signaling is promoted by IFN-I and poly I:C, which increases expression and nuclear translocation of NRF2 as described in L. amazonensis. Poly I:C mimics LRV1 infection by inducing TLR-3 signaling. LRV1 has been described to be present in L. guyanensis and L. braziliensis but not in L. amazonensis [123]. Therefore, L. amazonensis is able to promote PKR independently of the presence of dsRNA, which could be linked to the ROS production by NOX2 [124].
Additionally, the flavonoid quercetin, which has antioxidant properties, also acts as an anti-leishmanial drug by reducing the parasite burden within macrophages. The NRF2/HO-1 pathway induced by quercetin results in upregulation of the ferritin complex, which controls the bioavailability of labile iron pool, impeding the uptake of this metal by L. braziliensis. Depletion of available iron decreases parasite replication and survival within macrophages [125].
Thus, NRF2 presents a dual role favoring both the parasite and the host by the induction of the antioxidant and anti-inflammatory response. Parasite contact with the macrophage seems sufficient to reprogram macrophage metabolism in response to Leishmania which induces the SRC-family kinase (SFK) signaling cascade triggering the activation of the NRF2 pathway (Figure 2) [122]. To sum up, the tripartite mutualism of Leishmania, LRV1, and macrophages favors parasite survival and therefore induces disease exacerbation. Leishmania parasites take advantage of the induced host detoxification machinery via the NRF2 pathway. In addition, the NRF2 pathway confers protection to LRV1/Leishmania by limiting the proinflammatory response. These results put into evidence the challenges in the design of specific drugs against Leishmania parasites and the important role of oncogenic kinases in leishmaniasis.
8. Perspectives
Leishmania parasites modulate host cell metabolism just 15 min post initial exposure [126]. The nature of the contact between Leishmania and the host cell surface molecules is not known. It could be relevant to define whether it is the flagellum that is responsible for the initial contact and to initiate the NRF2 pathway. Leishmania promastigotes possibly enter macrophages in a polarized manner through their flagellar tip and are then internalized into the host lysosomal compartments [126]. Interestingly, the initial parasite contact with the host cell is sufficient to mount the antioxidant response prior to phagocytosis [122].
In every Leishmania infection, the NRF2 pathway is induced by NOX2 signaling. Despite the large knowledge of NRF2 and its relationship with ROS, this is the first report of the link between NRF2 and the superoxide-producing NOX2 [122]. It also indicates that the activation of the antioxidant response is a general mechanism conserved among species.
Oxidative stimuli have been described to promote NRF2 via an SFK/PKCδ signaling circuit [127]. Consistently, in Lgy infection of macrophages, the expression of NRF2 depends on the NOX2/SFK/PKCδ axis. The NRF2 pathway is similarly activated by both LgyLRV1+ and LgyLRV1- parasites in macrophages and therefore the antioxidant response is promoted independently of the presence of LRV1 [122]. Furthermore, no TLRs are likely involved in NRF2 activation as shown by using mice deficient in the TLRs MyD88 and TRIF adaptors proteins. These findings reveal that Leishmania parasites could modulate the activation of the NRF2 pathway in macrophages by contact with a non-TLR pattern-recognition receptor such as DECTIN-1, which could mediate SFK signaling [128,129]. DECTIN-1 activation has already been described in the context of L. amazonensis [130]. Further studies are required to confirm the role of DECTIN-1 in this activation pathway.
Contrary to LgyLRV1- parasites, which only activate NRF2, LgyLRV1+ parasites induce both NRF2 and TLR-3-dependent inflammatory cytokines. This crosstalk between NRF2 and NF-κB pathways prevents hyper-inflammation due to high oxidative levels within the cell [89]. These data revealed that LgyLRV1+ parasites not only take advantage of the antioxidant response from activation of the NRF2 pathway but also from the NRF2-dependent anti-inflammatory response [131]. In this regard, other Leishmania spp. impair NF-κB signaling and dampen host immune response. For example, L. mexicana promastigotes favor the cleavage of p65 NF-κB subunit generating a smaller p35 protein [132] and L. amazonensis activates the NF-κB p50/p50 repressor complex [133]. Similarly, mice deficient in NRF2 have significantly reduced disease pathology when infected with LgyLRV1+, but not when infected with LgyLRV1-. This validates that NRF2 participates in the control of the host pro-inflammatory response provoked by LgyLRV1+ parasites. Nrf2−/− mice have been shown to present higher levels of proinflammatory cytokines [134]. In this context, pro-inflammatory cytokine TNF-α levels are higher in Nrf2−/− cells infected with LgyLRV1+ parasites, and TNF-α levels are highly increased in MCL. TNF-α constitutes a clear risk factor for disease development and immunotherapies directed to its production, such as TNF-α blockers [135], have been proposed as a treatment against leishmaniasis [136]. Thus, TNF- α levels are crucial for the outcome of leishmaniasis and could be of interest to investigate the impact of anti-leishmania drugs on this pro-inflammatory cytokine.
The NRF2 pathway could confer tissue damage control and diseases tolerance to systemic infections [118]. Wounds produce large amounts of ROS to combat invading pathogens, as ocurred in Leishmania infection, resulting in immune cells attraction. NRF2 has been shown to become activated in tissue damage favoring wound repair [137]. Nrf2−/− mice do not have an obvious skin phenotype under LgyLRV1+ infection but NRF2 could play a role in chronic leishmaniasis as assessed in a metastatic model of leishmaniasis. The deficiency of the NRF2 protein in Ifng−/− mice, and Nrf2xIfng double knock-out mice (dKO), promotes disease exacerbation and hyper-inflammation in LgyLRV1+ infected mice. Additionally, the tails of these dKO mice show augmented cartilage destruction and increased cellular infiltration at the footpads, which could be induced by higher expression of matrix metalloproteases (MMPs) [122]. Indeed, studies with Nrf2−/− mice with spinal cord injury display higher MMP9 activity [138]. Thus, IFN-γ is essential for controlling LgyLRV1+ infection and the NRF2-mediated anti-inflammatory response and tissue damage control. Additionally, the hyperinflammatory response observed in dKO could be linked to the IL-17 cytokine, which has been described to be negatively regulated by NRF2 in autoimmune encephalomyelitis [96]. IL-17 secretion favors the dissemination of LgyLRV1+ parasites over LgyLRV1- and inversely correlates with IFN-γ inducing disease exacerbation [106]. The link between Il-17 and NRF2 would require further attention.
