Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery


Council of Scientific and Industrial Research, Institute of Microbial Technology, Chandigarh 160036, India
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Pathogens 2018, 7(1), 24; https://doi.org/10.3390/pathogens7010024
Submission received: 11 January 2018
/
Revised: 29 January 2018
/
Accepted: 31 January 2018
/
Published: 23 February 2018
(This article belongs to the Special Issue Mechanisms of Mycobacterium tuberculosis Pathogenesis)
Abstract
:Mycobacterium tuberculosis (Mtb) exhibits remarkable metabolic flexibility that enables it to survive a plethora of host environments during its life cycle. With the advent of bedaquiline for treatment of multidrug-resistant tuberculosis, oxidative phosphorylation has been validated as an important target and a vulnerable component of mycobacterial metabolism. Exploiting the dependence of Mtb on oxidative phosphorylation for energy production, several components of this pathway have been targeted for the development of new antimycobacterial agents. This includes targeting NADH dehydrogenase by phenothiazine derivatives, menaquinone biosynthesis by DG70 and other compounds, terminal oxidase by imidazopyridine amides and ATP synthase by diarylquinolines. Importantly, oxidative phosphorylation also plays a critical role in the survival of persisters. Thus, inhibitors of oxidative phosphorylation can synergize with frontline TB drugs to shorten the course of treatment. In this review, we discuss the oxidative phosphorylation pathway and development of its inhibitors in detail.
1. Introduction
Tuberculosis (TB) remains a leading cause of death worldwide, with an estimated 1.3 million mortalities in 2016. The synergy of human immunodeficiency virus (HIV)-TB co-infection has further aggravated this as a major public health problem. TB treatment is riddled with the use of multiple drugs for at least 6 months. Such lengthy treatment has resulted in the rise of multidrug resistant (MDR) and extremely drug resistant (XDR) strains of Mycobacterium tuberculosis (Mtb). Treatment of MDR TB requires administration of a multitude of second-line TB drugs for 18–24 months; this leads to cure rates of 60–70%. Of even more concern, cure rates for XDR-TB range from 40–50%. These low cure rates could be further confounded by other factors, such as co-infections and poor nutrition. Given these observations, new drugs with novel mechanisms of action are urgently required. Such innovative drugs could synergize with current treatment regimens to improve recovery rates and shorten treatment times. Extensive research efforts have been made in this direction, and the U.S. Food and Drug Administration has recently approved two new drugs. These drugs are bedaquiline (BDQ), a diarylquinoline ATP synthase inhibitor, and delamanid, a nitro-dihydro-imidazooxazole derivative that inhibits mycolic acid biosynthesis. Accelerated approval of these drugs has raised hope for a new regimen that could improve the outcome of treatment and reduce daily dose burden.
The myth that targeting Mtb bioenergetics would be a poor therapeutic strategy due to the presence of parallel and alternative metabolic pathways has been dispelled by the discovery of BDQ. Given the success of this compound, several research groups have focused on targeting oxidative phosphorylation for the discovery of new drugs. It is important to note that, unlike many bacteria that can utilize substrate-level phosphorylation for ATP synthesis and growth, Mtb relies upon oxidative phosphorylation for its viability. During oxidative phosphorylation, electrons are harvested from central metabolic pathways, and then fed into the electron transport chain (ETC) through reduction of menaquinone. Later on, the quinones are re-oxidized by either the cytochrome bc1-aa3 complex or by the cytochrome bd-type terminal oxidase. The transport of electrons between membrane-bound primary dehydrogenase and terminal reductase is coupled to the generation of the proton motive force (PMF). The energy generated as PMF from this process is utilized by ATP synthase in the production of ATP. In this review, we have provided an overview of the machinery involved in oxidative phosphorylation and the development of inhibitors (listed in Table 1) targeting the components of oxidative phosphorylation.
2. Feeding the Electrons into the Electron Transport Chain: Reduction of Menaquinone
During the infection cycle, Mtb cells harvest reductive energy from host-derived carbohydrates and fatty acids using NADH/NAD+ and/or FADH2/FAD redox pairs [1,2]. NAD+ acts as the primary electron sink and is converted into NADH, which feeds electrons into the ETC through the NADH dehydrogenase complex. Besides NADH dehydrogenase, a number of other primary dehydrogenases pump electrons directly into the ETC, such as succinate dehydrogenase (SDH), proline dehydrogenase, and L-lactate cytochrome c oxidoreductase. In this review, we have primarily focused on NADH dehydrogenase and SDH.
2.1. NADH/Menaquinone Oxidoreductase
NADH/menaquinone oxidoreductases are the primary port of entry for electrons into the ETC. There are three types of these oxidoreductases in bacteria: (i) the highly-complex, multi-subunit-proton-pumping type I NADH dehydrogenase (NDH-1), (ii) a simple, single subunit flavoenzyme-non-proton-pumping type II NADH dehydrogenase (NDH-2), and (iii) a sodium-pumping NADH dehydrogenase (NQR) that is unique to bacteria [3]. In 2005, using a genome mining approach, Weinstein et al. identified NDH-1 and NDH-2 in Mtb [4]. NDH-1 is encoded by the NuoABCDEFGHIJKLMN operon and is predicted to be the energy efficient NADH dehydrogenase that translocates protons, while transferring electrons to menaquinone to generate PMF. Interestingly, genes encoding NDH-1 are not essential in Mtb [5], as exemplified by their loss from Mycobacterium leprae through genome reductive evolution [4]. NDH-1 is downregulated by acidic pH [6], nutrient starvation [7], and in murine lungs [8]. This expression profile suggests that NDH-1 plays a minor role in Mtb respiration, and thus, is not a good target for the development of antimycobacterials. This view is further strengthened by the observation that rotenone (an NDH-1 inhibitor) does not kill hypoxia-adapted Mtb cells [9]. However, recently developed reporter strains for imaging cellular NADH/NAD+ levels [10] has demonstrated that exposure of aerobic Mtb cultures to rotenone leads to accumulation of cellular NADH levels, suggesting a role for NDH-1 in the ETC during aerobic conditions. Furthermore, infection of macrophages with an Mtb strain that has a deletion of nuoG (which encodes an NDH-1 subunit) accelerates their apoptotic death. Moreover, this mutant has reduced survival in a murine model of TB infection [11].
The non-proton pumping NDH-2 isoforms are encoded by ndh (Rv1854c) and ndhA (Rv0392c). Both the two isoforms were found to be functional in biochemical assays. NDH-2 has a stringent catalytic function, thereby minimizing leakage of electrons to oxygen during their transfer from NADH to menaquinone. Importantly, Mtb NDH-2 utilizes only NADH, in contrast to NDH-2 of Saccharomyces and Corynebacterium, which uses NAD(P)H [12]. Mtb NDH-2 transfers electrons to the quinone pool by a ping-pong mechanism [13]. Sena et al. demonstrated that NDH-2 has two distinct substrate binding sites (i.e., NADH and quinone). NDH-2 first interacts with NADH to receive electrons, and then releases NAD+ before interacting with the quinone [14]. It is assumed that the reduction of quinone/menaquinone is a rate-limiting step in the overall reaction [15]. Both the isoforms of NDH-2 are conserved in slow-growing mycobacterial species, with the exception of M. leprae, in which ndhA is absent [4]. ndh is essential for Mtb survival [5], whereas ndhA disruption using transposon mutagenesis is tolerated [16]. Importantly, disruption of NDH-2 through inhibitors, such as phenothiazine analogues, leads to mycobacterial death in aerobic cultures and in animals [4]. In agreement with these findings, the NDH-2 inhibitor thioridazine (TZ) kills Mtb that are in a hypoxia-induced non-replicating state [9]. ndh is also induced upon Mtb infection of macrophages [17]. Interestingly, mammalian mitochondrion lacks an NDH-2 orthologue, which provides a significant therapeutic window. Earlier studies have also validated NDH-2 as a target to control Plasmodium growth [18].
2.2. Inhibitors of NADH Dehydrogenase
The above cited literature strongly suggests that NDH-2 is an attractive target whose inhibition would elicit a choke point in the mycobacterial respiratory chain. In an effort to discover additional respiratory inhibitors, Harvey and colleagues found that phenothiazine analogues have antimycobacterial activity and demonstrated that these compounds inhibit NDH-2 [4]. They also demonstrated that classical inhibitors of NDH-1 (piericidinA, rotenone, and pyridaben) do not inhibit Mtb respiration. Phenothiazines were also effective against MDR strains, suggesting a new mode of action [19,20]. Trifluoperazine (TPZ), a phenothiazine analogue, can effectively inhibit Mtb growth, and synergizes with rifampicin (RIF) to kill intracellular Mtb residing in macrophages [21,22]. A recent report indicates that TPZ significantly inhibits ATP synthesis in M. leprae [23]. Phenothiazines also inhibit efflux pumps and calcium-binding protein. Similar antimycobacterial activity (both in drug-sensitive and drug-resistant strains) was observed with the NDH-2 specific inhibitor, thioridazine (TZ), in Mtb-infected mice [24]. TZ has been used for 60 years to control psychosis and is considered therapeutically safe. The drug targets NDH-2 to block the ETC of mycobacteria and is effective against latent TB [13,25]. Given the critical role of NDH-2 and the potent activity of TPZ and TZ against Mtb cells, high throughput screening (HTS) was performed with 11,000 compounds rationally selected from a commercial library (Biofocus, DPI). This effort resulted in the identification of lead compound 42a (MTC420) that had a minimum inhibitory concentration (MIC) in the nanomolar range. This lead compound is capable of killing drug-sensitive and -resistant strains of Mtb in aerobic and hypoxic cultures. Furthermore, 42a also has favorable pharmacokinetic and toxicological profiles [26].
Clofazimine (CFZ) is derived from phenazines, and has potent anti-tuberculosis activity [27]. The drug effectively kills M. leprae, and is therefore used for the treatment of leprosy [28]. The main side effect of this drug is skin pigmentation. Recently developed CFZ derivatives namely, B746 and B4157, have increased anti-mycobacterial activity and elicit less pigmentation, and are therefore suitable for testing for treatment of mycobacterial infection in animal models [29,30]. An eminent study from the Harvey group demonstrated that the bactericidal action of CFZ depends upon its reduction by NDH-2. The reduced form of CFZ is highly unstable, and spontaneously reacts with oxygen to generate reactive oxygen species (ROS) that leads to the killing of mycobacterial cells [31]. This cycle of CFZ-mediated ROS generation continues under aerobic conditions. Importantly, the NDH-1 present in mitochondrial and bacterial respiratory chains does not reduce CFZ. Therefore, Gram-negative bacteria, including Escherichia coli, Pseudomonas denitrificans, and P. aeruginosa do not produce bactericidal ROS upon exposure to CFZ [32,33]. CFZ is also active against MDR TB strains, is metabolized slowly, and is associated with a low frequency of resistance [34]. CFZ synergizes with BDQ and Q203 to rapidly kill Mtb in vitro and inside macrophages [35].
Since NDH-2 is a respiratory choke point of Mtb, AstraZeneca (India) performed an HTS against Mtb NDH-2 with 100,000 compounds. This effort identified quinolinyl pyrimidines as a new class of NDH-2 inhibitors. A good correlation was observed between enzyme inhibition (nanomolar) and anti-mycobacterial activity (micromolar) of quinolinyl pyrimidines [36]. These compounds are non-toxic and have desirable absorption, distribution, metabolism, and excretion (ADME) profiles, making them suitable for further development. In line with these findings, compounds belonging to 7-phenyl benzoxaborole series exhibited potent anti-Mtb activity [37].
2.3. Succinate Dehydrogenase
The succinate dehydrogenase (SDH) or succinate/menaquinone oxidoreductase forms complex II of the respiratory chain. Importantly, this enzyme complex is also an integral part of the tricarboxylic acid (TCA) cycle. SDH links the respiratory chain with central metabolism [38]. The enzyme oxidizes succinate to fumarate in the cytoplasm, and during this process, it simultaneously reduces menaquinone to menaquinol in the membrane. This enzyme complex is comprised of four subunits: SdhA, SdhB, SdhC, and SdhD. SdhA is a flavoprotein (flavin is bound here covalently, and not as a cofactor) that catalyzes the conversion of succinate into fumarate. SdhB possesses three Fe−S clusters that facilitate the transfer of electrons from succinate to menaquinone. The SdhC and SdhD subunits anchor SdhA and SdhB to the membrane, facilitating the transfer of electrons from succinate to menaquinone using haem. Notably, fumarate reductase (FRD) is a paralogue of SDH that can catalyze the reverse reaction. FRD (encoded by the FrdA-Rv1556 operon) is primarily functional in anaerobic conditions. Mtb utilizes menaquinone (MQH2 E′0 = −74 mV) to reduce fumarate in the last step of the anaerobic ETC [39,40,41]. Interestingly, Mtb possesses two isoforms of SDH; Sdh1 (encoded by Rv0247c–Rv0249c) and Sdh2 (encoded by Rv3316–Rv3319). It is crucial to note that under hypoxic conditions, Mtb cells utilize the reverse TCA cycle to accumulate and secrete succinate into the culture medium [36,42]. The excretion of succinate has been associated with the upregulation of genes involved in the reverse TCA cycle. Addition of succinate to the culture medium resulted in membrane potential depolarization and cell death, suggesting that secretion of succinate is critical for surviving hypoxic stress [42,43]. A role for SDH in this adaption had been predicted, since inhibition of SDH function using 3-nitropropionate (3NP) results in decreased survival of Mtb [42]. Individual Sdh1 and Sdh2 deletion mutants were generated in order to dissect the role of both Sdh1 and Sdh2 during hypoxic adaptation. This revealed that survival of the Sdh1 mutant was impaired in stationary phase, while the survival of the Sdh2 mutant was not affected [44]. The function of Sdh1 as the catalytic center during aerobic conditions was demonstrated through the use of stable isotope labelling and mass spectroscopy [44]. Sdh2 was not required for this activity during aerobic conditions. Furthermore, depletion of Sdh1 resulted in increased levels of the reduced lipogenic electron carrier, menaquinol, which was associated with an increased rate of respiration, suggesting that SDH is a regulator of respiration [44]. Importantly, survival of the Sdh1 mutant was compromised in the Mtb-infected C3HeB/FeJ mouse model system, which produces lesions similar to the hypoxic granulomas found in humans upon Mtb infection [44]. These observations suggest that inhibition of SDH can help in the elimination of hypoxia-induced persisters in the infected lungs. Sdh1 is, thus, a potential drug target, however, stringent selectivity will be required to avoid a general inhibition of mitochondrial respiration.
