
Space–Time Patterns of Poultry Pathogens in the USA: A Case Study of Ornithobacterium rhinotracheale and Pasteurella multocida in Turkey Populations
 , , , and
, , , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Period, Study Area and Sample Collection
2.2. Molecular Characterization
2.3. Statistical Analysis
3. Results
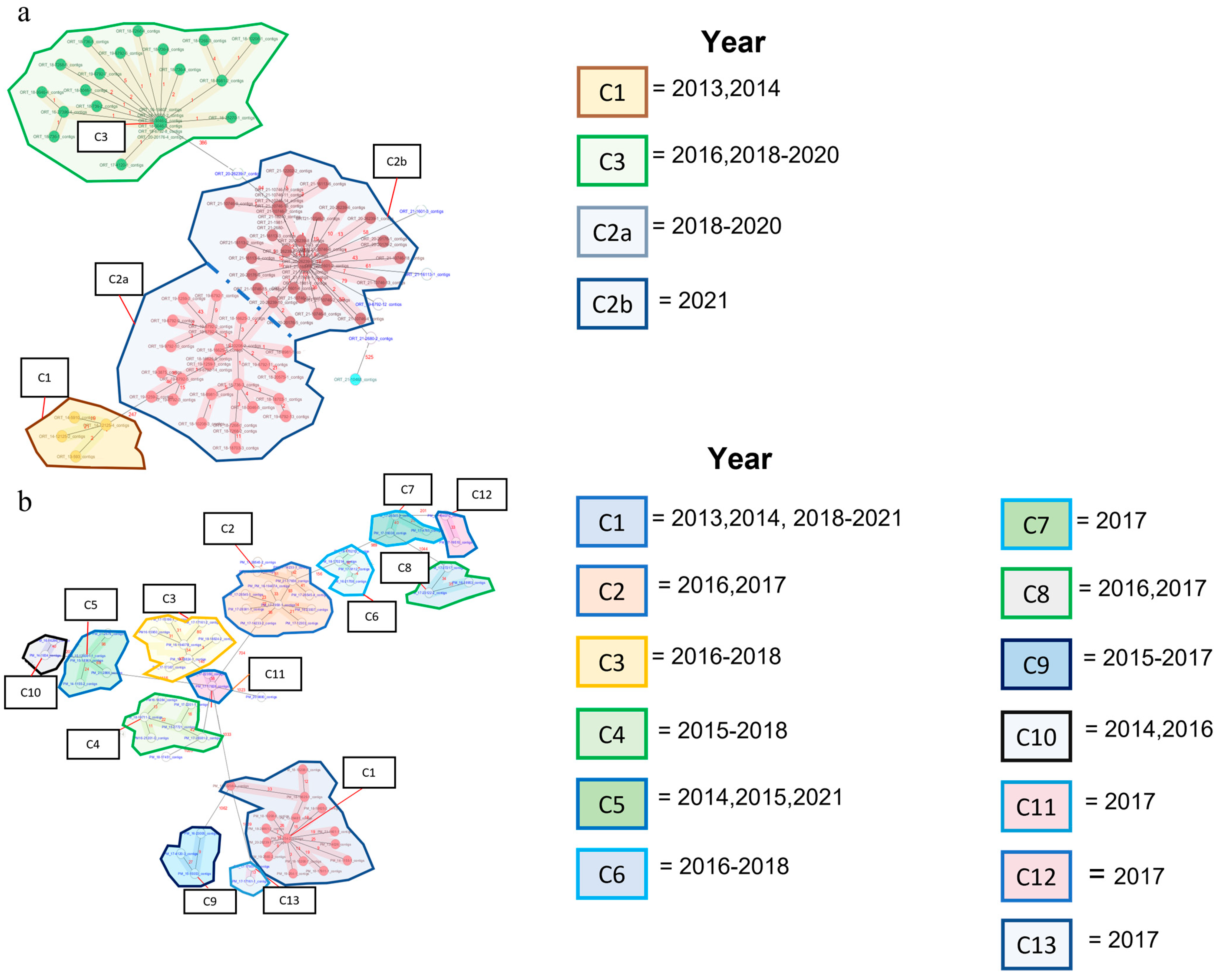
3.1. Allelic Clusters and Diversity
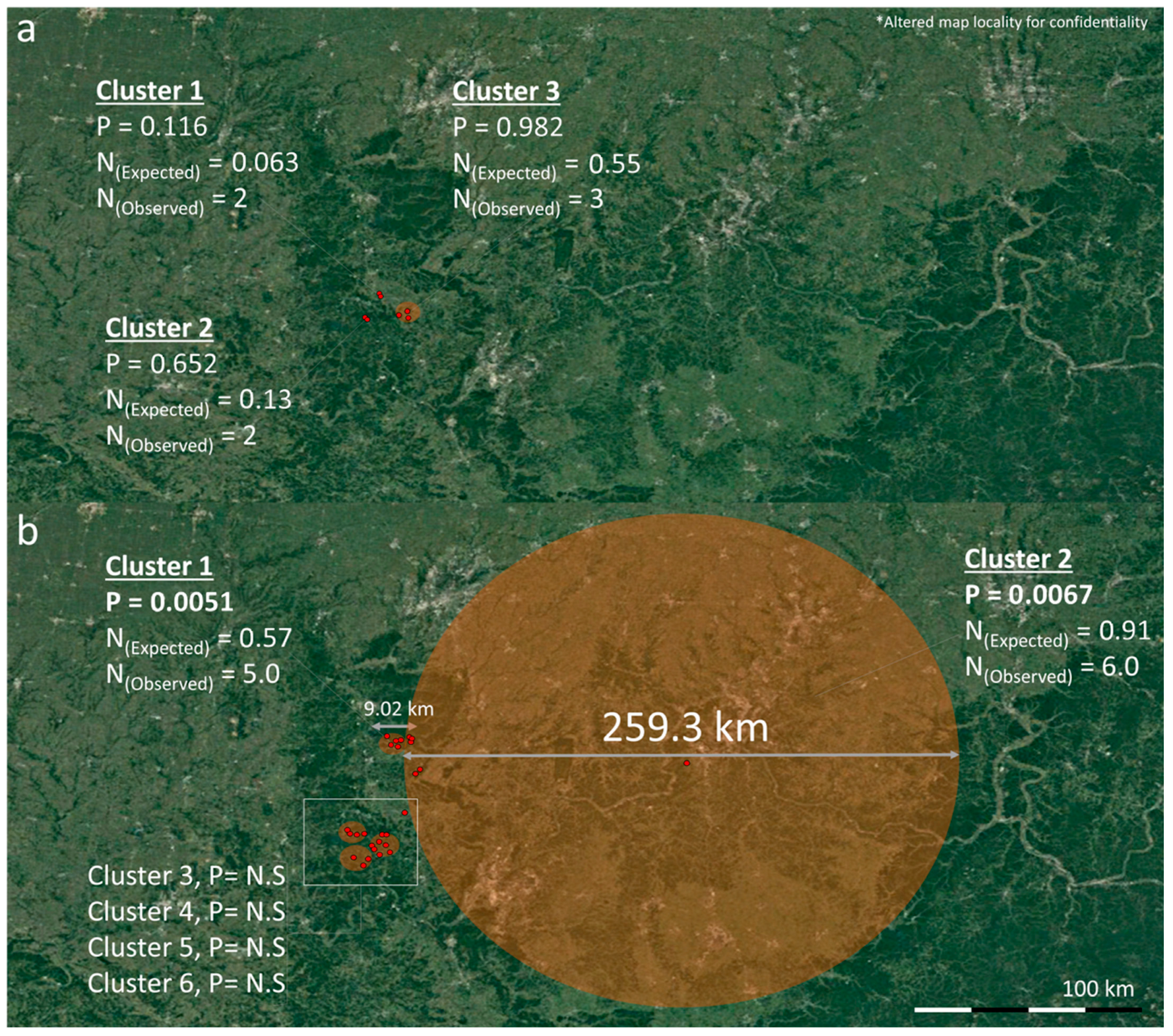
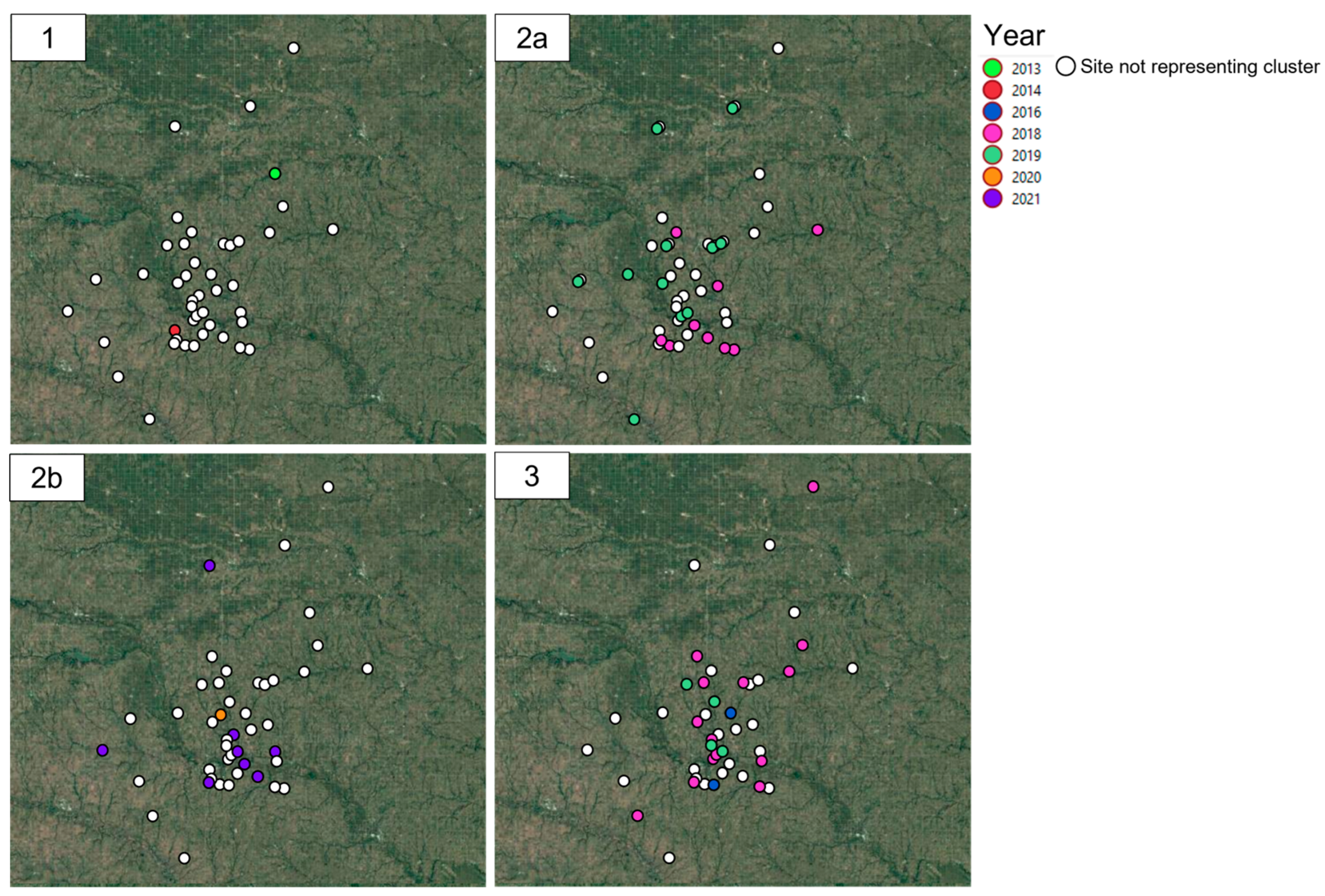
3.2. Spatio-Temporal Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Numee, S.; Hauck, R.; Hafez, H.M. Detection and Typing of Ornithobacterium rhinotracheale from German Poultry Flocks. Avian Dis. 2012, 56, 654–658. [Google Scholar] [CrossRef]
- Smith, E.; Miller, E.; Aguayo, J.M.; Figueroa, C.F.; Nezworski, J.; Studniski, M.; Wileman, B.; Johnson, T. Genomic diversity and molecular epidemiology of Pasteurella multocida. PLoS ONE 2021, 16, e0249138. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.; Ahlmeyer, V. Current Health & Industry Issues Facing the Turkey Industry: 2018; United States Animal Health Association: Kansas City, MO, USA, 2018. [Google Scholar]
- Boulianne, M.; Blackall, P.J.; Hofacre, C.L.; Ruiz, J.A.; Sandhu, T.S.; Hafez, H.M.; Chin, R.P.; Register, K.B.; Jackwood, M.W. Pasteurellosis and Other Respiratory Bacterial Infections. Dis. Poult. 2020, 831–889. [Google Scholar] [CrossRef]
- van Empel, P.C.M.; Hafez, H.M. Ornithobacterium rhinotracheale: A review. Avian Pathol. 1999, 28, 217–227. [Google Scholar] [CrossRef]
- Ewers, C.; Lubkebecker, A.; Bethe, A.; Kießling, S.; Filter, M.; Wieler, L.H. Virulence genotype of Pasteurella multocida strains isolated from different hosts with various disease status. Vet. Microbiol. 2006, 114, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Z.H.; Zhang, T.Y.; Guo, Y.X.; Chu, J.; Qu, G.G.; Miao, L.Z.; Shen, Z.Q.; He, C. Serosurvey of Avian metapneumovirus, Orithobacterium rhinotracheale, and Chlamydia psittaci and Their Potential Association with Avian Airsacculitis. Biomed. Environ. Sci. 2018, 31, 403–406. [Google Scholar] [PubMed]