Overall, NRF2 presents a dual role in Leishmania infection favoring both the parasite and the host by the induction of the antioxidant and anti-inflammatory response. Initial contact of the parasite and the host is sufficient to reprogram macrophage metabolism in response to Leishmania which induces the SFK signaling cascade resulting in the NRF2 pathway activation. In this respect, developing targeted drugs towards inhibition of the SFK family could offer potential therapeutic candidates to treat and prevent leishmaniasis. These dangerous liaisons, the tripartite mutualism of Leishmania, LRV1, and macrophages favor parasite survival and therefore induce disease exacerbation. Leishmania parasites benefit from the increased survival of macrophages via the LRV1 induced AKT signaling pathway. Furthermore, Leishmania parasites take advantage of the induced host detoxification machinery via the NRF2 pathway and the NRF2 pathway confers protection to LRV1/Leishmania by limiting the proinflammatory response. Thus, this association benefits the parasite and the infected cell but not the host.
Author Contributions
Original draft preparation, M.R.; writing, reviewing and editing, T.S. and N.F. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Swiss National fund for research to NF (Grant No. 310030_173180, and IZRJZ3_164176/1).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Available Statement
Not applicable.
Acknowledgments
The authors are grateful to all the past members of the group and to the administrative support who contributed in one way or another to this publication and to former publications which made possible this review. We thank specially Matteo Rossi for giving us the template for Figure 1.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Kaye, P.; Scott, P. Leishmaniasis: Complexity at the host-pathogen interface. Nat. Rev. Microbiol. 2011, 9, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Real, F.; Mortara, R.A. The diverse and dynamic nature of Leishmania parasitophorous vacuoles studied by multidimensional imaging. PLoS Negl. Trop. Dis. 2012, 6, e1518. [Google Scholar] [CrossRef] [Green Version]
- Weiss, G.; Schaible, U.E. Macrophage defense mechanisms against intracellular bacteria. Immunol. Rev. 2015, 264, 182–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seguin, O.; Descoteaux, A. Leishmania, the phagosome, and host responses: The journey of a parasite. Cell Immunol. 2016, 309, 1–6. [Google Scholar] [CrossRef]
- Scorza, B.M.; Carvalho, E.M.; Wilson, M.E. Cutaneous Manifestations of Human and Murine Leishmaniasis. Int. J. Mol. Sci. 2017, 18, 1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, W.N.; Ribeiro, L.E.; Schrieffer, A.; Machado, P.; Carvalho, E.M.; Bacellar, O. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of human tegumentary leishmaniasis. Cytokine 2014, 66, 127–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, P. IFN-gamma modulates the early development of Th1 and Th2 responses in a murine model of cutaneous leishmaniasis. J. Immunol. 1991, 147, 3149–3155. [Google Scholar] [PubMed]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Antonelli, L.R.; Dutra, W.O.; Almeida, R.P.; Bacellar, O.; Carvalho, E.M.; Gollob, K.J. Activated inflammatory T cells correlate with lesion size in human cutaneous leishmaniasis. Immunol. Lett. 2005, 101, 226–230. [Google Scholar] [CrossRef] [PubMed]
- Novais, F.O.; Carvalho, L.P.; Graff, J.W.; Beiting, D.P.; Ruthel, G.; Roos, D.S.; Betts, M.R.; Goldschmidt, M.H.; Wilson, M.E.; de Oliveira, C.I.; et al. Cytotoxic T Cells Mediate Pathology and Metastasis in Cutaneous Leishmaniasis. PLoS Pathog. 2013, 9, e1003504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, V.S.; Tuon, F.F.; Siqueira, A.M.; Nicodemo, A.C.; Neto, V.A. Treatment of mucosal leishmaniasis in Latin America: Systematic review. Am. J. Trop. Med. Hyg. 2007, 77, 266–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaze, S.T.; Dutra, W.O.; Lessa, M.; Lessa, H.; Guimarães, L.H.; Jesus, A.R.; Carvalho, L.P.; Machado, P.; Carvalho, E.M.; Gollob, K.J. Mucosal leishmaniasis patients display an activated inflammatory T-cell phenotype associated with a nonbalanced monocyte population. Scand. J. Immunol. 2006, 63, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Bacellar, O.; Faria, D.; Nascimento, M.; Cardoso, T.M.; Gollob, K.J.; Dutra, W.O.; Scott, P.; Carvalho, E.M. Interleukin 17 production among patients with American cutaneous leishmaniasis. J. Infect. Dis. 2009, 200, 75–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvalho, E.M.; Barral, A.; Costa, J.M.L.; Bittencourt, A.; Marsden, P. Clinical and immunopathological aspects of disseminated cutaneous leishmaniasis. Acta Trop. 1994, 56, 315–325. [Google Scholar] [CrossRef]
- Turetz, M.L.; Machado, P.R.; Ko, A.I.; Alves, F.; Bittencourt, A.; Almeida, R.P.; Mobashery, N.; Johnson, W.D., Jr.; Carvalho, E.M. Disseminated Leishmaniasis: A New and Emerging Form of Leishmaniasis Observed in Northeastern Brazil. J. Infect. Dis. 2002, 186, 1829–1834. [Google Scholar] [CrossRef] [PubMed]
- Machado, P.R.; Rosa, M.E.; Costa, D.; Mignac, M.; Silva, J.S.; Schriefer, A.; Teixeira, M.M.; Bacellar, O.; Carvalho, E.M. Reappraisal of the immunopathogenesis of disseminated leishmaniasis: In situ and systemic immune response. Trans. R. Soc. Trop. Med. Hyg. 2011, 105, 438–444. [Google Scholar] [CrossRef] [Green Version]