2.4. Menaquinone Biosynthesis and Inhibitors of Menaquinone Biosynthesis Pathway in Mtb
Ubiquinone (Q) and menaquinone (MK) are the primary lipid-soluble electron carriers that transport electrons in the ETC [1]. In most Gram-positive bacteria and in mycobacteria, MK is the primary electron carrier, while Gram-negative bacteria possess both Q and MK [45]. Mycobacterial species, including Mtb, possess MK-9(H2) (hereafter MK) [46,47]. Besides playing the role as an electron carrier, MK assists in the proper folding of secreted proteins through vitamin K epoxide reductase or VKOR [48]. The ratio of oxidized to reduced MK is sensed by the DosRST system to control mycobacterial metabolism [49,50]. Since MK is the only electron carrier in mycobacteria, and because humans do not synthesize this vitamin, it is believed to be an excellent target for the development of drugs against mycobacterial pathogens. MK is synthesized in bacteria through a classical pathway and a second, alternate pathway. Both pathways begin with a chorismate molecule, which is then metabolized to a different MK precursor, depending on whether the classical or alternate pathway is engaged. Although the alternate pathway is operational in evolutionarily related Streptomyces, it has not been reported in mycobacteria. In regard to the classical pathway, chorismate is converted into MK through a series of reactions carried out by enzymes encoded by the MenFDHCEBA cluster [5], as summarized in Figure 1. Some of these enzymes are essential for mycobacterial growth [5,51] suggesting that MK biosynthesis is a valid drug target.
Since cholesterol metabolism is important for mycobacterial survival within the host, the effect of exposure of Ro 48-8071 (an inhibitor of cholesterol biosynthesis) on mycobacterial growth was analyzed. This compound killed Mycobacterium bovis Bacillus Calmette Guerin (BCG) cells at low micromolar concentrations [52]. Further metabolic labelling experiments identified MK biosynthesis to be the target of Ro 48-8071. These findings were supported by the observation that Ro 48-8071 was capable of reducing oxygen consumption during growth. Further scrutiny led to the identification of MenA (Rv0534c) as the target of Ro 48-8071. The same group utilized a rational drug design approach to target MenA. They screened around 100 molecules, and established that an allylaminomethonone class of compounds are potent inhibitors of Mtb strains, including those which are drug-resistant [53]. This class of MenA inhibitors was also highly effective against hypoxia-induced drug-tolerant persisters [54]. Interestingly, aurachin RE, a new quinoline antibiotic isolated from Rhodococcus erythropolis JCM 6824 is highly effective against Gram-positive bacteria [55]. The aurachin RE molecules were modified to specifically target MenA, and validated as MenA inhibitors in functional in vitro assays. These compounds were low micromolar inhibitors of aerobic growth and hypoxic survival of Mtb [56]. Additionally, 7-methoxy-2-naphthol-based molecules have been utilized as lead structures for the development of non-traditional inhibitors of MenA of Mtb [57]. These efforts led to the development of bicyclic inhibitors that exert potent growth-suppressing activity against Mtb [57]
High-density phenotypic profiling has identified menB (Rv0548c) as essential for mycobacterial growth [58]. MenB catalyzes the formation of 1,4-dihydroxy-2-naphthoyl-CoA from o-succinylbenzoyl CoA through a Claisen condensation reaction. Li and coworkers performed an HTS with more than 100,000 compounds, which identified a few hundred leads. Subsequent elaboration yielded multiple 1,4-benzoxazine compounds that were potent inhibitors of MenB activity, and consequently blocked mycobacterial growth [59]. These leads were then utilized for the development of 4-oxo-4-chlorophenylbutenoyl methyl ester derivatives with potent mycobactericidal activity against replicating and non-replicating Mtb [60].
MenE (Rv0542c) is the fifth enzyme in the MK biosynthesis pathway, and catalyzes the formation of OSB-CoA from O-succinyl-1-benzoate (OSB) through two mechanistic steps. The first is the ATP-dependent adenylation of OSB, and the second step is thioesterification with CoA. Given the structural and functional similarity of MenE with adenylate-forming enzymes, it is a promising target of acyl-AMP intermediates. Considering the mechanistic details, Tan and coworkers employed OSB-AMP analogues to inhibit MenE in Mtb [61]. The IC50 of OSB-AMP analogues MeOSB-AMS, MeOSB-AMSN, and MeOSB-AVSN for MenE are in the low micromolar range [61]. Further development of these analogues resulted in the identification of OSB-AMS with an IC50 for MenE in the nanomolar range; these compounds are competitive inhibitors of the binding of ATP and OSB [62]. Paradoxically, these inhibitors had poor mycobacterial growth inhibitory activity, perhaps due to their instability, and the OSB-AMP analogues were, therefore, further modified. This resulted in the development of OSB-AMS. Importantly, the difluoroindanediol analogue 11 of OSB-AMS was an effective mycobactericidal compound [63].
In an effort to identify inhibitors of oxidative phosphorylation, Alland and coworkers created a cell-based screen [64]. In this screen, the mWasabi reporter was fused to the promoter of the cydAB operon (coding for part of cytochrome bd oxidase) to monitor inhibition of respiration in the presence of 168 known inhibitors of mycobacterial growth. This led to the identification of DG70 as an inhibitor of bacterial respiration. DG70 is a biphenyl benzamide that kills both drug-sensitive and -resistant Mtb cells [64]. Interestingly, this novel inhibitor is highly specific against Mtb, and does not kill non-tuberculous mycobacteria (NTMs), Gram-positive, Gram-negative, or ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumonia, Acinetobacter baumannii, P. aeruginosa, and Enterobacter) pathogens. Sequencing of two DG70-resistant spontaneous mutant strains suggested the presence of a unique single nucleotide polymorphism (SNP) in menG. MenG (Rv0558, also known as MenH) catalyzes the conversion of demethylmenaquinol to menaquinol. Importantly MK4, an analogue of MK9, rescued the DG70-mediated killing of Mtb cells, thus validating that MK biosynthesis is the target of DG70. Importantly, DG70 synergizes with BDQ, isoniazid (INH), and PA824 to kill Mtb [64]. In summary, all these observations indicate that small molecule targeting of MK biosynthesis (summarized in Figure 1) is a valid therapeutic strategy.
3. Oxidation of Menaquinone: A Tale of Two Terminal Oxidases
Mycobacteria possess a branched ETC. During aerobic respiration, electrons are fed into the menaquinone(reduced/oxidized) pool of respiratory chains. Electrons are then transferred to oxygen through two branches, each of which possesses a terminal oxidase. One is cytochrome bd-type menaquinol oxidase (encoded by cydABDC), wherein electrons are transferred directly from MK (reduced) to oxygen. The other branch runs through a supercomplex of menaquinol-cytochrome c oxidoreductase or the bc1 complex and aa3 type cytochrome oxidase [1,65,66,67]. The bc1-aa3 branch pumps protons out of the cell during transfer of electrons to oxygen, whereas the bd-type oxidase does not pump protons, but has a higher affinity for oxygen [68]. It is assumed that similar to E. coli, the two terminal oxidases of mycobacteria have different affinities for oxygen; however, this awaits experimental validation.
3.1. bc1-aa3 Pathway
The bc1 complex is a key component of the bc1-aa3 pathway, and is required for the bulk of the electron transfer to oxygen during normoxia [69]. bc1 is encoded by the qcrCAB operon in Mtb. It consists of redox groups comprising a 2Fe/2S centre, located on a Rieske protein (QcrA), two b-type haems (low and high potential) located on a single polypeptide (QcrB), and the haem of cytochrome c1 (QcrC). QcrA of Mtb has three transmembrane helices and characteristic sequence motifs (CSHLGC and CPCH) of 2Fe–2S Rieske iron-sulfur proteins, QcrB has a 120 amino acid extension at the C-terminus, and the QcrC subunit consists of two haem binding motifs (CVSCH and CASCH) for c-type cytochromes, suggesting a covalent di-haem (bcc) configuration [70]. According to Q-cycle model, oxidation of quinol molecules occurs at the interface of cytochrome b and the 2Fe–2S cluster carrying domain of the Rieske protein, which forms the catalytic center P at the positively charged membrane side of the enzyme [71]. Each quinol molecule oxidizes at the center P, liberating two electrons that move towards different acceptors. One electron proceeds to the iron-sulfur Rieske protein, and is then transferred to cytochrome c, while the other moves to another quinone-binding site on the opposite side of the membrane through haem molecules of variable redox potentials. Complex III releases four protons for every two electrons transferred from menaquinol to cytochrome c into the periplasmic side of the membrane [69].
The Mtb aa3-type cytochrome c oxidase is comprised of four subunits: CtaB (a cytochrome c oxidase assembly factor), CtaD (cytochrome c oxidase subunit I containing haem a, a3, and copperB (CuB), CtaC (cytochrome c oxidase subunit II containing copperA (CuA) and CtaE (subunit III). The aa3 cytochrome c oxidase of Mtb is encoded by ctaBCDE, which is dispersed over different locations of the Mtb genome [72]. Interestingly, the ctaE gene is located immediately upstream of the qcrCAB operon. The putative fourth subunit is encoded by ctaF (Rv2199c). This subunit is co-purified with other subunits in Corynebacterium. Importantly, the deletion mutants display the phenotype similar to CtaC mutant [73]. Four protons are taken up, while two protons are released into the periplasm for every two electrons passing through complex IV [72]. There are three ctaD alleles in Mycobacterium smegmatis, while only one in Mtb, indicating the presence of multiple isoforms of cytochrome c oxidase in M. smegmatis [65]. The proton-pumping type (and energetically more efficient) bc1-aa3 branch of ETC is essential for mycobacteria, as its deletion by homologous recombination is lethal [66]. This observation indicates that the bc1-aa3 branch of ETC is an important drug target.
Unlike E. coli, which utilizes membrane-soluble cytochrome C for transferring electrons between bc1 complex and aa3 cytochrome c oxidase, Mycobacterium, Corynebacterium glutamicum, and Rhodococcus do not possess cytochrome C [70,74,75]. Importantly, in C. glutamicum, the QcrC contains an extra binding site for another haem c that may function as a merged cytochrome C [76]. Moreover, subunit II of the oxidase contains an additional 30 amino acids that could participate in the direct interaction between complexes bc1 and aa3, to create a respiratory “supercomplex” [66], obviating the requirement for free cytochrome c [74,76]. Indeed, in C. glutamicum, bc1 and aa3 are isolated as a complex that is resistant to detergent treatment [73]. A similar scenario was predicted for mycobacteria [74], and was validated following the isolation of a detergent-resistant supercomplex in M. smegmatis [77]. The interaction between the bc1 and aa3 complexes is guided by hydrophobic interactions, while ionic interactions facilitate the alignment between the two complexes for efficient electron transfer from menaquinol to oxygen [77]. Recently, overexpression of the Mtb complex III in M. smegmatis was shown to yield a functional, stable, hybrid supercomplex in the presence of dodecyl maltoside detergent [78]. These observations strongly suggest that, in mycobacterial cells, the bc1 complex and aa3 cytochrome c oxidase interact with each other to facilitate the flow of electrons from MK to oxygen, without the requirement of cytochrome C.
3.2. Cytochrome bd-Oxidase
Cytochrome bd-type menaquinol oxidase (bd-oxidase) in Mtb is non-proton pumping, and is therefore a less energetically efficient terminal oxidase. E. coli bd-oxidase is the prototypical member of this enzyme class and is encoded by two separate operons, cydAB and cydDC. This is contrary to Mtb, in which all the genes are transcribed from a single cydABDC operon. cydAB encodes the functional cytochromes, while the products of cydDC contribute to cytochrome bd assembly [79]. Furthermore, in E. coli mutants of cydDC, the periplasmic space is more oxidized than in the wild type bacteria [79]. The function of cydDC in mycobacteria is largely unknown, but it plays an important role in mycobacterial persistence in vivo. cydDC mutation reduces the ability of Mtb to survive the transition from acute to chronic infection in mice [12]. Another report suggests that cydDC supports mycobacterial persistence in INH-treated mice [13]. Compared to their wild type counterparts, cytochrome bd mutants of E. coli are sensitive to stress induced by temperature alterations, nitric oxide, H2O2, and iron (III) chelators. Additionally, they are unable to resume growth upon entering into the stationary phase [80,81,82,83]. Importantly, the growth of cydA mutant M. smegmatis is normal at ambient oxygen levels, but is severely impaired during hypoxia (0.5–1% air saturation). These mutants are also sensitive to cyanide, and are outcompeted when co-cultured with wild type M. smegmatis in its presence [65]. In E. coli, bd-oxidase has a higher affinity for oxygen, and is induced in response to low oxygen tension [84]. Although the affinity of the Mtb bd-oxidase for oxygen has not been determined, its expression is also induced during hypoxia [65]. The expression of cydAB is regulated by a SenX3-RegX3 two-component system [85], which also acts as an oxygen-controlled replication switch in Mtb [86]. It was recently demonstrated that Mtb cydAB mutants are sensitive to H2O2 and antibiotic stress [87]. Importantly, drugs inhibiting mycobacterial respiration enhance the expression of cydAB [88]. Interestingly in M. smegmatis, inactivation of bc1 complex results in the upregulation of the bd-type terminal oxidase; however, the bc1 complex does not compensate for the loss of the bd-type oxidase [87]. Genetic inactivation of cytochrome bd-oxidase in various pathogenic microorganisms like Shigella flexneri, Brucella abortus, and Salmonella enterica serovar Typhimurium, impairs their intracellular survival and virulence [89,90,91].