- Harper, M.; Boyce, J.D. The Myriad Properties of Pasteurella multocida Lipopolysaccharide. Toxins 2017, 9, 254. [Google Scholar] [CrossRef] [Green Version]
- Hauck, R.; Chin, R.P.; Shivaprasad, H. Retrospective Study on the Isolation of Ornithobacterium rhinotracheale from Chickens and Turkeys in Central California: 294 cases (2000–12). Avian Dis. 2015, 59, 130–137. [Google Scholar] [CrossRef]
- Van Loock, M.; Geens, T.; De Smit, L.; Nauwynck, H.; Van Empel, P.; Naylor, C.; Hafez, H.; Goddeeris, B.; Vanrompay, D. Key role of Chlamydophila psittaci on Belgian turkey farms in association with other respiratory pathogens. Vet. Microbiol. 2005, 107, 91–101. [Google Scholar] [CrossRef]
- Marien, M.; Decostere, A.; Martel, A.; Chiers, K.; Froyman, R.; Nauwynck, H. Synergy between avian pneumovirus and Ornithobacterium rhinotracheale in turkeys. Avian Pathol. 2005, 34, 204–211. [Google Scholar] [CrossRef] [Green Version]
- Pan, Q.; Liu, A.; Zhang, F.; Ling, Y.; Ou, C.; Hou, N.; He, C. Co-infection of broilers with Ornithobacterium rhinotracheale and H9N2 avian influenza virus. BMC Vet. Res. 2012, 8, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehrabanpour, M.; Bushehri, M.R.; Mehrabanpour, D. Molecular and serological diagnosis of Newcastle and Ornithobacterium rhinotracheale broiler in chicken in Fars Province, Iran. Int. J. Anim. Vet. Sci. 2017, 11, 224–227. [Google Scholar]
- Taylor, K.J.M.; Ngunjiri, J.M.; Abundo, M.C.; Jang, H.; Elaish, M.; Ghorbani, A.; Kc, M.; Weber, B.P.; Johnson, T.J.; Lee, C.-W. Respiratory and Gut Microbiota in Commercial Turkey Flocks with Disparate Weight Gain Trajectories Display Differential Compositional Dynamics. Appl. Environ. Microbiol. 2020, 86, e00431-20. [Google Scholar] [CrossRef]
- Watteyn, A.; Devreese, M.; De Baere, S.; Wyns, H.; Plessers, E.; Boyen, F.; Haesebrouck, F.; De Backer, P.; Croubels, S. Pharmacokinetic and pharmacodynamic properties of gamithromycin in turkey poults with respect to Ornithobacterium rhinotracheale. Poult. Sci. 2015, 94, 2066–2074. [Google Scholar] [CrossRef] [PubMed]