- Hartley, M.-A.; Bourreau, E.; Rossi, M.; Castiglioni, P.; Eren, R.O.; Prevel, F.; Couppié, P.; Hickerson, S.M.; Launois, P.; Beverley, S.M.; et al. Leishmaniavirus-Dependent Metastatic Leishmaniasis Is Prevented by Blocking IL-17A. PLoS Pathog. 2016, 12, e1005852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ives, A.; Ronet, C.; Prevel, F.; Ruzzante, G.; Fuertes-Marraco, S.; Schutz, F.; Zangger, H.; Revaz-Breton, M.; Lye, L.F.; Hickerson, S.M.; et al. Leishmania RNA virus controls the severity of mucocutaneous leishmaniasis. Science 2011, 331, 775–778. [Google Scholar] [CrossRef] [Green Version]
- Fernández-Presas, A.M.; Padilla-Noriega, L.; Becker, I.; Robert, L.; Jiménez, J.A.; Solano, S.; Delgado, J.; Tato, P.; Molinari, J.L. Enveloped and non-enveloped viral-like particles in Trypanosoma cruzi epimastigotes. Rev. Inst. Med. Trop. Sao Paulo 2017, 59, e46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grybchuk, D.; Akopyants, N.S.; Kostygov, A.Y.; Konovalovas, A.; Lye, L.F.; Dobson, D.E.; Zangger, H.; Fasel, N.; Butenko, A.; Frolov, A.O.; et al. Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite Leishmania. Proc. Natl. Acad. Sci. USA 2018, 115, E506–E515. [Google Scholar] [CrossRef] [Green Version]
- Wang, A.L.; Wang, C.C. Viruses of parasitic protozoa. Parasitol. Today 1991, 7, 76–80. [Google Scholar] [CrossRef]
- Barrow, P.; Dujardin, J.C.; Fasel, N.; Greenwood, A.D.; Osterrieder, K.; Lomonossoff, G.; Fiori, P.L.; Atterbury, R.; Rossi, M.; Lalle, M. Viruses of protozoan parasites and viral therapy: Is the time now right? Virol. J. 2020, 17, 142. [Google Scholar] [CrossRef] [PubMed]
- Stuart, K.D.; Weeks, R.; Guilbride, L.; Myler, P.J. Molecular organization of Leishmania RNA virus 1. Proc. Natl. Acad. Sci. USA 1992, 89, 8596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tarr, P.I.; Aline, R.F.; Jr Smiley, B.L.; Scholler, J.; Keithly, J.; Stuart, K. LR1: A candidate RNA virus of Leishmania. Proc. Natl. Acad. Sci. USA 1988, 85, 9572–9575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zangger, H.; Ronet, C.; Desponds, C.; Kuhlmann, F.M.; Robinson, J.; Hartley, M.-A.; Prevel, F.; Castiglioni, P.; Pratlong, F.; Bastien, P.; et al. Detection of Leishmania RNA Virus in Leishmania Parasites. PLoS Negl. Trop. Dis. 2013, 7, e2006. [Google Scholar] [CrossRef] [Green Version]
- Scheffter, S.M.; Ro, Y.T.; Chung, I.K.; Patterson, J.L. The Complete Sequence of Leishmania RNA Virus LRV2-1, a Virus of an Old World Parasite Strain. Virology 1995, 212, 84–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajjaran, H.; Mahdi, M.; Mohebali, M.; Samimi-Rad, K.; Ataei-Pirkooh, A.; Kazemi-Rad, E.; Naddaf, S.R.; Raoofian, R. Detection and molecular identification of leishmania RNA virus (LRV) in Iranian Leishmania species. Arch. Virol. 2016, 161, 3385–3390. [Google Scholar] [CrossRef]
- Zangger, H.; Hailu, A.; Desponds, C.; Lye, L.F.; Akopyants, N.S.; Dobson, D.E.; Ronet, C.; Ghalib, H.; Beverley, S.M.; Fasel, N. Leishmania aethiopica field isolates bearing an endosymbiontic dsRNA virus induce pro-inflammatory cytokine response. PLoS Negl. Trop. Dis. 2014, 8, e2836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhlmann, F.M.; Robinson, J.I.; Bluemling, G.R.; Ronet, C.; Fasel, N.; Beverley, S.M. Antiviral screening identifies adenosine analogs targeting the endogenous dsRNA Leishmania RNA virus 1 (LRV1) pathogenicity factor. Proc. Natl. Acad. Sci. USA 2017, 114, E811–E819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adaui, V.; Lye, L.F.; Akopyants, N.S.; Zimic, M.; Llanos-Cuentas, A.; Garcia, L.; Maes, I.; De Doncker, S.; Dobson, D.E.; Arevalo, J.; et al. Association of the Endobiont Double-Stranded RNA Virus LRV1 With Treatment Failure for Human Leishmaniasis Caused by Leishmania braziliensis in Peru and Bolivia. J. Infect. Dis. 2016, 213, 112–121. [Google Scholar] [CrossRef] [Green Version]
- Bourreau, E.; Ginouves, M.; Prevot, G.; Hartley, M.A.; Gangneux, J.P.; Robert-Gangneux, F.; Dufour, J.; Sainte-Marie, D.; Bertolotti, A.; Pratlong, F.; et al. Presence of Leishmania RNA Virus 1 in Leishmania guyanensis Increases the Risk of First-Line Treatment Failure and Symptomatic Relapse. J. Infect. Dis. 2016, 213, 105–111. [Google Scholar] [CrossRef] [Green Version]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, K.; Mehendale, H.M. Oxidative Stress. In Encyclopedia of Toxicology, 3rd ed.; Wexler, P., Ed.; Academic Press: Oxford, UK, 2014; pp. 735–737. [Google Scholar]
- Bedard, K.; Krause, K.-H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Ferrari, C.K.B. Oxidative Stress and Antioxidants in Host Defense in Leishmaniasis. In Oxidative Stress in Microbial Diseases; Chakraborti, S., Chakraborti, T., Chattopadhyay, D., Shaha, C., Eds.; Springer: Singapore, 2019; pp. 245–256. [Google Scholar]
- Rossi, M.; Fasel, N. How to master the host immune system? Leishmania parasites have the solutions! Int. Immunol. 2017, 30, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Burton, G.J.; Jauniaux, E. Oxidative stress. Best Pract. Res. Clin. Obstet. Gynaecol. 2011, 25, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging11This article is dedicated to the memory of our dear friend, colleague, and mentor Lars Ernster (1920–1998), in gratitude for all he gave to us. Free. Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: Mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, T.B.; Moon, K.A.; Kim, T.J.; Shin, D.; Cho, Y.S.; Moon, H.B.; Lee, K.Y. Regulation of pro-inflammatory responses by lipoxygenases via intracellular reactive oxygen species in vitro and in vivo. Exp. Mol. Med. 2008, 40, 461–476. [Google Scholar] [CrossRef] [Green Version]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases: Molecular mechanisms of activation. Free. Radic. Biol. Med. 2014, 76, 208–226. [Google Scholar] [CrossRef] [PubMed]