3.3. Supercomplex Inhibitors
The essentiality of Mtb cytochrome bc1, along with the large differences in structure and function from its mammalian counterpart, makes it a good target for therapeutic intervention [78]. A variety of antibiotics and chemical compounds have long been known to inhibit the bc1-aa3 complex. The aurachin class of compounds contains quinone analogues, which are reported to inhibit a variety of cytochrome oxidases [56,92,93]. Aurachin D is well-known among this class of compounds that inhibit E. coli cytochrome bd-oxidase [92]. It inhibits oxygen consumption in M. smegmatis in a dose-dependent manner [87]. These observations demonstrate the importance of cytochrome bd inhibitors as drug molecules. Optimized derivatives of aurachin D, with better ability to penetrate the mycobacterial cell wall, have a great potential as a new class of antitubercular drugs [87].
Similar to aurachin D, myxothiazol-based compounds, which are isolated from Myxococcus fulvus, target mitochondrial cytochrome b. Myxothiazol interacts both with cytochrome b and an iron-sulfur protein of the complex; this displaces quinone from the high-affinity binding site of the iron-sulfur protein [94]. Importantly, myxothiazol inhibits the growth of Mycobacterium sp. GBF 3 at an MIC value of 6.3 μg/mL [95], indicating that respiratory inhibitors can successfully target mycobacterial species. Interestingly, myxothiazol does not kill Gram-negative or -positive bacteria, but does exhibit strong antifungal activity [95]. A few other antifungal antibiotics, such as mucidin (from Basidiomycetes Oudemansiella mucida) and strobilurin A (from Strobilurin stenacellus) also inhibit the bc1 complex by binding to the same site as myxothiazol [96]. However, antimycin A (another antibiotic) binds to a different location, inhibiting the oxidation of cytochrome b subunit [97]. A screen in search of new antibiotics identified a novel compound, stigmatellin, from Stigmatella aurantiaca strain Sg a15, that showed activity against various Gram-positive bacteria, yeasts, and filamentous fungi [98]. Subsequently, it was found to block mitochondrial electron transport by inhibiting the cytochrome bc1 complex as effectively as antimycin A and myxothiazol [99].
The capability of a number of small molecules to specifically target energy production in Mtb has validated oxidative phosphorylation as a viable drug target. Since energy production through oxidative phosphorylation plays an important role in Mtb survival during hypoxia-induced non-replicating persistence [9,100], blockade of oxidative phosphorylation could be pursued, therapeutically, to kill persisters. A number of small molecule inhibitors that specifically target the bc1-aa3 complex have been identified. Several lines of evidence suggest that these inhibitors, which are structural analogues of quinone, target the quinone binding catalytic domains of the bc1 complex [101]; prominent examples are the imidazo[1,2-a]pyridines (IPs). Using HTS, IPs were identified as potent inhibitors of Mtb and BCG [101]. Importantly, four IP inhibitors were shown to specifically target Mtb strains and BCG, but were not able to kill a number of other Gram-positive and Gram-negative bacteria, or human cell lines, including HepG2 and Neuro2A. Whole genome sequencing of the spontaneous resistant mutants established that these IPs targeted QcrB. These findings were further supported by induction of drug resistance upon overexpression of qcrB [101]. HTS of 100,997 compounds led to the discovery of phenoxyalkylbenzimidazole (PAB) class of compounds showing activity against Mtb with MICs in the nanomolar range, and low cytotoxicity against eukaryotic cells [102,103]. A recent study identified the probable target of PABS to be the QcrB subunit of cytochrome bc1 oxidase, further confirming it to be an important drug target [104].
An independent screen of more than 100,000 compounds to identify inhibitors of macrophage-resident bacterial growth led to the isolation of two highly active IP compounds with less toxicity than previous molecules of the same class. Further lead optimization resulted in the development of IP Q203, which is specifically active against Mtb (MIC50 of 2.7 nM in broth cultures and 0.28 nM for intracellular bacteria) [105]. Furthermore, whole genome sequencing of spontaneously resistant mutants revealed the target of Q203 to be the cytochrome bc1 complex (qcrB) (as depicted in Figure 2). Mutation of Thr313 to either alanine or isoleucine was specifically involved in Q203 resistance [34]. Importantly, this compound was highly active against MDR and XDR clinical isolates [106]. This leading drug candidate has recently progressed to Phase I clinical trials (trial identifier: NCT02858973) under a U.S. FDA Investigational New Drug application. Notably, Q203 is synthesized through a one-pot reaction of 2-aminopyridines or 2-(or 4-)aminopyrimidines, respectively, with 1,2-bis(benzotriazolyl)-1,2-(dialkylamino)-ethanes [107]. These studies were followed by derivatization of imidazo[1,2-a]pyridine 3 carboxyamides into imidazo[1,2-b]thiazole-5-carboxyamides. Structure–activity relationship (SAR)-based assays were utilized for optimizing the imidazo[1,2-b]thiazole-5-carboxyamides to yield three lead compounds that have potent antitubercular activity at low nanomolar concentrations. These compounds specifically inhibit QcrB, and thus, block the growth of replicating Mtb and intracellular mycobacteria, and display very low toxicity [108]. These compounds were also active against large numbers of NTMs, including Mycobacterium avium in murine lungs [108,109]. In another parallel drug discovery effort, an HTS campaign by Novartis Institute for Tropical Diseases identified several potential hit molecules with potent activity against Mtb [110]. However, the presence of ester linkages raised concerns over their metabolic instability [111]. Several compounds were thus optimized for metabolic stability through pharmacokinetic studies in the mouse. These studies led to the development of pyrrolo[3,4-c]pyridine-1,3(2H)-diones having an MIC90 in the micromolar range [111]. These compounds are hyperactive against cytochrome bd-oxidase mutants, suggesting that they target the bc1-aa3 complex of the respiratory chain. The target for these molecules was identified as qcrB, and a point mutation (Ala317Thr) in qcrB results in resistance to these compounds [111]. Importantly, the target site is identical to that bound by Q203, suggesting that this site may be promiscuous for molecules targeting the bc1 complex.
Interestingly, in an effort to build a strong pipeline for antimycobacterial drugs, a pool of 1280 FDA approved drugs was screened for growth inhibition of Mtb residing in MRC-5 lung fibroblasts. This screen resulted revealed that the gastric proton pump inhibitor lansoprazole (LPZ, Prevacid) exerted potent activity against Mtb. Importantly, other proton pump inhibitors, such as omeprazole and pantoprazole, were not able to inhibit Mtb growth. It was further demonstrated that LPZ is rapidly converted to the potent antimycobacterial agent, lansoprazole sulfide (LPZS). Thus, LPZ acts as a prodrug that is converted into LPZS within the host cell. As the conversion of LPZ into LPZS is inefficient, a 22-fold higher drug dose was required to kill Mtb cells in broth cultures. LPZS selectively kills Mtb; it is not toxic to closely related NTMs or Gram-positive and-negative bacteria [112]. Target identification studies revealed that LPZS targets the cytochrome bc1 complex (as depicted in Figure 2) of the respiratory chain, and conversion of leucine-176 to proline in qcrB confers drug resistance. Furthermore, superimposition of the mutant mycobacterial protein structure onto the published QcrB protein from R. sphaeroides [113] revealed that both L176P (a mutation leading to LPZS resistance) and T313A (which confers IPA resistance) were localized to the same site, i.e., the P site, at which ubiquinol oxidation occurs. However, L176P mutants remained susceptible to various IPA, whereas T313A mutants were susceptible to LPZS, revealing different binding mechanisms for each compound [112]. There are conflicting reports on the oral bioavailability of this drug. For example, oral administration of the drug is reported to significantly reduce mycobacterial burden [112], whereas others have shown that intraperitoneal injection of LPZS molecules, rather than the LPZ prodrug, is required to acquire plasma and lung tissue concentrations of the active molecule sufficient to kill Mtb [114]. Recently, the use of LPZ has been associated with reduced incidence of TB when compared with the use of omeprazole or pantoprazole [115].
The ATP synthase inhibitor BDQ and Q203 both result in an increase in mycobacterial oxygen consumption rate (OCR) over a range of physiologically relevant oxygen concentrations [35]. It was suggested that bacteria are compelled to utilize cytochrome bd-oxidase as the terminal oxidase upon disruption of the flow of electrons through the bc1-aa3 pathway by Q203. This postulate is supported by the observation that Q203 completely inhibits respiration in the cytochrome bd-oxidase knockout mutant (cydKO), while the Q203 resistant SNP remains unaffected [35]. Furthermore, cydKO is hypersusceptible to Q203 [116,117]. BDQ and Q203 are relatively slow acting drugs with regard to mycobacterial cytotoxicity. Importantly, BDQ both alone or in combination with Q203, show similar slow kill kinetics, but the combination of CFZ with either BDQ or Q203 results in increased OCR and synergistic killing. The triple combination of CFZ, BDQ, and Q203 kills even faster, and results in complete sterility within 5 days. This combination of drugs is far more effective and rapid than the combination of first-line drugs INH and RIF in culture broths and in infected macrophages. BDQ and Q203 push the cell into reductive stress, during which NADH levels increase, and this potentiates the ROS-generating activity of CFZ, leading to mycobacterial killing [35]. These observations suggest that a combination of drugs targeting different components of the respiratory chain may help in the development of more effective therapeutic regimens. Targeting the bacterium at multiple steps of oxidative phosphorylation may be a better strategy for tackling drug-susceptible as well as MDR and XDR-TB. The role of efflux pumps in Mtb drug resistance is well established [118], and therefore, targeting them may help to resensitize Mtb to antibacterial agents. In support of this, verapamil (an efflux pump inhibitor) increases the potency of Q203 [119].
4. ATP Synthase
During the transfer of electrons from NADH or succinate to a terminal electron acceptor (such as oxygen), protons are pumped out to generate PMF. ATP synthase can utilize PMF to generate ATP from ADP and inorganic phosphate [120,121]. ATP synthase is highly conserved from bacteria to mammals; it consists of a transmembrane Fo complex that is used for ion translocation, and a peripheral F1 complex that catalyzes ATP generation. The membrane-embedded Fo complex is made up of a, b, and c subunits that are present in a variable stoichiometry of ab2c8–15. On the other hand, the hydrophilic F1 complex consists of 5 subunits in the stoichiometry α3β3γδε [122]. The proton movement is utilized by the Fo subunit to generate rotation of the oligomeric c ring, and this rotation is coupled with the rotation of the attached γ and ε subunits of the F1 complex [123,124,125].
During its infection cycle, mycobacteria must survive host-generated stress [126,127]. In order to do so, Mtb utilizes its metabolic flexibility to maintain ATP homeostasis [1,128]. Regulated synthesis of ATP through ATP synthase is critical for the survival of pathogens inside the host. This enzyme is even more important in mycobacterial cells, since they require it for growth on fermentable carbon sources [129]. It must be noted that Mtb downregulates its metabolic rate upon entering macrophages or lung tissue [1,130], and primarily depends on lipids for energy [131]. ATP levels are also reduced in the pathogen upon oxygen and nutrient deficit [9,132]. In the case of Streptococcus, ATP synthase is downregulated during biofilm formation [133], which is believed to harbor dormant bacteria. However, the level of ATP in biofilm-resident Mtb is not significantly different to that observed in planktonic bacteria [134]. Also, single cell microscopy has revealed that antibiotic exposure tremendously reduces the ATP levels in M. smegmatis cells [135]. ATP synthase in mycobacteria is encoded by the atpBEFHAGDC operon (Rv1303–1312). The expression of this operon is tightly regulated to match the cellular ATP requirements, and is therefore closely linked to the respiratory chain activity. This operon is downregulated in hypoxic cultures and in murine lung tissue [8]. Recently, a transcriptional regulator (blaI, Rv1846c) of the atp operon has been identified [136].
The subunit composition of mycobacterial ATP synthase and the mechanism of ATP generation is similar to that of other bacteria, such as E. coli and humans (Figure 3A). However, there are subtle differences in Mtb ATP synthase that contribute to its survival in humans [137]. These differences, however, can be exploited for the development of Mtb-specific drugs. These differences include the capability to catalyze the ATP biosynthesis at low PMF of ~100 mV [9] compared with ~200 mV of E. coli [138], and the ability to block ATPase activity [139,140] in order to maintain an optimal level of PMF, as seen in many other bacteria [141]. Mycobacterial ATP synthase has several structural differences compared with the human orthologue. This includes a 36 amino acid C-terminal domain extension of subunit α [142], which is not seen in any other prokaryotes or eukaryotes. Interestingly, this extension is critical for the ability of mycobacterial ATP synthase to reduce ATPase activity. The Mtb ATP synthase γ subunit possesses a unique loop (γ165–178) of 13 amino acids [143] that also inhibits ATP hydrolysis-driven proton pumping [144]. Interestingly, due to the capability of ATP generation at low PMF, Mtb ATP synthase was expected to have a large c ring with more than 10 monomers, as seen in some alkaliphilic bacteria [145,146,147]. However, the crystal structure of the mycobacterial c ring revealed that it contains 9 monomers [148]. This is the smallest bacterial c ring reported with a H+/ATP ratio of 3.
Given the essentiality of ATP synthase for the survival of mycobacteria on fermentable and non-fermentable carbon sources, and its subtle but peculiar structural differences from the mitochondrial ATP synthase present in humans, this enzyme can be utilized as a potential drug target. However, drugs like oligomycin and N,N′-dicyclohexylcarbodiimide (DCCD) (that target ATP synthase) are non-selective, and also inhibit generation of ATP in mitochondria and bacteria [149,150], suggesting the conserved nature of this enzyme. Nonetheless, the recent discovery of BDQ as a specific inhibitor of mycobacterial ATP synthase has validated this enzyme complex as a drug target.