- Kehrenberg, C.; Schulze-Tanzil, G.; Martel, J.-L.; Chaslus-Dancla, E.; Schwarz, S. Antimicrobial resistance in Pasteurella and Mannheimia: Epidemiology and genetic basis. Vet. Res. 2001, 32, 323–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Zhao, Z.; Hu, J.; Wu, B.; Cai, X.; He, Q.; Chen, H. Isolation, antimicrobial resistance, and virulence genes of Pasteurella multocida strains from swine in China. J. Clin. Microbiol. 2009, 47, 951–958. [Google Scholar] [CrossRef] [Green Version]
- Hashish, A.; Johnson, T.J.; Smith, E.; Chundru, D.; Williams, M.L.; Macedo, N.R.; Sato, Y.; Ghanem, M.; El-Gazzar, M. Complete Genome Sequences of Three Ornithobacterium rhinotracheale Strains from Avian Sources, Using Hybrid Nanopore-Illumina Assembly. Genome Announc. 2023, 12, e0105922. [Google Scholar] [CrossRef]
- Alispahic, M.; Endler, L.; Hess, M.; Hess, C. Ornithobacterium rhinotracheale: MALDI-TOF MS and Whole Genome Sequencing Confirm That Serotypes K, L and M Deviate from Well-Known Reference Strains and Numerous Field Isolates. Microorganisms 2021, 9, 1006. [Google Scholar] [CrossRef]
- Nisar, M.; Thieme, S.; Hafez, H.M.; Sentíes-Cúe, G.; Chin, R.P.; Muhammad, S.P.; Aboubakr, H.; Goyal, S.M.; Nagaraja, K.V. Genetic Diversity of Ornithobacterium rhinotracheale Isolated from Chickens and Turkeys in the United States. Avian Dis. 2020, 64, 324–329. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, X.; Zhou, R.; Chen, H.; Wilson, B.A.; Wu, B. Pasteurella multocida: Genotypes and Genomics. Microbiol. Mol. Biol. Rev. 2019, 83, 10-1128. [Google Scholar] [CrossRef]
- Hashish, A.; Sinha, A.; Sato, Y.; Macedo, N.R.; El-Gazzar, M. Development and Validation of a New TaqMan Real-Time PCR for the Detection of Ornithobacterium rhinotracheale. Microorganisms 2022, 10, 341. [Google Scholar] [CrossRef] [PubMed]