- Babior, B.M.; Lambeth, J.D.; Nauseef, W. The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 2002, 397, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Groemping, Y.; Rittinger, K. Activation and assembly of the NADPH oxidase: A structural perspective. Biochem. J. 2005, 386 Pt 3, 401–416. [Google Scholar] [CrossRef] [Green Version]
- Fang, F.C. Antimicrobial reactive oxygen and nitrogen species: Concepts and controversies. Nat. Rev. Microbiol. 2004, 2, 820–832. [Google Scholar] [CrossRef]
- Aktan, F. iNOS-mediated nitric oxide production and its regulation. Life Sci. 2004, 75, 639–653. [Google Scholar] [CrossRef]
- Stuehr, D.J. Mammalian nitric oxide synthases. Biochim. Biophys. Acta (BBA)—Bioenerg. 1999, 1411, 217–230. [Google Scholar] [CrossRef] [Green Version]
- Calegari-Silva, T.C.; Vivarini, A.C.; Pereira, R.M.S.; Dias-Teixeira, K.L.; Rath, C.T.; Pacheco, A.S.S.; Silva, G.B.L.; Pinto, C.A.S.; Dos Santos, J.V.; Saliba, A.M.; et al. Leishmania amazonensis downregulates macrophage iNOS expression via Histone Deacetylase 1 (HDAC1): A novel parasite evasion mechanism. Eur. J. Immunol. 2018, 48, 1188–1198. [Google Scholar] [CrossRef] [Green Version]
- Gaur, U.; Roberts, S.C.; Dalvi, R.P.; Corraliza, I.; Ullman, B.; Wilson, M.E. An effect of parasite-encoded arginase on the outcome of murine cutaneous leishmaniasis. J. Immunol. 2007, 179, 8446–8453. [Google Scholar] [CrossRef]
- Bogdan, C. Macrophages as host, effector and immunoregulatory cells in leishmaniasis: Impact of tissue micro-environment and metabolism. Cytokine X 2020, 2, 100041. [Google Scholar] [CrossRef]
- Colotti, G.; Ilari, A. Polyamine metabolism in Leishmania: From arginine to trypanothione. Amino Acids 2011, 40, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Pohanka, M. Role of oxidative stress in infectious diseases. A review. Folia Microbiol. 2013, 58, 503–513. [Google Scholar] [CrossRef]
- Carneiro, M.B.H.; Roma, E.H.; Ranson, A.J.; Doria, N.A.; Debrabant, A.; Sacks, D.L.; Vieira, L.Q.; Peters, N.C. NOX2-Derived Reactive Oxygen Species Control Inflammation during Leishmania amazonensis Infection by Mediating Infection-Induced Neutrophil Apoptosis. J. Immunol. 2018, 200, 196–208. [Google Scholar] [CrossRef] [Green Version]
- McNeely, T.B.; Rosen, G.; Londner, M.V.; Turco, S.J. Inhibitory effects on protein kinase C activity by lipophosphoglycan fragments and glycosylphosphatidylinositol antigens of the protozoan parasite Leishmania. Biochem. J. 1989, 259, 601–604. [Google Scholar] [CrossRef] [Green Version]
- Ramezani, A.; Nahad, M.P.; Faghihloo, E. The role of Nrf2 transcription factor in viral infection. J. Cell. Biochem. 2018, 119, 6366–6382. [Google Scholar] [CrossRef]
- Tomiotto-Pellissier, F.; Alves, D.R.; Miranda-Sapla, M.M.; de Morais, S.M.; Assolini, J.P.; da Silva Bortoleti, B.T.; Gonçalves, M.D.; Cataneo, A.H.D.; Kian, D.; Madeira, T.B.; et al. Caryocar coriaceum extracts exert leishmanicidal effect acting in promastigote forms by apoptosis-like mechanism and intracellular amastigotes by Nrf2/HO-1/ferritin dependent response and iron depletion: Leishmanicidal effect of Caryocar coriaceum leaf exracts. Biomed. Pharmacother. 2018, 98, 662–672. [Google Scholar] [PubMed]
- Moi, P.; Chan, K.; Asunis, I.; Cao, A.; Kan, Y.W. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc. Natl. Acad. Sci. USA 1994, 91, 9926–9930. [Google Scholar] [CrossRef] [Green Version]
- Venugopal, R.; Jaiswal, A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H: Quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. USA 1996, 93, 14960. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Chiba, T.; Takahashi, S.; Ishii, T.; Igarashi, K.; Katoh, Y.; Oyake, T.; Hayashi, N.; Satoh, K.; Hatayama, I.; et al. An Nrf2/Small Maf Heterodimer Mediates the Induction of Phase II Detoxifying Enzyme Genes through Antioxidant Response Elements. Biochem. Biophys. Res. Commun. 1997, 236, 313–322. [Google Scholar] [CrossRef]
- Fuse, Y.; Kobayashi, M. Conservation of the Keap1-Nrf2 System: An Evolutionary Journey through Stressful Space and Time. Molecules 2017, 22, 436. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Bohmann, D. Stress-activated cap’n’collar transcription factors in aging and human disease. Sci. Signal. 2010, 3, re3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baird, L.; Yamamoto, M. The Molecular Mechanisms Regulating the KEAP1-NRF2 Pathway. Mol. Cell. Biol. 2020, 40, e00099-20. [Google Scholar] [CrossRef] [PubMed]
- He, F.; Antonucci, L.; Karin, M. NRF2 as a regulator of cell metabolism and inflammation in cancer. Carcinogenesis 2020, 41, 405–416. [Google Scholar] [CrossRef]
- Maicas, N.; Ferrándiz, M.L.; Brines, R.; Ibáñez, L.; Cuadrado, A.; Koenders, M.I.; van den Berg, W.B.; Alcaraz, M.J. Deficiency of Nrf2 Accelerates the Effector Phase of Arthritis and Aggravates Joint Disease. Antioxid. Redox Signal. 2011, 15, 889–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- IIizuka, T.; Ishii, Y.; Itoh, K.; Kiwamoto, T.; Kimura, T.; Matsuno, Y.; Morishima, Y.; Hegab, A.E.; Homma, S.; Nomura, A.; et al. Nrf2-deficient mice are highly susceptible to cigarette smoke-induced emphysema. Genes Cells 2005, 10, 1113–1125. [Google Scholar] [CrossRef]