4.1. Bedaquiline
Several naturally occurring and synthetic ATP synthase inhibitors (such as DCCD) have been described [151]. However, it was only in 2004 [152,153], that an ATP synthase-targeting lead compound of the diarylquinoline class (R207910) was shown to have potent antimycobacterial activity. Four decades after the development of rifabutin in 1975, a new class of antibiotic that specifically targeted tuberculosis was developed by the Andries group [152]. This compound was later called BDQ, or TMC207, and is sold under the brand name of Sirturo®.
Importantly, this compound specifically inhibited the growth of mycobacterial species, and was non-inhibitory to Gram-positive bacteria, such as Nocardia, and Gram-negative bacteria such as E. coli. More importantly, this compound was able to kill MDR and XDR Mtb strains with MICs in the range of 0.03–0.12 µg/mL. Interestingly, usage of 10× MIC for 12 days resulted in a 3-log reduction of bacterial colony forming units (cfu). This killing was time dependent, rather than being concentration dependent. The delayed onset of killing could be explained by the metabolic remodeling of mycobacterial cells upon BDQ exposure. This includes upregulation of the dormancy regulon, ATP synthase, isocitrate lyase, and cytochrome bd oxidase. Additionally, there was a downregulation of DNA/protein biosynthesis and efflux pump expression, in order to conserve energy. The drug also demonstrated significant in vivo activity in a murine model of TB infection. Importantly, its use alone led to a similar reduction of mycobacterial load in the lungs as that of a combination of frontline drugs (RIF + INH + pyrazinamide (PZA)). Moreover, its use in combination with these drugs resulted in a 2-log greater reduction in mycobacterial load compared to the “no BDQ” group. Importantly, BDQ also showed desirable pharmacokinetic properties [152]. Furthermore, use of the Wayne model demonstrated that BDQ efficiently targets both replicating and non-replicating mycobacteria [9]. The drug is posited to be more effective against non-replicating bacteria than those that are actively replicating; this could be due to 5–10-fold lower ATP levels in the non-replicating dormant bacteria [154]. The ability to kill non-replicating bacteria correlates with BDQ’s exceptionally high bactericidal activity in mouse TB models [155,156,157], and also with the reduced time for sputum culture conversion in TB patients [158,159].
In an effort to identify the target of BDQ, whole genome sequencing was performed. Mutations in atpE, which codes for the Fo subunit of ATP synthase, were associated with drug resistance [152]. In another target identification effort, mycobacterial membrane extract was passed through a column of BDQ-coated Sepharose beads. This approach also identified subunits of ATP synthase. These experiments were followed by surface plasmon resonance assays, which demonstrated that atpE binds to a BDQ analogue with a Kd of 500 nM [160]. The MIC50 in the case of human mitochondrial ATP synthase was more than 20,000-fold higher than that of the bacterial enzyme, underscoring the high degree of selectivity afforded by BDQ [161]. A high-resolution crystal structure of mycobacterial ATP synthase, with and without BDQ, has provided insights into the mechanism of BDQ binding, and will also help in structure-based drug design [148]. It was suggested that one or more BDQ molecules could specifically associate with ion binding site of ATP synthase in the c ring of the Fo rotor. BDQ interacts with ~135 Å2 of the c ring of the Fo rotor consisting of nine residues (Gly62, Leu63, Glu65, Ala66, Ala67, Tyr68, Phe69, Ile70, and Leu72) through van der Waals interactions (Figure 3B). BDQ’s dimethylamino moiety extends into the ion-binding site of C-rings. The interaction is then stabilized by an intermolecular H-bond between the dimethylamino group and Glu65 [148]. BDQ has also been tested against NTM, M. abscessus, which affects cystic fibrosis patients [162], and against M. avium and M. intracellulare [163]. Additionally, it was found to be active against M. abscessus in a zebrafish model of infection [164]. Furthermore, BDQ was effective against M. leprae [165], but bacteriostatic in the case of M. avium [166], and inactive in M. abscessus mouse infection [167,168].
Moreover, BDQ, when combined with other TB drugs, helped to reduce the time of treatment [156,157,169]. A synergistic effect of BDQ and PZA was seen in a mouse model of TB [170]. BDQ was also synergistic with the cell wall synthesis inhibitor, SQ109, as it improved the killing rate and lowered the MIC [171]. Another cell wall synthesis targeting compound, BTZ043, was also found to be synergistic with BDQ in vitro [172]. BTZ043 is a benzothiazinone which inhibits DprE1, an enzyme involved in arabinan biosynthesis [173]. The increased efficacy of BDQ, when used in combination with cell wall biosynthesis inhibitors, could be due to its increased penetration. Furthermore, clinical trials are ongoing for the combination therapy of BDQ with another recently approved drug, delamanid [174]. BDQ results in prolonged QT interval, however, co-administration of RIFor rifapentine with BDQ increases its clearance, likely due to their activity as CYP3A4 inducers, which is involved in BDQ metabolism [175]. Thus, we infer that the currently approved BDQ–RIF combination therapy should be re-evaluated. Furthermore, the addition of ketoconazole, a CYP3A4 inhibitor with BDQ increased the QT interval [176]. A study conducted in human hepatocytes to understand the BDQ metabolism identified N-dealkylation, a novel metabolic pathway. In this study, CYP3A4, CYP2C8, and CYP2C19 were shown to be involved in BDQ N-demethylation [177]. This kind of study is important to understand and prevent BDQ associated adverse drug reactions.
The rate of generation of BDQ-resistant mutants is one in 108 bacteria. Certain mycobacteria, including M. novocastrense, M. xenopi, and M. shimoidei, are naturally resistant to BDQ, due to polymorphisms at the atpE locus [168,178]. BDQ resistance is also acquired, and is present in bacteria with cross-resistance to CFZ. This cross-resistance is due to mutations in Rv0678, a transcriptional repressor of the MmpL5 efflux pump [179,180,181]. BDQ is very effective against drug-susceptible MDR and XDR-TB; however, due to its toxicity (attributed to the increased QT interval) and the development of BDQ-resistant strains [182], its use is currently limited to MDR and XDR-TB patients.
4.2. Squaramides
After the discovery of BDQ, several studies have been conducted in order to identify novel molecules that target the mycobacterial oxidative phosphorylation pathway [100,101,103,105]. In one example, AstraZeneca employed a luminescence-based method in which inverted membrane vesicles from mycobacteria were charged with NADH and formation of ATP was monitored. Following HTS of 900,000 compounds, two lead classes, namely squaramide and imidazo [1, 2-a] pyridine ethers (IPE), were identified. Given their poor solubility, pharmacokinetics, and bacteriostatic nature, IPE were deprioritized, and squaramides were further developed [183]. This resulted in the identification of 31f, which exhibited potent antituberculosis activity (IC50 of 0.03 µM in proliferation assays). Similar to the case with BDQ, docking studies and the generation of spontaneous mutants identified ATP synthase as the target of squaramide. However, the mode of binding employed by squaramides is different to that of BDQ. 31f is predicted to bind to the interface of ATP synthase subunits a and c. This prediction was supported by the observation that the K179N mutation of subunit a and the D28N mutation of subunit c of ATP synthase engendered resistance to 31f. Importantly, squaramides were also found to be active against single drug resistant clinical strains, including BDQ. Furthermore, squaramides show a similar MIC for BDQ-resistant mutant strains and do not show cross-resistance, suggesting that they interact with a different binding site. Lead compound 31f was also effective in a mouse model of acute TB infection. Further studies are required to see whether squaramides can potentiate the activity of current TB drugs, and whether they will be effective in a chronic TB infection model.
5. Uncouplers of Proton Motive Force: Pyrazinamide and SQ109
PMF generated by ETC plays a critical role in bacterial growth and survival. During hypoxic growth, dissipation of PMF by uncouplers, such as valinomycin and nigericin, can cause death in the hypoxic cultures [9]. The importance of PMF as a drug target is further validated by the observation that efflux of a number of drugs from mycobacterial cells is dependent on PMF [184]. However, general uncouplers of PMF are not sufficiently selective to be used as antimycobacterial agents, and the development of specific PMF uncouplers remains an area of interest. Importantly, PZA, which is currently being used as a first-line anti-TB drug along with RIF and INH, can disrupt the PMF in mycobacterial cells. Another lead compound, SQ109, also targets the PMF and oxidative phosphorylation [185].
PZA was discovered in the early 1940s, and is effective in mouse models of TB [186] and in TB patients [187]. Consequently, the drug is used as a frontline agent to treat TB. PZA is a prodrug that requires acidic conditions [188] in order to be hydrolyzed by pyrazinamidase or nicotinamidase to pyrazinoic acid (POA), the active form of the drug [189]. PZA-resistant mutants often have variations in the pyrazinamidase gene, which is encoded by pcnA [190,191,192]. PZA is a multitarget drug that dissipates the PMF, inhibits ATP synthesis [193], inhibits membrane transport [194], and also reduces the activity of other proteins [195]. The first clue that PZA could dissipate PMF was the requirement of acidic pH for its bactericidal activity [196]. PZA requires 4–5 fold higher concentration (850 μg/mL) at pH 6.5 compared to pH 5.5 (200 μg/mL) [194]. The active form of the drug, POA, is a weak acid that accumulates inside the cell in acidic conditions [196]. This weakly acidic property helps POA (and the structurally unrelated benzoic acid) to disrupt the membrane potential of Mtb under acidic conditions [194]. In line with this observation, PZA is more active against non-replicating dormant bacteria, as compared to actively replicating bacteria [197] and old cultures [198]. These findings are further supported by the fact that PMF uncouplers synergize with PZA in order to deplete ATP depletion and enhance mycobacterial killing [194]. However, the exact mechanism by which PZA dissipates the membrane potential remains unknown.
SQ109 is structurally related to the frontline TB drug ethambutol, and was developed by Sequella Incorporation in collaboration with Laboratory of Host Defenses, NIH. It has successfully completed phase 2 clinical trials, and is active against drug-susceptible MDR and XDR-TB [199]. Although in TB patients, SQ109 monotherapy for 14 days did not reduce bacterial burden [200], a recent phase 2b-3 clinical study suggested an 80% increase in the culture conversion rate by addition of SQ109 to the standard regimen [201]. Initially, SQ109 was reported to interfere with the assembly of mycolic acid in the mycobacterial cell wall. This was attributed to its ability to target MmpL3, a membrane transporter involved in uptake of trehalose monomycolate [202,203]. However, SQ109 is also active against other organisms, including Helicobacter pylori [204], Trypanosoma cruzi [205], Neisseria gonorrhoeae, Candida albicans [206], and P. falciparum [185]. Since these organisms lack a functional homolog of MmpL3, it is highly likely that MmpL3 is not the primary target of SQ109. This hypothesis is further supported by the absence of spontaneously resistant mutants, a phenomenon observed with many drugs that inhibit multiple targets. Recently, Li et al., have demonstrated that SQ109 and its analogues inhibit MK biosynthesis, and interfere with respiration and generation of PMF [185,205]. It acts as an uncoupler, collapsing both ΔpH and Δψ, leading to a decrease in ATP synthesis [185]. Importantly, SQ109 synergizes with BDQ and CFZ, further suggesting that it affects oxidative phosphorylation [206]. Further studies are required to completely understand the complex mechanism by which SQ109 inhibits mycobacterial cell growth.
6. Conclusions
Despite the presence of alternate respiratory complexes, oxidative phosphorylation is an excellent target for the development of new antimycobacterial drugs (Figure 4). This statement is validated by the presence of a number of its inhibitors in the current TB drug pipeline. Most notable among these inhibitors are Q203 (an inhibitor of bc1-aa3 complex) that has entered clinical trials, and BDQ (ATP synthase inhibitor) that has been approved for treatment of MDR TB. It is worth noting that inhibitors of other components of oxidative phosphorylation, such TPZ&QPs (NDH-2 inhibitor), Ro 48-8071 & DG70 (inhibitors of MK biosynthesis) and SQ109 (PMF disruptor) are potent blockers of mycobacterial growth, in vitro and in vivo. We believe that co-inhibition of parallel respiratory components could significantly shorten the duration of TB treatment. This belief is supported by the enhanced susceptibility of cydAB deletion mutant to BDQ or accelerated killing of Mtb cells by the combination of CFZ with BDQ or Q203. Such combinations shall be tested, in vivo, to further test this hypothesis.
Competing Financial Interests
The authors declare that they do not have any competing financial interests.
Acknowledgments
AK is supported through Swarnajayanti Fellowship by Department of Science and Technology and CSIR (OLP-0652 and OLP-0141). IKI, SB, are grateful to the CSIR for funding SRF and JRF respectively. AKA is SRF funded by DBT.
Conflicts of Interest
The authors declare that they do not have any conflict of interest.