- Hashish, A.; Smith, E.; Ghanem, M.; Johnson, T.; Sato, Y.; El-Gazzar, M. Development of Core Genome Multilocus Sequence Typing Scheme (cgMLST) for Ornithobacterium rhinotracheale. In Proceedings of the 72th North Central Avian Disease Conference-2021, Virtual, 11–13 April 2023. [Google Scholar]
- El-Gazzar, M. Development of Core Genome Multilocus Sequence Typing (cgMLST) Scheme for Genotyping of Pasteurella multocida. In Proceedings of the American Association of Avian Pathologists Annual Meeting, Philadelphia, PA, USA, 29 July–2 August 2022; p. 073122. [Google Scholar]
- Barbosa, E.V.; Cardoso, C.V.; Silva, R.d.C.F.; Cerqueira, A.d.M.F.; Liberal, M.H.T.; Castro, H.C. Ornithobacterium rhinotracheale: An Update Review about An Emerging Poultry Pathogen. Vet. Sci. 2019, 7, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado, R.; Maturrano, L.; Azevedo, V.; Aburjaile, F. Pathogenomics insights for understanding Pasteurella multocida adaptation. Int. J. Med. Microbiol. 2020, 310, 151417. [Google Scholar] [CrossRef] [PubMed]
- Wilkie, I.W.; Harper, M.; Boyce, J.D.; Adler, B. Pasteurella multocida: Diseases and Pathogenesis. Poxviruses 2012, 361, 1–22. [Google Scholar] [CrossRef]
- Almoheer, R.; Abd Wahid, M.E.; Zakaria, H.A.; Jonet, M.A.B.; Al-Shaibani, M.M.; Al-Gheethi, A.; Addis, S.N.K. Spatial, Temporal, and Demographic Patterns in the Prevalence of Hemorrhagic Septicemia in 41 Countries in 2005–2019: A Systematic Analysis with Special Focus on the Potential Development of a New-Generation Vaccine. Vaccines 2022, 10, 315. [Google Scholar] [CrossRef]
- Becker, T.A.; Ahlers, A.A.; Hesting, S.; Haukos, D.A. Spatiotemporal Distribution of Waterfowl Disease Outbreaks in Kansas, USA. Prairie Nat. 2018, 50, 5–15. [Google Scholar]
- Blakey, J.; Shivaprasad, H.L.; Crispo, M.; Ochoa, J.; Stoute, S. Retrospective Study of Pasteurella multocida Diagnosed in Commercial Turkeys Submitted to California Animal Health and Food Safety Laboratory System; 1991–2017. Avian Dis. 2018, 62, 364–373. [Google Scholar] [CrossRef]
- Carpenter, T.E.; Hird, D.W.; Snipes, K.P. A time-space investigation of the epidemiology of fowl cholera. Prev. Vet. Med. 1996, 28, 159–163. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [Green Version]