- Reddy, N.M.; Kleeberger, S.R.; Kensler, T.W.; Yamamoto, M.; Hassoun, P.M.; Reddy, S.P. Disruption of Nrf2 Impairs the Resolution of Hyperoxia-Induced Acute Lung Injury and Inflammation in Mice. J. Immunol. 2009, 182, 7264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taguchi, K.; Motohashi, H.; Yamamoto, M. Molecular mechanisms of the Keap1–Nrf2 pathway in stress response and cancer evolution. Genes Cells 2011, 16, 123–140. [Google Scholar] [CrossRef]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional Regulation by Nrf2. Antioxid. Redox Signal. 2017, 29, 1727–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; An, C.; Gao, Y.; Leak, R.K.; Chen, J.; Zhang, F. Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 2013, 100, 30–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, K.; Wakabayashi, N.; Katoh, Y.; Ishii, T.; Igarashi, K.; Engel, J.D.; Yamamoto, M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999, 13, 76–86. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Lamark, T.; Sjøttem, E.; Larsen, K.B.; Awuh, J.A.; Øvervatn, A.; McMahon, M.; Hayes, J.D.; Johansen, T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J. Biol. Chem. 2010, 285, 22576–22591. [Google Scholar] [CrossRef] [Green Version]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.S.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Lau, A.; Wang, X.J.; Zhao, F.; Villeneuve, N.F.; Wu, T.; Jiang, T.; Sun, Z.; White, E.; Zhang, D.D. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: Direct interaction between Keap1 and p62. Mol. Cell. Biol. 2010, 30, 3275–3285. [Google Scholar] [CrossRef] [Green Version]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-resident macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bautista-Hernández, L.A.; Gómez-Olivares, J.L.; Buentello-Volante, B.; Bautista-de Lucio, V.M. Fibroblasts: The Unknown Sentinels Eliciting Immune Responses Against Microorganisms. Eur. J. Microbiol. Immunol. 2017, 7, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Larsen, S.B.; Cowley, C.J.; Fuchs, E. Epithelial cells: Liaisons of immunity. Curr. Opin. Immunol. 2020, 62, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atri, C.; Guerfali, F.Z.; Laouini, D. Role of Human Macrophage Polarization in Inflammation during Infectious Diseases. Int. J. Mol. Sci. 2018, 19, 1801. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, M.; Takeda, K. Current Views of Toll-Like Receptor Signaling Pathways. Gastroenterol. Res. Pract. 2010, 2010, 240365. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [Green Version]
- Karin, M.; Ben-Neriah, Y. Phosphorylation Meets Ubiquitination: The Control of NF-κB Activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Scheidereit, C. IkappaB kinase complexes: Gateways to NF-kappaB activation and transcription. Oncogene 2006, 25, 6685–6705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, V.F.-S.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFκB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Häcker, H.; Karin, M. Regulation and Function of IKK and IKK-Related Kinases. Science’s STKE 2006, 2006, re13. [Google Scholar]
- Hayden, M.S.; Ghosh, S. Signaling to NF-kappaB. Genes Dev. 2004, 18, 2195–2224. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.G.; Boone, D.L.; Chai, S.; Libby, S.L.; Chien, M.; Lodolce, J.P.; Ma, A. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science 2000, 289, 2350–2354. [Google Scholar] [CrossRef]
- Ahmed, S.M.U.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2017, 1863, 585–597. [Google Scholar] [CrossRef]
- Lin, W.; Wu, R.T.; Wu, T.; Khor, T.O.; Wang, H.; Kong, A.N. Sulforaphane suppressed LPS-induced inflammation in mouse peritoneal macrophages through Nrf2 dependent pathway. Biochem. Pharmacol. 2008, 76, 967–973. [Google Scholar] [CrossRef] [Green Version]
- Thimmulappa, R.K.; Lee, H.; Rangasamy, T.; Reddy, S.P.; Yamamoto, M.; Kensler, T.W.; Biswal, S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Investig. 2006, 116, 984–995. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, K.; Karin, M. NF-kappaB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef]
- Seldon, M.P.; Silva, G.; Pejanovic, N.; Larsen, R.; Gregoire, I.P.; Filipe, J.; Anrather, J.; Soares, M.P. Heme oxygenase-1 inhibits the expression of adhesion molecules associated with endothelial cell activation via inhibition of NF-κB RelA phosphorylation at serine 276. J. Immunol. 2007, 179, 7840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rushworth, S.A.; MacEwan, D.J.; Connell, M.A. Lipopolysaccharide-Induced Expression of NAD(P)H: Quinone Oxidoreductase 1 and Heme Oxygenase-1 Protects against Excessive Inflammatory Responses in Human Monocytes. J. Immunol. 2008, 181, 6730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, W. Limiting inflammation with p62. Sci. Signal. 2016, 9, ec52. [Google Scholar] [CrossRef]
- Pareek, T.K.; Belkadi, A.; Kesavapany, S.; Zaremba, A.; Loh, S.L.; Bai, L.; Cohen, M.L.; Meyer, C.; Liby, K.T.; Miller, R.H.; et al. Triterpenoid modulation of IL-17 and Nrf-2 expression ameliorates neuroinflammation and promotes remyelination in autoimmune encephalomyelitis. Sci. Rep. 2011, 1, 201. [Google Scholar] [CrossRef]
- Morito, N.; Yoh, K.; Itoh, K.; Hirayama, A.; Koyama, A.; Yamamoto, M.; Takahashi, S. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene 2003, 22, 9275–9281. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Murakami, S.; Biswal, S.S.; Sakaguchi, S.; Harigae, H.; Yamamoto, M.; Motohashi, H. Systemic Activation of NRF2 Alleviates Lethal Autoimmune Inflammation in Scurfy Mice. Mol. Cell. Biol. 2017, 37, e00063-17. [Google Scholar] [CrossRef] [Green Version]