Abbreviations
| 3NP | 3-Nitropropionate |
| ADP | Adenosine diphosphate |
| ATP | Adenosine triphosphate |
| BCG | Bacillus Calmette Guerin |
| BDQ | Bedaquiline |
| cfu | Colony forming units |
| CFZ | Clofazimine |
| CO2 | Carbon dioxide |
| cydKO | Cytochrome bd-oxidase knockout mutant |
| DCCD | N,N′-dicyclohexylcarbodiimide |
| DprE1 | Decaprenylphosphoryl-D-ribose oxidase |
| ETC | Electron transport chain |
| FADH2 | Reduced flavin adenine dinucleotide |
| FRD | Fumarate reductase |
| H+ | Proton |
| H2O | Water |
| HIV | Human immunodeficiency virus |
| HTS | High throughput screening |
| INH | Isoniazid |
| IP | Imidazo[1,2-a]pyridines |
| LPZ | Lansoprazole |
| LPZS | Lansoprazole sulphide |
| MDR | Multi drug resistant |
| MIC | Minimum inhibitory concentration |
| MK | Menaquinone |
| MKH2 | Menaquinol |
| Mtb | Mycobacterium tuberculosis |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Reduced nicotinamide adenine dinucleotide |
| NDH-1 | Type I NADH dehydrogenase |
| NDH-2 | Type II NADH dehydrogenase |
| NQR | Sodium-pumping NADH dehydrogenase |
| NTMs | Non-tuberculous mycobacteria |
| OCR | Oxygen consumption rate |
| OSB | O-succinyl-1-benzoate |
| PAB | Phenoxyalkylbenzimidazole |
| Pi | Inorganic phosphate |
| PMF | Proton motive force |
| POA | Pyrazinoic acid |
| PZA | Pyrazinamide |
| Q | Ubiquinone |
| RIF | Rifampicin |
| ROS | Reactive oxygen species |
| SAR | Structure–activity relationship |
| SDH | Succinate dehydrogenase |
| SNP | Single nucleotide polymorphism |
| TB | Tuberculosis |
| TCA cycle | Tricarboxylic acid cycle |
| TPZ | Trifluoperazine |
| TZ | Thioridazine |
| XDR | Extremely drug resistant |
References
- Boshoff, H.I.; Barry, C.E., 3rd. Tuberculosis—Metabolism and respiration in the absence of growth. Nat. Rev. Microbiol. 2005, 3, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Russell, D.G.; Vander Ven, B.C.; Lee, W.; Abramovitch, R.B.; Kim, M.J.; Homolka, S.; Niemann, S.; Rohde, K.H. Mycobacterium tuberculosis wears what it eats. Cell Host Microbe 2010, 8, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, S.; Dröse, S.; Zickermann, V.; Brandt, U. The three families of respiratory NADH dehydrogenases. Results Probl. Cell Differ. 2008, 45, 185–222. [Google Scholar] [PubMed]
- Weinstein, E.A.; Yano, T.; Li, L.S.; Avarbock, D.; Avarbock, A.; Helm, D.; McColm, A.A.; Duncan, K.; Lonsdale, J.T.; Rubin, H. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc. Natl. Acad. Sci. USA 2005, 102, 4548–4553. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.A.; Plikaytis, B.B.; Shinnick, T.M. Microarray analysis of the Mycobacterium tuberculosis transcriptional response to the acidic conditions found in phagosomes. J. Bacteriol. 2002, 184, 4025–4032. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Sohaskey, C.D.; Kana, B.D.; Dawes, S.; North, R.J.; Mizrahi, V.; Gennaro, M.L. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. USA 2005, 102, 15629–15634. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.P.; Alonso, S.; Rand, L.; Dick, T.; Pethe, K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2008, 105, 11945–11950. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.A.; Iqbal, I.K.; Kumar, A. Imaging the NADH:NAD(+) Homeostasis for Understanding the Metabolic Response of Mycobacterium to Physiologically Relevant Stresses. Front. Cell. Infect. Microbiol. 2016, 6, 145. [Google Scholar] [CrossRef] [PubMed]
- Velmurugan, K.; Chen, B.; Miller, J.L.; Azogue, S.; Gurses, S.; Hsu, T.; Glickman, M.; Jacobs, W.R., Jr.; Porcelli, S.A.; Briken, V. Mycobacterium tuberculosis nuoG is a virulence gene that inhibits apoptosis of infected host cells. PLoS Pathog. 2007, 3, e110. [Google Scholar] [CrossRef] [PubMed]
- Nantapong, N.; Otofuji, A.; Migita, C.T.; Adachi, O.; Toyama, H.; Matsushita, K. Electron transfer ability from NADH to menaquinone and from NADPH to oxygen of type II NADH dehydrogenase of Corynebacterium glutamicum. Biosci. Biotechnol. Biochem. 2005, 69, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Li, L.S.; Weinstein, E.; Teh, J.S.; Rubin, H. Steady-state kinetics and inhibitory action of antitubercular phenothiazines on Mycobacterium tuberculosis type-II NADH-menaquinone oxidoreductase (NDH-2). J. Biol. Chem. 2006, 281, 11456–11463. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yamashita, T.; Nakamaru-Ogiso, E.; Hashimoto, T.; Murai, M.; Igarashi, J.; Miyoshi, H.; Mori, N.; Matsuno-Yagi, A.; Yagi, T.; et al. Reaction mechanism of single subunit NADH-ubiquinone oxidoreductase (Ndi1) from Saccharomyces cerevisiae: Evidence for a ternary complex mechanism. J. Biol. Chem. 2011, 286, 9287–9297. [Google Scholar] [CrossRef] [PubMed]
- Sena, F.V.; Batista, A.P.; Catarino, T.; Brito, J.A.; Archer, M.; Viertler, M.; Madl, T.; Cabrita, E.J.; Pereira, M.M. Type-II NADH:quinone oxidoreductase from Staphylococcus aureus has two distinct binding sites and is rate limited by quinone reduction. Mol. Microbiol. 2015, 98, 272–288. [Google Scholar] [CrossRef] [PubMed]
- McAdam, R.A.; Quan, S.; Smith, D.A.; Bardarov, S.; Betts, J.C.; Cook, F.C.; Hooker, E.U.; Lewis, A.P.; Woollard, P.; Everett, M.J.; et al. Characterization of a mycobacterium tuberculosis h37rv transposon library reveals insertions in 351 orfs and mutants with altered virulence. Microbiology 2002, 148, 2975–2986. [Google Scholar] [CrossRef] [PubMed]
- Schnappinger, D.; Ehrt, S.; Voskuil, M.I.; Liu, Y.; Mangan, J.A.; Monahan, I.M.; Dolganov, G.; Efron, B.; Butcher, P.D.; Nathan, C.; et al. Transcriptional adaptation of mycobacterium tuberculosis within macrophages: Insights into the phagosomal environment. J. Exp. Med. 2003, 198, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Pidathala, C.; Amewu, R.; Pacorel, B.; Nixon, G.L.; Gibbons, P.; Hong, W.D.; Leung, S.C.; Berry, N.G.; Sharma, R.; Stocks, P.A.; et al. Identification, design and biological evaluation of bisaryl quinolones targeting plasmodium falciparum type ii nadh:Quinone oxidoreductase (pfndh2). J. Med. Chem. 2012, 55, 1831–1843. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.; Kristiansen, J.E.; Viveiros, M.; Atouguia, J. Activity of phenothiazines against antibiotic-resistant mycobacterium tuberculosis: A review supporting further studies that may elucidate the potential use of thioridazine as anti-tuberculosis therapy. J. Antimicrob. Chemother. 2001, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Gadre, D.V.; Talwar, V.; Gupta, H.C.; Murthy, P.S. Effect of trifluoperazine, a potential drug for tuberculosis with psychotic disorders, on the growth of clinical isolates of drug resistant mycobacterium tuberculosis. Int. Clin. Psychopharmacol. 1998, 13, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Crowle, A.J.; Douvas, G.S.; May, M.H. Chlorpromazine: A drug potentially useful for treating mycobacterial infections. Chemotherapy 1992, 38, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.V.; Nadadhur, G.; Gangadharam, P.R. In-vitro and intracellular antimycobacterial activity of trifluoperazine. J. Antimicrob. Chemother. 1996, 37, 196–197. [Google Scholar] [CrossRef] [PubMed]
- Katoch, V.M.; Saxena, N.; Shivannavar, C.T.; Sharma, V.D.; Katoch, K.; Sharma, R.K.; Murthy, P.S. Effect of trifluoperazine on in vitro atp synthesis by mycobacterium leprae. FEMS Immunol. Med. Microbiol. 1998, 20, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Van Soolingen, D.; Hernandez-Pando, R.; Orozco, H.; Aguilar, D.; Magis-Escurra, C.; Amaral, L.; van Ingen, J.; Boeree, M.J. The antipsychotic thioridazine shows promising therapeutic activity in a mouse model of multidrug-resistant tuberculosis. PLoS ONE 2010, 5, e12640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teh, J.S.; Yano, T.; Rubin, H. Type ii nadh: Menaquinone oxidoreductase of mycobacterium tuberculosis. Infect. Disord. Drug Targets 2007, 7, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.D.; Gibbons, P.D.; Leung, S.C.; Amewu, R.; Stocks, P.A.; Stachulski, A.; Horta, P.; Cristiano, M.L.S.; Shone, A.E.; Moss, D.; et al. Rational design, synthesis, and biological evaluation of heterocyclic quinolones targeting the respiratory chain of mycobacterium tuberculosis. J. Med. Chem. 2017, 60, 3703–3726. [Google Scholar] [CrossRef] [PubMed]
- Barry, V.C.; Belton, J.G.; Conalty, M.L.; Denneny, J.M.; Edward, D.W.; O'Sullivan, J.F.; Twomey, D.; Winder, F. A new series of phenazines (rimino-compounds) with high antituberculosis activity. Nature 1957, 179, 1013–1015. [Google Scholar] [CrossRef] [PubMed]
- Garrelts, J.C. Clofazimine: A review of its use in leprosy and mycobacterium avium complex infection. DICP Ann. Pharmacother. 1991, 25, 525–531. [Google Scholar] [CrossRef]
- O’Connor, R.; O’Sullivan, J.F.; O’Kennedy, R. The pharmacology, metabolism, and chemistry of clofazimine. Drug Metab. Rev. 1995, 27, 591–614. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.M.; Nadadhur, G.; Daneluzzi, D.; O’Sullivan, J.F.; Gangadharam, P.R. Antituberculosis activities of clofazimine and its new analogs b4154 and b4157. Antimicrob. Agents Chemother. 1996, 40, 633–636. [Google Scholar] [PubMed]
- Yano, T.; Kassovska-Bratinova, S.; Teh, J.S.; Winkler, J.; Sullivan, K.; Isaacs, A.; Schechter, N.M.; Rubin, H. Reduction of clofazimine by mycobacterial type 2 nadh: Quinone oxidoreductase: A pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 2011, 286, 10276–10287. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Ohnishi, T. The origin of cluster n2 of the energy-transducing nadh-quinone oxidoreductase: Comparisons of phylogenetically related enzymes. J. Bioenerg. Biomembr. 2001, 33, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.M.; Bandeiras, T.M.; Teixeira, M. New insights into type ii nad(p)h: Quinone oxidoreductases. Microbiol. Mol. Biol. Rev. 2004, 68, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.M.; O’Sullivan, J.F.; Gangadharam, P.R. Antimycobacterial activities of riminophenazines. J. Antimicrob. Chemother. 1999, 43, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Lamprecht, D.A.; Finin, P.M.; Rahman, M.A.; Cumming, B.M.; Russell, S.L.; Jonnala, S.R.; Adamson, J.H.; Steyn, A.J. Turning the respiratory flexibility of mycobacterium tuberculosis against itself. Nat. Commun. 2016, 7, 12393. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Paul, B.; Roy Choudhury, N.; Kedari, C.; Bandodkar, B.; Ugarkar, B.G. Quinolinyl pyrimidines: Potent inhibitors of ndh-2 as a novel class of anti-tb agents. ACS Med. Chem. Lett. 2012, 3, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Korkegian, A.; O’Malley, T.; Xia, Y.; Zhou, Y.; Carter, D.S.; Sunde, B.; Flint, L.; Thompson, D.; Ioerger, T.R.; Sacchettini, J.; et al. The 7-phenyl benzoxaborole series is active against Mycobacterium tuberculosis. Tuberculosis 2018, 108, 96–98. [Google Scholar] [CrossRef]
- Cecchini, G. Respiratory complex II: Role in cellular physiology and disease. Biochim. Biophys. Acta 2013, 1827, 541–542. [Google Scholar]
- Unden, G.; Bongaerts, J. Alternative respiratory pathways of Escherichia coli: Energetics and transcriptional regulation in response to electron acceptors. Biochim. Biophys. Acta 1997, 1320, 217–234. [Google Scholar] [CrossRef]
- Unden, G.; Schirawski, J. The oxygen-responsive transcriptional regulator FNR of Escherichia coli: The search for signals and reactions. Mol. Microbiol. 1997, 25, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Kroger, A.; Biel, S.; Simon, J.; Gross, R.; Unden, G.; Lancaster, C.R. Fumarate respiration of wolinella succinogenes: Enzymology, energetics and coupling mechanism. Biochim. Biophys. Acta 2002, 1553, 23–38. [Google Scholar] [CrossRef]
- Eoh, H.; Rhee, K.Y. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, 6554–6559. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Zimmermann, M.; Goodwin, M.B.; Sauer, U.; Barry, C.E., 3rd; Boshoff, H.I. Fumarate reductase activity maintains an energized membrane in anaerobic mycobacterium tuberculosis. PLoS Pathog. 2011, 7, e1002287. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.; Weinrick, B.; Vilcheze, C.; Berney, M.; Tufariello, J.; Cook, G.M.; Jacobs, W.R., Jr. Succinate dehydrogenase is the regulator of respiration in Mycobacterium tuberculosis. PLoS Pathog 2014, 10, e1004510. [Google Scholar] [CrossRef] [PubMed]
- Nowicka, B.; Kruk, J. Occurrence, biosynthesis and function of isoprenoid quinones. Biochim. Biophys. Acta 2010, 1797, 1587–1605. [Google Scholar] [CrossRef] [PubMed]