- Schmid, D.; Allerberger, F.; Huhulescu, S.; Pietzka, A.; Amar, C.; Kleta, S.; Prager, R.; Preußel, K.; Aichinger, E.; Mellmann, A. Whole genome sequencing as a tool to investigate a cluster of seven cases of listeriosis in Austria and Germany, 2011–2013. Clin. Microbiol. Infect. 2014, 20, 431–436. [Google Scholar] [CrossRef] [Green Version]
- Leopold, S.R.; Goering, R.V.; Witten, A.; Harmsen, D.; Mellmann, A. Bacterial Whole-Genome Sequencing Revisited: Portable, Scalable, and Standardized Analysis for Typing and Detection of Virulence and Antibiotic Resistance Genes. J. Clin. Microbiol. 2014, 52, 2365–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellmann, A.; Harmsen, D.; Cummings, C.A.; Zentz, E.B.; Leopold, S.R.; Rico, A.; Prior, K.; Szczepanowski, R.; Ji, Y.; Zhang, W.; et al. Prospective Genomic Characterization of the German Enterohemorrhagic Escherichia coli O104:H4 Outbreak by Rapid Next Generation Sequencing Technology. PLoS ONE 2011, 6, e22751. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, M.; Wang, L.; Zhang, Y.; Edwards, S.; Lu, A.; Ley, D.; El-Gazzar, M. Core Genome Multilocus Sequence Typing: A Standardized Approach for Molecular Typing of Mycoplasma gallisepticum. J. Clin. Microbiol. 2018, 56, e01145-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashish, A.; Johnson, T.J.; Chundru, D.; Williams, M.L.; Sato, Y.; Macedo, N.R.; Clessin, A.; Gantelet, H.; Bost, C.; Tornos, J.; et al. Complete Genome Sequences of Two Pasteurella multocida Isolates from Seabirds. Genome Announc. 2023, 12, e0136522. [Google Scholar] [CrossRef]
- Lay, J.O. MALDI-TOF mass spectrometry of bacteria. Mass Spectrom. Rev. 2001, 20, 172–194. [Google Scholar] [CrossRef]
- Kulldorff, M.; Heffernan, R.; Hartman, J.; Assunção, R.; Mostashari, F. A Space–Time Permutation Scan Statistic for Disease Outbreak Detection. PLoS Med. 2005, 2, e59. [Google Scholar] [CrossRef] [Green Version]