- Rockwell, C.E.; Zhang, M.; Fields, P.E.; Klaassen, C.D. Th2 skewing by activation of Nrf2 in CD4+ T cells. J. Immunol. 2012, 188, 1630. [Google Scholar] [CrossRef] [Green Version]
- Yarosz, E.L.; Chang, C.-H. The Role of Reactive Oxygen Species in Regulating T Cell-mediated Immunity and Disease. Immune Netw. 2018, 18, e14. [Google Scholar] [CrossRef] [PubMed]
- Scott, P.; Novais, F.O. Cutaneous leishmaniasis: Immune responses in protection and pathogenesis. Nat. Rev. Immunol. 2016, 16, 581–592. [Google Scholar] [CrossRef]
- Maspi, N.; Abdoli, A.; Ghaffarifar, F. Pro- and anti-inflammatory cytokines in cutaneous leishmaniasis: A review. Pathog. Glob. Health 2016, 110, 247–260. [Google Scholar] [CrossRef] [Green Version]
- Takeda, K.; Akira, S. Toll-like receptors. Curr. Protoc. Immunol. 2015, 109, 14.2.1–14.2.10. [Google Scholar] [CrossRef] [PubMed]
- de Veer, M.J.; Curtis, J.M.; Baldwin, T.M.; DiDonato, J.A.; Sexton, A.; McConville, M.J.; Handman, E.; Schofield, L. MyD88 is essential for clearance of Leishmania major: Possible role for lipophosphoglycan and Toll-like receptor 2 signaling. Eur. J. Immunol. 2003, 33, 2822–2831. [Google Scholar] [CrossRef] [PubMed]
- Ronet, C.; Passelli, K.; Charmoy, M.; Scarpellino, L.; Myburgh, E.; Hauyon La Torre, Y.; Turco, S.; Mottram, J.C.; Fasel, N.; Luther, S.A.; et al. TLR2 Signaling in Skin Nonhematopoietic Cells Induces Early Neutrophil Recruitment in Response to Leishmania major Infection. J. Investig. Dermatol. 2019, 139, 1318–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eren, R.O.; Reverte, M.; Rossi, M.; Hartley, M.A.; Castiglioni, P.; Prevel, F.; Martin, R.; Desponds, C.; Lye, L.F.; Drexler, S.K.; et al. Mammalian Innate Immune Response to a Leishmania-Resident RNA Virus Increases Macrophage Survival to Promote Parasite Persistence. Cell Host Microbe 2016, 20, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Rossi, M.; Castiglioni, P.; Hartley, M.A.; Eren, R.O.; Prevel, F.; Desponds, C.; Utzschneider, D.T.; Zehn, D.; Cusi, M.G.; Kuhlmann, F.M.; et al. Type I interferons induced by endogenous or exogenous viral infections promote metastasis and relapse of leishmaniasis. Proc. Natl. Acad. Sci. USA 2017, 114, 4987–4992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parmentier, L.; Cusini, A.; Muller, N.; Zangger, H.; Hartley, M.A.; Desponds, C.; Castiglioni, P.; Dubach, P.; Ronet, C.; Beverley, S.M.; et al. Severe Cutaneous Leishmaniasis in a Human Immunodeficiency Virus Patient Coinfected with Leishmania braziliensis and Its Endosymbiotic Virus. Am. J. Trop. Med. Hyg. 2016, 94, 840–843. [Google Scholar] [CrossRef] [Green Version]
- Hartley, M.A.; Eren, R.O.; Rossi, M.; Prevel, F.; Castiglioni, P.; Isorce, N.; Desponds, C.; Lye, L.F.; Beverley, S.M.; Drexler, S.K.; et al. Leishmania guyanensis parasites block the activation of the inflammasome by inhibiting maturation of IL-1beta. Microb. Cell 2018, 5, 137–149. [Google Scholar] [CrossRef]
- Dixit, E.; Kagan, J.C. Chapter Four—Intracellular Pathogen Detection by RIG-I-Like Receptors. In Advances in Immunology; Alt, F.W., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 117, pp. 99–125. [Google Scholar]
- Stäger, S.; Rafati, S. CD8(+) T cells in leishmania infections: Friends or foes? Front. Immunol. 2012, 3, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos Cda, S.; Boaventura, V.; Ribeiro Cardoso, C.; Tavares, N.; Lordelo, M.J.; Noronha, A.; Costa, J.; Borges, V.M.; de Oliveira, C.I.; Van Weyenbergh, J.; et al. CD8(+) granzyme B(+)-mediated tissue injury vs. CD4(+)IFNγ(+)-mediated parasite killing in human cutaneous leishmaniasis. J. Investig. Dermatol. 2013, 133, 1533–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Carvalho, R.V.H.; Lima-Junior, D.S.; da Silva, M.V.G.; Dilucca, M.; Rodrigues, T.S.; Horta, C.V.; Silva, A.L.N.; da Silva, P.F.; Frantz, F.G.; Lorenzon, L.B.; et al. Leishmania RNA virus exacerbates Leishmaniasis by subverting innate immunity via TLR3-mediated NLRP3 inflammasome inhibition. Nat. Commun. 2019, 10, 5273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alenquer, M.; Amorim, M.J. Exosome Biogenesis, Regulation, and Function in Viral Infection. Viruses 2015, 7, 5066–5083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atayde, V.D.; da Silva Lira Filho, A.; Chaparro, V.; Zimmermann, A.; Martel, C.; Jaramillo, M.; Olivier, M. Exploitation of the Leishmania exosomal pathway by Leishmania RNA virus 1. Nat. Microbiol. 2019, 4, 714–723. [Google Scholar] [CrossRef]
- Olivier, M.; Zamboni, D.S. Leishmania Viannia guyanensis, LRV1 virus and extracellular vesicles: A dangerous trio influencing the faith of immune response during muco-cutaneous leishmaniasis. Curr. Opin. Immunol. 2020, 66, 108–113. [Google Scholar] [CrossRef] [PubMed]
- Vivarini, A.d.C.; Lopes, U.G. The Potential Role of Nrf2 Signaling in Leishmania Infection Outcomes. Front. Cell. Infect. Microbiol. 2020, 9, 453. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.; Ribeiro, A.M. Nrf2 as a master regulator of tissue damage control and disease tolerance to infection. Biochem. Soc. Trans. 2015, 43, 663–668. [Google Scholar] [CrossRef] [Green Version]
- Page, A.; Volchkova, V.A.; Reid, S.P.; Mateo, M.; Bagnaud-Baule, A.; Nemirov, K.; Shurtleff, A.C.; Lawrence, P.; Reynard, O.; Ottmann, M.; et al. Marburgvirus Hijacks Nrf2-Dependent Pathway by Targeting Nrf2-Negative Regulator Keap1. Cell Rep. 2014, 6, 1026–1036. [Google Scholar] [CrossRef] [PubMed]