- Knapczyk, J. Kinetics of sodium nitrite decomposition. Acta Poloniae Pharm. 1975, 32, 683–689. [Google Scholar] [PubMed]
- Azerad, R.; Cyrot-Pelletier, M.O. Structure and configuration of the polyprenoid side chain of dihydromenaquinones from Myco- and Corynebacteria. Biochimie 1973, 55, 591–603. [Google Scholar] [CrossRef]
- Dutton, R.J.; Wayman, A.; Wei, J.R.; Rubin, E.J.; Beckwith, J.; Boyd, D. Inhibition of bacterial disulfide bond formation by the anticoagulant warfarin. Proc. Natl. Acad. Sci. USA 2010, 107, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Toledo, J.C.; Patel, R.P.; Lancaster, J.R., Jr.; Steyn, A.J. Mycobacterium tuberculosis doss is a redox sensor and dost is a hypoxia sensor. Proc. Natl. Acad. Sci. USA 2007, 104, 11568–11573. [Google Scholar] [CrossRef] [PubMed]
- Honaker, R.W.; Dhiman, R.K.; Narayanasamy, P.; Crick, D.C.; Voskuil, M.I. Doss responds to a reduced electron transport system to induce the Mycobacterium tuberculosis DosR regulon. J. Bacteriol. 2010, 192, 6447–6455. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Rubin, E.J. Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. USA 2003, 100, 12989–12994. [Google Scholar] [CrossRef] [PubMed]
- Dhiman, R.K.; Mahapatra, S.; Slayden, R.A.; Boyne, M.E.; Lenaerts, A.; Hinshaw, J.C.; Angala, S.K.; Chatterjee, D.; Biswas, K.; Narayanasamy, P.; et al. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol. Microbiol. 2009, 72, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, M.; Narayanasamy, P.; Biswas, K.; Dhiman, R.; Crick, D.C. Discovery of 1,4-dihydroxy-2-naphthoate [corrected] prenyltransferase inhibitors: New drug leads for multidrug-resistant gram-positive pathogens. J. Med. Chem. 2007, 50, 3973–3975. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, M.; Crick, D.C. MenA is a promising drug target for developing novel lead molecules to combat Mycobacterium tuberculosis. Med. Chem. 2009, 5, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, W.; Tamura, T. A quinoline antibiotic from Rhodococcus erythropolis JCM 6824. J. Antibiot. 2008, 61, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Siricilla, S.; Wan, B.; Crick, D.C.; Lenaerts, A.J.; Franzblau, S.G.; Kurosu, M. Discovery of selective menaquinone biosynthesis inhibitors against mycobacterium tuberculosis. J. Med. Chem. 2012, 55, 3739–3755. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.R.; Larson, M.A.; Hinrichs, S.H.; Bartling, A.M.; Frandsen, J.; Narayanasamy, P. Discovery of bicyclic inhibitors against menaquinone biosynthesis. Future Med. Chem. 2016, 8, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.E.; Gawronski, J.D.; Dejesus, M.A.; Ioerger, T.R.; Akerley, B.J.; Sassetti, C.M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, N.; Zhang, H.; Knudson, S.E.; Slayden, R.A.; Tonge, P.J. Synthesis and sar studies of 1,4-benzoxazine menb inhibitors: Novel antibacterial agents against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett. 2010, 20, 6306–6309. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, N.; Zhang, H.; Knudson, S.E.; Li, H.J.; Lai, C.T.; Simmerling, C.; Slayden, R.A.; Tonge, P.J. Coa adducts of 4-oxo-4-phenylbut-2-enoates: Inhibitors of menb from the m. Tuberculosis menaquinone biosynthesis pathway. ACS Med. Chem. Lett. 2011, 2, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhang, H.; Tonge, P.J.; Tan, D.S. Mechanism-based inhibitors of mene, an acyl-coa synthetase involved in bacterial menaquinone biosynthesis. Bioorg. Med. Chem. Lett. 2008, 18, 5963–5966. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhou, R.; Sharma, I.; Li, X.; Kumar, G.; Swaminathan, S.; Tonge, P.J.; Tan, D.S. Stable analogues of OSB-AMP: Potent inhibitors of MenE, the o-succinylbenzoate-CoA synthetase from bacterial menaquinone biosynthesis. Chembiochem Eur. J. Chem. Biol. 2012, 13, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Matarlo, J.S.; Evans, C.E.; Sharma, I.; Lavaud, L.J.; Ngo, S.C.; Shek, R.; Rajashankar, K.R.; French, J.B.; Tan, D.S.; Tonge, P.J. Mechanism of mene inhibition by acyl-adenylate analogues and discovery of novel antibacterial agents. Biochemistry 2015, 54, 6514–6524. [Google Scholar] [CrossRef] [PubMed]
- Sukheja, P.; Kumar, P.; Mittal, N.; Li, S.G.; Singleton, E.; Russo, R.; Perryman, A.L.; Shrestha, R.; Awasthi, D.; Husain, S.; et al. A Novel Small-Molecule Inhibitor of the Mycobacterium tuberculosis Demethylmenaquinone Methyltransferase MenG Is Bactericidal to Both Growing and Nutritionally Deprived Persister Cells. mBio 2017, 8, e02022-16. [Google Scholar] [CrossRef] [PubMed]
- Kana, B.D.; Weinstein, E.A.; Avarbock, D.; Dawes, S.S.; Rubin, H.; Mizrahi, V. Characterization of the cydab-encoded cytochrome bd oxidase from mycobacterium smegmatis. J. Bacteriol. 2001, 183, 7076–7086. [Google Scholar] [CrossRef] [PubMed]
- Matsoso, L.G.; Kana, B.D.; Crellin, P.K.; Lea-Smith, D.J.; Pelosi, A.; Powell, D.; Dawes, S.S.; Rubin, H.; Coppel, R.L.; Mizrahi, V. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J. Bacteriol. 2005, 187, 6300–6308. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.V.; Eiglmeier, K.; Gas, S.; Barry, C.E., 3rd; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, R.; Hill, S.; Poole, R.K. The cytochrome bd quinol oxidase in Escherichia coli has an extremely high oxygen affinity and two oxygen-binding haems: Implications for regulation of activity in vivo by oxygen inhibition. Microbiology 1996, 142 Pt 4, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Mulkidjanian, A.Y. Proton translocation by the cytochrome bc1 complexes of phototrophic bacteria: Introducing the activated Q-cycle. Photochem. Photobiol. Sci. 2007, 6, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Niebisch, A.; Bott, M. Molecular analysis of the cytochrome bc1-aa3 branch of the Corynebacterium glutamicum respiratory chain containing an unusual diheme cytochrome c1. Arch. Microbiol. 2001, 175, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Berry, E.A.; Guergova-Kuras, M.; Huang, L.S.; Crofts, A.R. Structure and function of cytochrome bc complexes. Annu. Rev. Biochem. 2000, 69, 1005–1075. [Google Scholar] [CrossRef] [PubMed]
- Cook, G.M.; Hards, K.; Vilcheze, C.; Hartman, T.; Berney, M. Energetics of Respiration and Oxidative Phosphorylation in Mycobacteria. Microbiol. Spectr. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Niebisch, A.; Bott, M. Purification of a cytochrome bc-aa3 supercomplex with quinol oxidase activity from Corynebacterium glutamicum. Identification of a fourth subunity of cytochrome aa3 oxidase and mutational analysis of diheme cytochrome c1. J. Biol. Chem. 2003, 278, 4339–4346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakamoto, J.; Shibata, T.; Mine, T.; Miyahara, R.; Torigoe, T.; Noguchi, S.; Matsushita, K.; Sone, N. Cytochrome c oxidase contains an extra charged amino acid cluster in a new type of respiratory chain in the amino-acid-producing Gram-positive bacterium Corynebacterium glutamicum. Microbiology 2001, 147, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Sone, N.; Fukuda, M.; Katayama, S.; Jyoudai, A.; Syugyou, M.; Noguchi, S.; Sakamoto, J. QcrCAB operon of a nocardia-form actinomycete Rhodococcus rhodochrous encodes cytochrome reductase complex with diheme cytochrome cc subunit. Biochim. Biophys. Acta 2003, 1557, 125–131. [Google Scholar] [CrossRef]
- Sone, N.; Nagata, K.; Kojima, H.; Tajima, J.; Kodera, Y.; Kanamaru, T.; Noguchi, S.; Sakamoto, J. A novel hydrophobic diheme c-type cytochrome. Purification from Corynebacterium glutamicum and analysis of the QcrCBA operon encoding three subunit proteins of a putative cytochrome reductase complex. Biochim. Biophys. Acta 2001, 1503, 279–290. [Google Scholar] [CrossRef]
- Megehee, J.A.; Hosler, J.P.; Lundrigan, M.D. Evidence for a cytochrome bcc-aa3 interaction in the respiratory chain of Mycobacterium smegmatis. Microbiology 2006, 152, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Jang, J.; Ab Rahman, N.B.; Pethe, K.; Berry, E.A.; Huang, L.S. Isolation and Characterization of a Hybrid Respiratory Supercomplex Consisting of Mycobacterium tuberculosis Cytochrome bcc and Mycobacterium smegmatis Cytochrome aa3. J. Biol. Chem. 2015, 290, 14350–14360. [Google Scholar] [CrossRef] [PubMed]
- Goldman, B.S.; Gabbert, K.K.; Kranz, R.G. The temperature-sensitive growth and survival phenotypes of Escherichia coli cydDC and cydAB strains are due to deficiencies in cytochrome bd and are corrected by exogenous catalase and reducing agents. J. Bacteriol. 1996, 178, 6348–6351. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.K.; Cook, G.M. Redundancy of aerobic respiratory chains in bacteria? Routes, reasons and regulation. Adv. Microb. Physiol. 2000, 43, 165–224. [Google Scholar] [PubMed]
- Borisov, V.B.; Gennis, R.B.; Hemp, J.; Verkhovsky, M.I. The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 2011, 1807, 1398–1413. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Borisov, V.B.; Davletshin, A.; Mastronicola, D.; Sarti, P.; Giuffre, A. Cytochrome bd oxidase and hydrogen peroxide resistance in mycobacterium tuberculosis. MBio 2013, 4, e01006-01013. [Google Scholar] [CrossRef] [PubMed]
- Giuffre, A.; Borisov, V.B.; Arese, M.; Sarti, P.; Forte, E. Cytochrome bd oxidase and bacterial tolerance to oxidative and nitrosative stress. Biochim. Biophys. Acta 2014, 1837, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Kita, K.; Konishi, K.; Anraku, Y. Terminal oxidases of Escherichia coli aerobic respiratory chain. II. Purification and properties of cytochrome b558-d complex from cells grown with limited oxygen and evidence of branched electron-carrying systems. J. Biol. Chem. 1984, 259, 3375–3381. [Google Scholar] [PubMed]
- Roberts, G.; Vadrevu, I.S.; Madiraju, M.V.; Parish, T. Control of CydB and GltA1 expression by the SenX3 RegX3 two component regulatory system of Mycobacterium tuberculosis. PLoS ONE 2011, 6, e21090. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Kumar, A. Virulence factor SenX3 is the oxygen-controlled replication switch of Mycobacterium tuberculosis. Antioxid. Redox Signal. 2015, 22, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Heineke, M.H.; Koul, A.; Andries, K.; Cook, G.M.; Lill, H.; van Spanning, R.; Bald, D. The cytochrome bd-type quinol oxidase is important for survival of Mycobacterium smegmatis under peroxide and antibiotic-induced stress. Sci. Rep. 2015, 5, 10333. [Google Scholar] [CrossRef] [PubMed]
- Boot, M.; Jim, K.K.; Liu, T.; Commandeur, S.; Lu, P.; Verboom, T.; Lill, H.; Bitter, W.; Bald, D. A fluorescence-based reporter for monitoring expression of mycobacterial cytochrome bd in response to antibacterials and during infection. Sci. Rep. 2017, 7, 10665. [Google Scholar] [CrossRef] [PubMed]
- Way, S.S.; Sallustio, S.; Magliozzo, R.S.; Goldberg, M.B. Impact of either elevated or decreased levels of cytochrome bd expression on Shigella flexneri virulence. J. Bacteriol. 1999, 181, 1229–1237. [Google Scholar] [PubMed]
- Endley, S.; McMurray, D.; Ficht, T.A. Interruption of the cydB locus in Brucella abortus attenuates intracellular survival and virulence in the mouse model of infection. J. Bacteriol. 2001, 183, 2454–2462. [Google Scholar] [CrossRef] [PubMed]
- Turner, A.K.; Barber, L.Z.; Wigley, P.; Muhammad, S.; Jones, M.A.; Lovell, M.A.; Hulme, S.; Barrow, P.A. Contribution of proton-translocating proteins to the virulence of Salmonella enterica serovars Typhimurium, Gallinarum, and Dublin in chickens and mice. Infect. Immun. 2003, 71, 3392–3401. [Google Scholar] [CrossRef] [PubMed]
- Meunier, B.; Madgwick, S.A.; Reil, E.; Oettmeier, W.; Rich, P.R. New inhibitors of the quinol oxidation sites of bacterial cytochromes bo and bd. Biochemistry 1995, 34, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Li, X.W.; Herrmann, J.; Zang, Y.; Grellier, P.; Prado, S.; Muller, R.; Nay, B. Synthesis and biological activities of the respiratory chain inhibitor aurachin D and new ring versus chain analogues. Beilstein J. Org. Chem. 2013, 9, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Von Jagow, G.; Ljungdahl, P.O.; Graf, P.; Ohnishi, T.; Trumpower, B.L. An inhibitor of mitochondrial respiration which binds to cytochrome b and displaces quinone from the iron-sulfur protein of the cytochrome bc1 complex. J. Biol. Chem. 1984, 259, 6318–6326. [Google Scholar] [PubMed]