- Hurtado, R.; Carhuaricra, D.; Soares, S.; Viana, M.V.C.; Azevedo, V.; Maturrano, L.; Aburjaile, F. Pan-genomic approach shows insight of genetic divergence and pathogenic-adaptation of Pasteurella multocida. Gene 2018, 670, 193–206. [Google Scholar] [CrossRef]
- Kursa, O.; Tomczyk, G.; Sawicka-Durkalec, A.; Adamska, K. Bacterial communities of the oviduct of turkeys. Sci. Rep. 2022, 12, 14884. [Google Scholar] [CrossRef]
- Amonsin, A.; Wellehan, J.F.; Li, L.L.; Vandamme, P.; Lindeman, C.; Edman, M.; A Robinson, R.; Kapur, V. Molecular epidemiology of Ornithobacterium rhinotracheale. J. Clin. Microbiol. 1997, 35, 2894–2898. [Google Scholar] [CrossRef]
- Thieme, S.; Mühldorfer, K.; Lüschow, D.; Hafez, H.M. Molecular characterization of the recently emerged poultry pathogen Ornithobacterium rhinotracheale by multilocus sequence typing. PLoS ONE 2016, 11, e0148158. [Google Scholar]
- van Empel, P.; van den Bosch, H.; Storm, P. Ornithobacterium rhinotracheale Infection by Egg Transmission. In Ornithobacterium rhinotracheale; Intervet International: Boxmeer, The Netherlands, 1998; pp. 49–56. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Campler, M.R.; Hashish, A.; Ghanem, M.; El-Gazzar, M.M.; Arruda, A.G. Space–Time Patterns of Poultry Pathogens in the USA: A Case Study of Ornithobacterium rhinotracheale and Pasteurella multocida in Turkey Populations. Pathogens 2023, 12, 1004. https://doi.org/10.3390/pathogens12081004
Campler MR, Hashish A, Ghanem M, El-Gazzar MM, Arruda AG. Space–Time Patterns of Poultry Pathogens in the USA: A Case Study of Ornithobacterium rhinotracheale and Pasteurella multocida in Turkey Populations. Pathogens. 2023; 12(8):1004. https://doi.org/10.3390/pathogens12081004
Chicago/Turabian StyleCampler, Magnus R., Amro Hashish, Mostafa Ghanem, Mohamed M. El-Gazzar, and Andréia G. Arruda. 2023. "Space–Time Patterns of Poultry Pathogens in the USA: A Case Study of Ornithobacterium rhinotracheale and Pasteurella multocida in Turkey Populations" Pathogens 12, no. 8: 1004. https://doi.org/10.3390/pathogens12081004