- Gjyshi, O.; Bottero, V.; Veettil, M.V.; Dutta, S.; Singh, V.V.; Chikoti, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces Nrf2 during de novo infection of endothelial cells to create a microenvironment conducive to infection. PLoS Pathog. 2014, 10, e1004460. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho Vivarini, Á.; Calegari-Silva, T.C.; Saliba, A.M.; Boaventura, V.S.; França-Costa, J.; Khouri, R.; Dierckx, T.; Dias-Teixeira, K.L.; Fasel, N.; Barral, A.M.P.; et al. Systems Approach Reveals Nuclear Factor Erythroid 2-Related Factor 2/Protein Kinase R Crosstalk in Human Cutaneous Leishmaniasis. Front. Immunol. 2017, 8, 1127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reverte, M.; Eren, R.O.; Jha, B.; Desponds, C.; Snaka, T.; Prevel, F.; Isorce, N.; Lye, L.F.; Owens, K.L.; Gazos Lopes, U.; et al. The antioxidant response favors Leishmania parasites survival, limits inflammation and reprograms the host cell metabolism. PLoS Pathog. 2021, 17, e1009422. [Google Scholar] [CrossRef] [PubMed]
- Ginouves, M.; Simon, S.; Bourreau, E.; Lacoste, V.; Ronet, C.; Couppie, P.; Nacher, M.; Demar, M.; Prevot, G. Prevalence and Distribution of Leishmania RNA Virus 1 in Leishmania Parasites from French Guiana. Am. J. Trop. Med. Hyg. 2016, 94, 102–106. [Google Scholar] [CrossRef] [Green Version]
- Gal-Ben-Ari, S.; Barrera, I.; Ehrlich, M.; Rosenblum, K. PKR: A Kinase to Remember. Front. Mol. Neurosci. 2019, 11, 480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cataneo, A.H.D.; Tomiotto-Pellissier, F.; Miranda-Sapla, M.M.; Assolini, J.P.; Panis, C.; Kian, D.; Yamauchi, L.M.; Colado Simão, A.N.; Casagrande, R.; Pinge-Filho, P.; et al. Quercetin promotes antipromastigote effect by increasing the ROS production and anti-amastigote by upregulating Nrf2/HO-1 expression, affecting iron availability. Biomed. Pharmacother. 2019, 113, 108745. [Google Scholar] [CrossRef]
- Forestier, C.L.; Machu, C.; Loussert, C.; Pescher, P.; Späth, G.F. Imaging host cell-Leishmania interaction dynamics implicates parasite motility, lysosome recruitment, and host cell wounding in the infection process. Cell Host Microbe 2011, 9, 319–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fão, L.; Mota, S.I.; Rego, A.C. c-Src regulates Nrf2 activity through PKCδ after oxidant stimulus. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2019, 1866, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D. Dectin-1: A signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 2006, 6, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Goodridge, H.S.; Reyes, C.N.; Becker, C.A.; Katsumoto, T.R.; Ma, J.; Wolf, A.J.; Bose, N.; Chan, A.S.; Magee, A.S.; Danielson, M.E.; et al. Activation of the innate immune receptor Dectin-1 upon formation of a ‘phagocytic synapse’. Nature 2011, 472, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Lima-Junior, D.S.; Mineo, T.W.P.; Calich, V.L.G.; Zamboni, D.S. Dectin-1 Activation during Leishmania amazonensis Phagocytosis Prompts Syk-Dependent Reactive Oxygen Species Production To Trigger Inflammasome Assembly and Restriction of Parasite Replication. J. Immunol. 2017, 199, 2055–2068. [Google Scholar] [CrossRef] [Green Version]
- Reinhard, K.; Huber, M.; Lohoff, M.; Visekruna, A. The role of NF-kappaB activation during protection against Leishmania infection. Int. J. Med. Microbiol. 2012, 302, 230–235. [Google Scholar] [CrossRef]
- Abu-Dayyeh, I.; Hassani, K.; Westra, E.R.; Mottram, J.C.; Olivier, M. Comparative study of the ability of Leishmania mexicana promastigotes and amastigotes to alter macrophage signaling and functions. Infect. Immun. 2010, 78, 2438. [Google Scholar] [CrossRef] [Green Version]
- Calegari-Silva, T.C.; Pereira, R.M.S.; De-Melo, L.D.B.; Saraiva, E.M.; Soares, D.C.; Bellio, M.; Lopes, U.G. NF-κB-mediated repression of iNOS expression in Leishmania amazonensis macrophage infection. Immunol. Lett. 2009, 127, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Wang, H.; Yan, W.; Xu, L.; Wang, X.; Zhao, X.; Yang, X.; Chen, G.; Ji, Y. Disruption of Nrf2 enhances upregulation of nuclear factor-kappaB activity, proinflammatory cytokines, and intercellular adhesion molecule-1 in the brain after traumatic brain injury. Mediat. Inflamm. 2008, 2008, 725174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch-Nicolau, P.; Ubals, M.; Salvador, F.; Sánchez-Montalvá, A.; Aparicio, G.; Erra, A.; Martinez de Salazar, P.; Sulleiro, E.; Molina, I. Leishmaniasis and tumor necrosis factor alpha antagonists in the Mediterranean basin. A switch in clinical expression. PLoS Negl. Trop. Dis. 2019, 13, e0007708. [Google Scholar] [CrossRef]
- Blackwell, J.M. Receptors and recognition mechanisms of Leishmania species. Trans. R. Soc. Trop. Med. Hyg. 1985, 79, 606–612. [Google Scholar] [CrossRef]
- Hiebert, P.; Werner, S. Regulation of Wound Healing by the NRF2 Transcription Factor-More Than Cytoprotection. Int. J. Mol. Sci. 2019, 20, 3856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, L.; Wang, H.; Qiao, L.; Wang, X. Disruption of Nrf2 enhances the upregulation of nuclear factor-kappaB activity, tumor necrosis factor-alpha, and matrix metalloproteinase-9 after spinal cord injury in mice. Mediat. Inflamm. 2010, 2010, 238321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Representation of the different pathways and enzymes leading to the production of parasitotoxic molecules and enzymes (red) and of detoxification enzymes (green) in macrophages infected by Leishmania parasites.