- Thierbach, G.; Reichenbach, H. Myxothiazol, a new antibiotic interfering with respiration. Antimicrob. Agents Chemother. 1981, 19, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Von Jagow, G.; Gribble, G.W.; Trumpower, B.L. Mucidin and strobilurin A are identical and inhibit electron transfer in the cytochrome bc1 complex of the mitochondrial respiratory chain at the same site as myxothiazol. Biochemistry 1986, 25, 775–780. [Google Scholar] [CrossRef] [PubMed]
- Kucera, I.; Hedbavny, R.; Dadak, V. Separate binding sites for antimycin and mucidin in the respiratory chain of the bacterium Paracoccus denitrificans and their occurrence in other denitrificans bacteria. Biochem. J. 1988, 252, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Kunze, B.; Kemmer, T.; Hofle, G.; Reichenbach, H. Stigmatellin, a new antibiotic from Stigmatella aurantiaca (Myxobacterales). I. Production, physico-chemical and biological properties. J. Antibiot. 1984, 37, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Thierbach, G.; Kunze, B.; Reichenbach, H.; Höfle, G. The mode of action of stigmatellin, a new inhibitor of the cytochrome b-c1 segment of the respiratory chain. Biochim. Biophys. Acta 1984, 765, 227–235. [Google Scholar] [CrossRef]
- Mak, P.A.; Rao, S.P.; Ping Tan, M.; Lin, X.; Chyba, J.; Tay, J.; Ng, S.H.; Tan, B.H.; Cherian, J.; Duraiswamy, J.; et al. A high-throughput screen to identify inhibitors of ATP homeostasis in non-replicating Mycobacterium tuberculosis. ACS Chem. Biol. 2012, 7, 1190–1197. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Cox, J.A.; Spivey, V.L.; Loman, N.J.; Pallen, M.J.; Constantinidou, C.; Fernandez, R.; Alemparte, C.; Remuinan, M.J.; Barros, D.; et al. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS ONE 2012, 7, e52951. [Google Scholar] [CrossRef] [PubMed]
- Ananthan, S.; Faaleolea, E.R.; Goldman, R.C.; Hobrath, J.V.; Kwong, C.D.; Laughon, B.E.; Maddry, J.A.; Mehta, A.; Rasmussen, L.; Reynolds, R.C.; et al. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis 2009, 89, 334–353. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekera, N.S.; Alling, T.; Bailey, M.A.; Files, M.; Early, J.V.; Ollinger, J.; Ovechkina, Y.; Masquelin, T.; Desai, P.V.; Cramer, J.W.; et al. Identification of Phenoxyalkylbenzimidazoles with Antitubercular Activity. J. Med. Chem. 2015, 58, 7273–7285. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekera, N.S.; Berube, B.J.; Shetye, G.; Chettiar, S.; O'Malley, T.; Manning, A.; Flint, L.; Awasthi, D.; Ioerger, T.R.; Sacchettini, J.; et al. Improved Phenoxyalkylbenzimidazoles with Activity against Mycobacterium tuberculosis Appear to Target QcrB. ACS Infect. Dis. 2017, 3, 898–916. [Google Scholar] [CrossRef] [PubMed]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Kim, R.Y.; Seo, M.J.; Lee, S.; Kim, Y.M.; Seo, M.; Seo, J.J.; Ko, Y.; Choi, I.; Jang, J.; et al. Lead optimization of a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug-resistant anti-tuberculosis agent. J. Med. Chem. 2014, 57, 5293–5305. [Google Scholar] [CrossRef] [PubMed]
- Katritzky, A.R.; Xu, Y.J.; Tu, H. Regiospecific synthesis of 3-substituted imidazo[1,2-a]pyridines, imidazo[1,2-a]pyrimidines, and imidazo[1,2-c]pyrimidine. J. Org. Chem. 2003, 68, 4935–4937. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Seeger, N.; Miller, P.A.; Oliver, A.G.; Boshoff, H.I.; Cho, S.; Mulugeta, S.; Anderson, J.R.; Franzblau, S.G.; Miller, M.J. Arrival of Imidazo[2,1-b]thiazole-5-carboxamides: Potent Anti-tuberculosis Agents That Target QcrB. ACS Infect. Dis. 2016, 2, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Cheng, Y.; Cho, S.; Cramer, J.W.; Godfrey, A.; Masquelin, T.; Franzblau, S.G.; Miller, M.J.; Schorey, J. Imidazo[1,2-a]Pyridine-3-Carboxamides Are Active Antimicrobial Agents against Mycobacterium avium Infection In Vivo. Antimicrob. Agents Chemother. 2016, 60, 5018–5022. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, U.H.; Smith, P.W. Perspective: Challenges and opportunities in TB drug discovery from phenotypic screening. Bioorgan. Med. Chem. 2015, 23, 5087–5097. [Google Scholar] [CrossRef] [PubMed]
- Van der Westhuyzen, R.; Winks, S.; Wilson, C.R.; Boyle, G.A.; Gessner, R.K.; Soares de Melo, C.; Taylor, D.; de Kock, C.; Njoroge, M.; Brunschwig, C.; et al. Pyrrolo[3,4-c]pyridine-1,3(2H)-diones: A Novel Antimycobacterial Class Targeting Mycobacterial Respiration. J. Med. Chem. 2015, 58, 9371–9381. [Google Scholar] [CrossRef] [PubMed]
- Rybniker, J.; Vocat, A.; Sala, C.; Busso, P.; Pojer, F.; Benjak, A.; Cole, S.T. Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat. Commun. 2015, 6, 7659. [Google Scholar] [CrossRef] [PubMed]
- Esser, L.; Elberry, M.; Zhou, F.; Yu, C.A.; Yu, L.; Xia, D. Inhibitor-complexed structures of the cytochrome bc1 from the photosynthetic bacterium Rhodobacter sphaeroides. J. Biol. Chem. 2008, 283, 2846–2857. [Google Scholar] [CrossRef] [PubMed]
- Mdanda, S.; Baijnath, S.; Shobo, A.; Singh, S.D.; Maguire, G.E.M.; Kruger, H.G.; Arvidsson, P.I.; Naicker, T.; Govender, T. Lansoprazole-sulfide, pharmacokinetics of this promising anti-tuberculous agent. Biomed. Chromatogr. BMC 2017, 31. [Google Scholar] [CrossRef] [PubMed]
- Yates, T.A.; Tomlinson, L.A.; Bhaskaran, K.; Langan, S.; Thomas, S.; Smeeth, L.; Douglas, I.J. Lansoprazole use and tuberculosis incidence in the United Kingdom Clinical Practice Research Datalink: A population based cohort. PLoS Med. 2017, 14, e1002457. [Google Scholar] [CrossRef] [PubMed]
- Kalia, N.P.; Hasenoehrl, E.J.; Ab Rahman, N.B.; Koh, V.H.; Ang, M.L.T.; Sajorda, D.R.; Hards, K.; Gruber, G.; Alonso, S.; Cook, G.M.; et al. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc. Natl. Acad. Sci. USA 2017, 114, 7426–7431. [Google Scholar] [CrossRef] [PubMed]
- Moosa, A.; Lamprecht, D.A.; Arora, K.; Barry, C.E., 3rd; Boshoff, H.I.M.; Ioerger, T.R.; Steyn, A.J.C.; Mizrahi, V.; Warner, D.F. Susceptibility of Mycobacterium tuberculosis Cytochrome bd Oxidase Mutants to Compounds Targeting the Terminal Respiratory Oxidase, Cytochrome c. Antimicrob. Agents Chemother. 2017, 61, e01338-17. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, P.E.; Von Groll, A.; Martin, A.; Palomino, J.C. Efflux as a mechanism for drug resistance in Mycobacterium tuberculosis. FEMS Immunol. Med. Microbiol. 2011, 63, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Kim, R.; Woo, M.; Jeong, J.; Park, D.E.; Kim, G.; Delorme, V. Efflux Attenuates the Antibacterial Activity of Q203 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Laubinger, W.; Dimroth, P. Characterization of the ATP synthase of Propionigenium modestum as a primary sodium pump. Biochemistry 1988, 27, 7531–7537. [Google Scholar] [CrossRef] [PubMed]
- Reidlinger, J.; Muller, V. Purification of ATP synthase from Acetobacterium woodii and identification as a Na(+)-translocating F1F0-type enzyme. Eur. J. Biochem. 1994, 223, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Foster, D.L.; Fillingame, R.H. Stoichiometry of subunits in the H+-ATPase complex of Escherichia coli. J. Biol. Chem. 1982, 257, 2009–2015. [Google Scholar] [PubMed]
- Stock, D.; Leslie, A.G.; Walker, J.E. Molecular architecture of the rotary motor in ATP synthase. Science 1999, 286, 1700–1705. [Google Scholar] [CrossRef] [PubMed]
- Sambongi, Y.; Iko, Y.; Tanabe, M.; Omote, H.; Iwamoto-Kihara, A.; Ueda, I.; Yanagida, T.; Wada, Y.; Futai, M. Mechanical rotation of the c subunit oligomer in ATP synthase (F0F1): Direct observation. Science 1999, 286, 1722–1724. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Ueno, H.; Mitome, N.; Suzuki, J.; Yoshida, M. F(0) of ATP synthase is a rotary proton channel. Obligatory coupling of proton translocation with rotation of c-subunit ring. J. Biol. Chem. 2002, 277, 13281–13285. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Farhana, A.; Guidry, L.; Saini, V.; Hondalus, M.; Steyn, A.J. Redox homeostasis in mycobacteria: The key to tuberculosis control? Expert Rev. Mol. Med. 2011, 13, e39. [Google Scholar] [CrossRef] [PubMed]
- Bhat, S.A.; Singh, N.; Trivedi, A.; Kansal, P.; Gupta, P.; Kumar, A. The mechanism of redox sensing in Mycobacterium tuberculosis. Free Radic. Biol. Med. 2012, 53, 1625–1641. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, A.; Singh, N.; Bhat, S.A.; Gupta, P.; Kumar, A. Redox biology of tuberculosis pathogenesis. Adv. Microb. Physiol. 2012, 60, 263–324. [Google Scholar] [PubMed]
- Tran, S.L.; Cook, G.M. The F1Fo-ATP synthase of Mycobacterium smegmatis is essential for growth. J. Bacteriol. 2005, 187, 5023–5028. [Google Scholar] [CrossRef] [PubMed]
- Wayne, L.G.; Sohaskey, C.D. Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 2001, 55, 139–163. [Google Scholar] [CrossRef] [PubMed]
- Timm, J.; Post, F.A.; Bekker, L.G.; Walther, G.B.; Wainwright, H.C.; Manganelli, R.; Chan, W.T.; Tsenova, L.; Gold, B.; Smith, I.; et al. Differential expression of iron-, carbon-, and oxygen-responsive mycobacterial genes in the lungs of chronically infected mice and tuberculosis patients. Proc. Natl. Acad. Sci. USA 2003, 100, 14321–14326. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, M.; Rao, S.P.S.; Pethe, K.; Dick, T. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology 2010, 156, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Shemesh, M.; Tam, A.; Steinberg, D. Differential gene expression profiling of Streptococcus mutans cultured under biofilm and planktonic conditions. Microbiology 2007, 153, 1307–1317. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, A.; Mavi, P.S.; Bhatt, D.; Kumar, A. Thiol reductive stress induces cellulose-anchored biofilm formation in Mycobacterium tuberculosis. Nat. Commun. 2016, 7, 11392. [Google Scholar] [CrossRef] [PubMed]
- Maglica, Z.; Ozdemir, E.; McKinney, J.D. Single-cell tracking reveals antibiotic-induced changes in mycobacterial energy metabolism. mBio 2015, 6, e02236-14. [Google Scholar] [CrossRef] [PubMed]
- Sala, C.; Haouz, A.; Saul, F.A.; Miras, I.; Rosenkrands, I.; Alzari, P.M.; Cole, S.T. Genome-wide regulon and crystal structure of BlaI (Rv1846c) from Mycobacterium tuberculosis. Mol. Microbiol. 2009, 71, 1102–1116. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Lill, H.; Bald, D. ATP synthase in mycobacteria: Special features and implications for a function as drug target. Biochim. Biophys. Acta 2014, 1837, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Kashket, E.R. Effects of aerobiosis and nitrogen source on the proton motive force in growing Escherichia coli and Klebsiella pneumoniae cells. J. Bacteriol. 1981, 146, 377–384. [Google Scholar] [PubMed]
- Haagsma, A.C.; Driessen, N.N.; Hahn, M.M.; Lill, H.; Bald, D. ATP synthase in slow- and fast-growing mycobacteria is active in ATP synthesis and blocked in ATP hydrolysis direction. FEMS Microbiol. Lett. 2010, 313, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Higashi, T.; Kalra, V.K.; Lee, S.H.; Bogin, E.; Brodie, A.F. Energy-transducing membrane-bound coupling factor-ATPase from Mycobacterium phlei. I. Purification, homogeneity, and properties. J. Biol. Chem. 1975, 250, 6541–6548. [Google Scholar] [PubMed]
- Von Ballmoos, C.; Wiedenmann, A.; Dimroth, P. Essentials for ATP synthesis by F1F0 ATP synthases. Annu. Rev. Biochem. 2009, 78, 649–672. [Google Scholar] [CrossRef] [PubMed]
- Ragunathan, P.; Sielaff, H.; Sundararaman, L.; Biukovic, G.; Subramanian Manimekalai, M.S.; Singh, D.; Kundu, S.; Wohland, T.; Frasch, W.; Dick, T.; et al. The uniqueness of subunit alpha of mycobacterial F-ATP synthases: An evolutionary variant for niche adaptation. J. Biol. Chem. 2017, 292, 11262–11279. [Google Scholar] [CrossRef] [PubMed]
- Priya, R.; Biukovic, G.; Manimekalai, M.S.; Lim, J.; Rao, S.P.; Gruber, G. Solution structure of subunit gamma (gamma(1-204)) of the Mycobacterium tuberculosis F-ATP synthase and the unique loop of gamma(165-178), representing a novel TB drug target. J. Bioenerg. Biomembr. 2013, 45, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Hotra, A.; Suter, M.; Biukovic, G.; Ragunathan, P.; Kundu, S.; Dick, T.; Gruber, G. Deletion of a unique loop in the mycobacterial F-ATP synthase gamma subunit sheds light on its inhibitory role in ATP hydrolysis-driven H(+) pumping. FEBS J. 2016, 283, 1947–1961. [Google Scholar] [CrossRef] [PubMed]