Figure 1.
Representation of the different pathways and enzymes leading to the production of parasitotoxic molecules and enzymes (red) and of detoxification enzymes (green) in macrophages infected by Leishmania parasites.

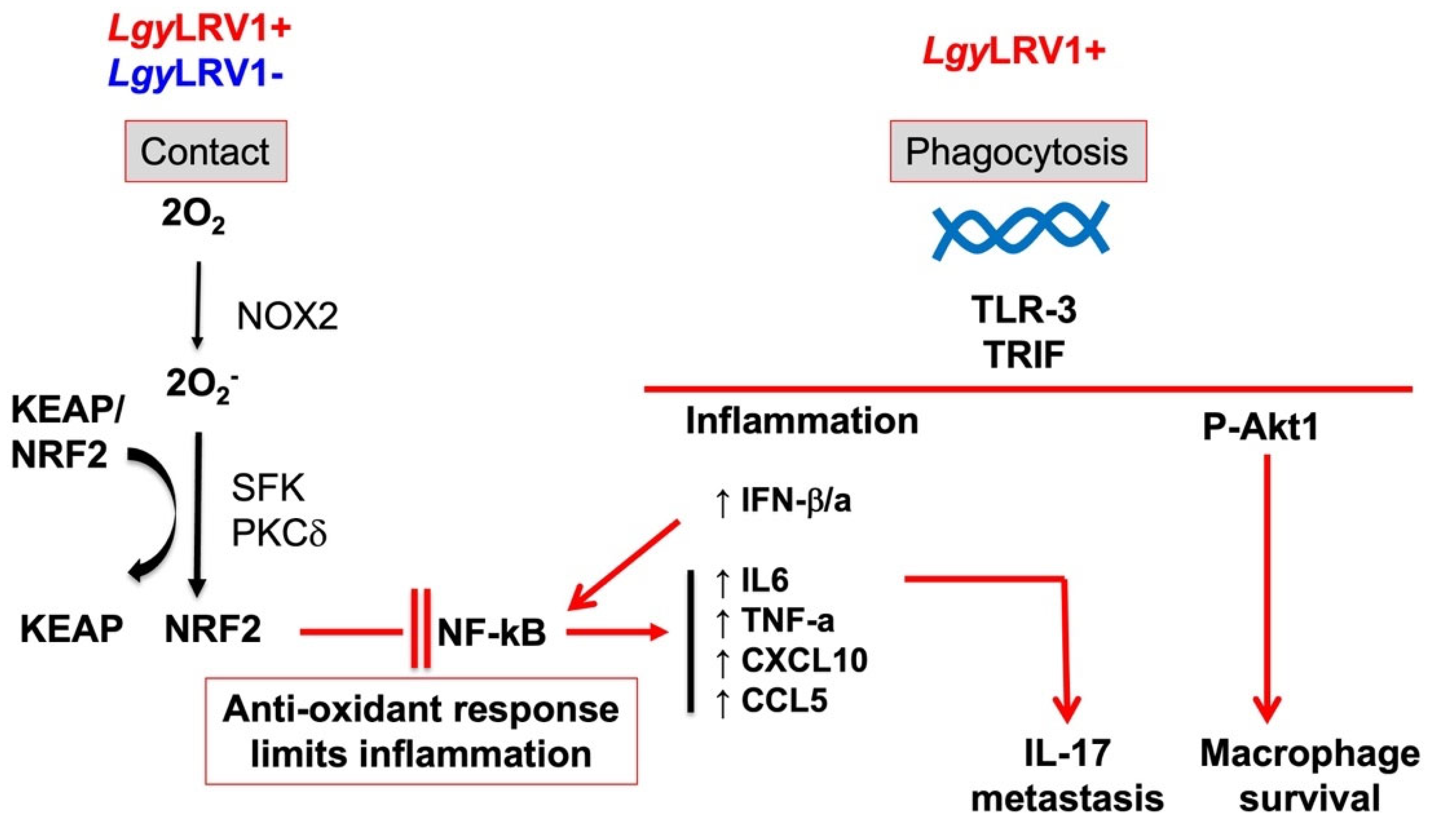
Figure 2.
The oxidative stress and the inflammatory response in Leishmania infection. Independently of the presence of LRV1, the NRF2 pathway is activated upon contact between the parasite and the macrophage producing oxygen species generated by NOX2 permitting the release of NRF2 from its negative regulator KEAP1 and phosphorylation via SFK and PKC. This anti-oxidant response limits the NF-kB inflammatory and the production of inflammatory chemokines and cytokines. In the presence of LRV1 in Leishmania parasites, survival of infected macrophages is increased and production of Type-I interferon and inflammatory chemokines and cytokines are induced leading to accelerated dissemination of the infection via IL-17.
Figure 2.
The oxidative stress and the inflammatory response in Leishmania infection. Independently of the presence of LRV1, the NRF2 pathway is activated upon contact between the parasite and the macrophage producing oxygen species generated by NOX2 permitting the release of NRF2 from its negative regulator KEAP1 and phosphorylation via SFK and PKC. This anti-oxidant response limits the NF-kB inflammatory and the production of inflammatory chemokines and cytokines. In the presence of LRV1 in Leishmania parasites, survival of infected macrophages is increased and production of Type-I interferon and inflammatory chemokines and cytokines are induced leading to accelerated dissemination of the infection via IL-17.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Reverte, M.; Snäkä, T.; Fasel, N. The Dangerous Liaisons in the Oxidative Stress Response to Leishmania Infection. Pathogens 2022, 11, 409. https://doi.org/10.3390/pathogens11040409
AMA Style
Reverte M, Snäkä T, Fasel N. The Dangerous Liaisons in the Oxidative Stress Response to Leishmania Infection. Pathogens. 2022; 11(4):409. https://doi.org/10.3390/pathogens11040409
Chicago/Turabian StyleReverte, Marta, Tiia Snäkä, and Nicolas Fasel. 2022. "The Dangerous Liaisons in the Oxidative Stress Response to Leishmania Infection" Pathogens 11, no. 4: 409. https://doi.org/10.3390/pathogens11040409
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.