- Matthies, D.; Preiss, L.; Klyszejko, A.L.; Muller, D.J.; Cook, G.M.; Vonck, J.; Meier, T. The c13 ring from a thermoalkaliphilic ATP synthase reveals an extended diameter due to a special structural region. J. Mol. Biol. 2009, 388, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Meier, T.; Morgner, N.; Matthies, D.; Pogoryelov, D.; Keis, S.; Cook, G.M.; Dimroth, P.; Brutschy, B. A tridecameric c ring of the adenosine triphosphate (ATP) synthase from the thermoalkaliphilic Bacillus sp. strain TA2.A1 facilitates ATP synthesis at low electrochemical proton potential. Mol. Microbiol. 2007, 65, 1181–1192. [Google Scholar] [CrossRef] [PubMed]
- Hicks, D.B.; Liu, J.; Fujisawa, M.; Krulwich, T.A. F1F0-ATP synthases of alkaliphilic bacteria: Lessons from their adaptations. Biochim. Biophys. Acta 2010, 1797, 1362–1377. [Google Scholar] [CrossRef] [PubMed]
- Preiss, L.; Langer, J.D.; Yildiz, O.; Eckhardt-Strelau, L.; Guillemont, J.E.; Koul, A.; Meier, T. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015, 1, e1500106. [Google Scholar] [CrossRef] [PubMed]
- Beechey, R.B.; Holloway, C.T.; Knight, I.G.; Roberton, A.M. Dicyclohexylcarbodiimide—An inhibitor of oxidative phosphorylation. Biochem. Biophys. Res. Commun. 1966, 23, 75–80. [Google Scholar] [CrossRef]
- Matsuno-Yagi, A.; Hatefi, Y. Studies on the mechanism of oxidative phosphorylation. Different effects of F0 inhibitors on unisite and multisite ATP hydrolysis by bovine submitochondrial particles. J. Biol. Chem. 1993, 268, 1539–1545. [Google Scholar] [PubMed]
- Hong, S.; Pedersen, P.L. ATP synthase and the actions of inhibitors utilized to study its roles in human health, disease, and other scientific areas. Microbiol. Mol. Biol. Rev. MMBR 2008, 72, 590–641. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Gohlmann, H.W.; Neefs, J.M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Janssen Pharmaceuticals, I. Quinoline Derivatives and Their Use as Mycobacterial Inhibitors. Patent EP1527050 A1, 2005. [Google Scholar]
- Koul, A.; Vranckx, L.; Dendouga, N.; Balemans, W.; Van den Wyngaert, I.; Vergauwen, K.; Gohlmann, H.W.; Willebrords, R.; Poncelet, A.; Guillemont, J.; et al. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 2008, 283, 25273–25280. [Google Scholar] [CrossRef] [PubMed]
- Veziris, N.; Ibrahim, M.; Lounis, N.; Andries, K.; Jarlier, V. Sterilizing activity of second-line regimens containing TMC207 in a murine model of tuberculosis. PLoS ONE 2011, 6, e17556. [Google Scholar] [CrossRef] [PubMed]
- Tasneen, R.; Li, S.Y.; Peloquin, C.A.; Taylor, D.; Williams, K.N.; Andries, K.; Mdluli, K.E.; Nuermberger, E.L. Sterilizing activity of novel TMC207- and PA-824-containing regimens in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2011, 55, 5485–5492. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Minkowski, A.; Amoabeng, O.; Peloquin, C.A.; Taylor, D.; Andries, K.; Wallis, R.S.; Mdluli, K.E.; Nuermberger, E.L. Sterilizing activities of novel combinations lacking first- and second-line drugs in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 3114–3120. [Google Scholar] [CrossRef] [PubMed]
- Diacon, A.H.; Pym, A.; Grobusch, M.P.; de los Rios, J.M.; Gotuzzo, E.; Vasilyeva, I.; Leimane, V.; Andries, K.; Bakare, N.; De Marez, T.; et al. Multidrug-resistant tuberculosis and culture conversion with bedaquiline. N. Engl. J. Med. 2014, 371, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Diacon, A.H.; Pym, A.; Grobusch, M.; Patientia, R.; Rustomjee, R.; Page-Shipp, L.; Pistorius, C.; Krause, R.; Bogoshi, M.; Churchyard, G.; et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Engl. J. Med. 2009, 360, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef] [PubMed]
- Haagsma, A.C.; Abdillahi-Ibrahim, R.; Wagner, M.J.; Krab, K.; Vergauwen, K.; Guillemont, J.; Andries, K.; Lill, H.; Koul, A.; Bald, D. Selectivity of TMC207 towards mycobacterial ATP synthase compared with that towards the eukaryotic homologue. Antimicrob. Agents Chemother. 2009, 53, 1290–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vesenbeckh, S.; Schonfeld, N.; Roth, A.; Bettermann, G.; Krieger, D.; Bauer, T.T.; Russmann, H.; Mauch, H. Bedaquiline as a potential agent in the treatment of Mycobacterium abscessus infections. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef] [PubMed]
- Vesenbeckh, S.; Schonfeld, N.; Krieger, D.; Bettermann, G.; Bauer, T.T.; Russmann, H.; Mauch, H. Bedaquiline as a potential agent in the treatment of M. intracellulare and M. avium infections. Eur. Respir. J. 2017, 49. [Google Scholar] [CrossRef]
- Dupont, C.; Viljoen, A.; Thomas, S.; Roquet-Baneres, F.; Herrmann, J.L.; Pethe, K.; Kremer, L. Bedaquiline Inhibits the ATP Synthase in Mycobacterium abscessus and Is Effective in Infected Zebrafish. Antimicrob. Agents Chemother. 2017, 61, e01225-17. [Google Scholar] [CrossRef] [PubMed]
- Gelber, R.; Andries, K.; Paredes, R.M.; Andaya, C.E.; Burgos, J. The diarylquinoline R207910 is bactericidal against Mycobacterium leprae in mice at low dose and administered intermittently. Antimicrob. Agents Chemother. 2009, 53, 3989–3991. [Google Scholar] [CrossRef] [PubMed]
- Lounis, N.; Gevers, T.; Van den Berg, J.; Vranckx, L.; Andries, K. ATP synthase inhibition of Mycobacterium avium is not bactericidal. Antimicrob. Agents Chemother. 2009, 53, 4927–4929. [Google Scholar] [CrossRef] [PubMed]
- Lerat, I.; Cambau, E.; Roth Dit Bettoni, R.; Gaillard, J.L.; Jarlier, V.; Truffot, C.; Veziris, N. In vivo evaluation of antibiotic activity against Mycobacterium abscessus. J. Infect. Dis. 2014, 209, 905–912. [Google Scholar] [CrossRef] [PubMed]
- Huitric, E.; Verhasselt, P.; Andries, K.; Hoffner, S.E. In vitro antimycobacterial spectrum of a diarylquinoline ATP synthase inhibitor. Antimicrob. Agents Chemother. 2007, 51, 4202–4204. [Google Scholar] [CrossRef] [PubMed]
- Lounis, N.; Veziris, N.; Chauffour, A.; Truffot-Pernot, C.; Andries, K.; Jarlier, V. Combinations of R207910 with drugs used to treat multidrug-resistant tuberculosis have the potential to shorten treatment duration. Antimicrob. Agents Chemother. 2006, 50, 3543–3547. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Andries, K.; Lounis, N.; Chauffour, A.; Truffot-Pernot, C.; Jarlier, V.; Veziris, N. Synergistic activity of R207910 combined with pyrazinamide against murine tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1011–1015. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.M.; Einck, L.; Andries, K.; Nacy, C.A. In vitro interactions between new antitubercular drug candidates SQ109 and TMC207. Antimicrob. Agents Chemother. 2010, 54, 2840–2846. [Google Scholar] [CrossRef] [PubMed]
- Lechartier, B.; Hartkoorn, R.C.; Cole, S.T. In vitro combination studies of benzothiazinone lead compound BTZ043 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 5790–5793. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Manina, G.; Mikusova, K.; Mollmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.J.; Lo, J.H. Delamanid: First global approval. Drugs 2014, 74, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.M.; Murray, S.; Karlsson, M.O.; Dooley, K.E. Rifampicin and rifapentine significantly reduce concentrations of bedaquiline, a new anti-TB drug. J. Antimicrob. Chemother. 2015, 70, 1106–1114. [Google Scholar] [CrossRef] [PubMed]
- Van Heeswijk, R.P.; Dannemann, B.; Hoetelmans, R.M. Bedaquiline: A review of human pharmacokinetics and drug-drug interactions. J. Antimicrob. Chemother. 2014, 69, 2310–2318. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Li, F.; Lu, J.; Liu, S.; Dorko, K.; Xie, W.; Ma, X. Bedaquiline metabolism: Enzymes and novel metabolites. Drug Metab. Dispos. Biol. Fate Chem. 2014, 42, 863–866. [Google Scholar] [CrossRef] [PubMed]
- Petrella, S.; Cambau, E.; Chauffour, A.; Andries, K.; Jarlier, V.; Sougakoff, W. Genetic basis for natural and acquired resistance to the diarylquinoline R207910 in mycobacteria. Antimicrob. Agents Chemother. 2006, 50, 2853–2856. [Google Scholar] [CrossRef] [PubMed]
- Hartkoorn, R.C.; Uplekar, S.; Cole, S.T. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2979–2981. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Villellas, C.; Coeck, N.; Thys, K.; Gevers, T.; Vranckx, L.; Lounis, N.; de Jong, B.C.; Koul, A. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS ONE 2014, 9, e102135. [Google Scholar] [CrossRef] [PubMed]
- Somoskovi, A.; Bruderer, V.; Homke, R.; Bloemberg, G.V.; Bottger, E.C. A mutation associated with clofazimine and bedaquiline cross-resistance in MDR-TB following bedaquiline treatment. Eur. Respir. J. 2015, 45, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Bloemberg, G.V.; Keller, P.M.; Stucki, D.; Trauner, A.; Borrell, S.; Latshang, T.; Coscolla, M.; Rothe, T.; Homke, R.; Ritter, C.; et al. Acquired Resistance to Bedaquiline and Delamanid in Therapy for Tuberculosis. N. Engl. J. Med. 2015, 373, 1986–1988. [Google Scholar] [CrossRef] [PubMed]
- Tantry, S.J.; Markad, S.D.; Shinde, V.; Bhat, J.; Balakrishnan, G.; Gupta, A.K.; Ambady, A.; Raichurkar, A.; Kedari, C.; Sharma, S.; et al. Discovery of Imidazo[1,2-a]pyridine Ethers and Squaramides as Selective and Potent Inhibitors of Mycobacterial Adenosine Triphosphate (ATP) Synthesis. J. Med. Chem. 2017, 60, 1379–1399. [Google Scholar] [CrossRef] [PubMed]
- Black, P.A.; Warren, R.M.; Louw, G.E.; van Helden, P.D.; Victor, T.C.; Kana, B.D. Energy metabolism and drug efflux in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2491–2503. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Schurig-Briccio, L.A.; Feng, X.; Upadhyay, A.; Pujari, V.; Lechartier, B.; Fontes, F.L.; Yang, H.; Rao, G.; Zhu, W.; et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 2014, 57, 3126–3139. [Google Scholar] [CrossRef] [PubMed]
- Malone, L.; Schurr, A.; Lindh, H.; Mc, K.D.; Kiser, J.S.; Williams, J.H. The effect of pyrazinamide (aldinamide) on experimental tuberculosis in mice. Am. Rev. Tuberc. 1952, 65, 511–518. [Google Scholar] [PubMed]
- Yeager, R.L.; Munroe, W.G.; Dessau, F.I. Pyrazinamide (aldinamide) in the treatment of pulmonary tuberculosis. Am. Rev. Tuberc. 1952, 65, 523–546. [Google Scholar] [PubMed]
- Mc, D.W.; Tompsett, R. Activation of pyrazinamide and nicotinamide in acidic environments in vitro. Am. Rev. Tuberc. 1954, 70, 748–754. [Google Scholar]
- Konno, K.; Nagayama, H.; Oka, S. Nicotinamidase in Mycobacteria: A method for distinguishing bovine type tubercle bacilli from other Mycobacteria. Nature 1959, 184 (Suppl. 22), 1743–1744. [Google Scholar] [CrossRef] [PubMed]
- Miotto, P.; Cabibbe, A.M.; Feuerriegel, S.; Casali, N.; Drobniewski, F.; Rodionova, Y.; Bakonyte, D.; Stakenas, P.; Pimkina, E.; Augustynowicz-Kopec, E.; et al. Mycobacterium tuberculosis pyrazinamide resistance determinants: A multicenter study. mBio 2014, 5, e01819-14. [Google Scholar] [CrossRef] [PubMed]
- Sheen, P.; Requena, D.; Gushiken, E.; Gilman, R.H.; Antiparra, R.; Lucero, B.; Lizarraga, P.; Cieza, B.; Roncal, E.; Grandjean, L.; et al. A multiple genome analysis of Mycobacterium tuberculosis reveals specific novel genes and mutations associated with pyrazinamide resistance. BMC Genom. 2017, 18, 769. [Google Scholar] [CrossRef] [PubMed]
- Scorpio, A.; Zhang, Y. Mutations in pncA, a gene encoding pyrazinamidase/nicotinamidase, cause resistance to the antituberculous drug pyrazinamide in tubercle bacillus. Nat. Med. 1996, 2, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Haagsma, A.C.; Pham, H.; Maaskant, J.J.; Mol, S.; Lill, H.; Bald, D. Pyrazinoic acid decreases the proton motive force, respiratory ATP synthesis activity, and cellular ATP levels. Antimicrob. Agents Chemother. 2011, 55, 5354–5357. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wade, M.M.; Scorpio, A.; Zhang, H.; Sun, Z. Mode of action of pyrazinamide: Disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Njire, M.; Wang, N.; Wang, B.; Tan, Y.; Cai, X.; Liu, Y.; Mugweru, J.; Guo, J.; Hameed, H.M.A.; Tan, S.; et al. Pyrazinoic Acid Inhibits a Bifunctional Enzyme in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e00070-17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Scorpio, A.; Nikaido, H.; Sun, Z. Role of acid pH and deficient efflux of pyrazinoic acid in unique susceptibility of Mycobacterium tuberculosis to pyrazinamide. J. Bacteriol. 1999, 181, 2044–2049. [Google Scholar] [PubMed]
- Zhang, Y.; Permar, S.; Sun, Z. Conditions that may affect the results of susceptibility testing of Mycobacterium tuberculosis to pyrazinamide. J. Med. Microbiol. 2002, 51, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mitchison, D. The curious characteristics of pyrazinamide: A review. Int. J. Tuberc. Lung Dis. 2003, 7, 6–21. [Google Scholar] [PubMed]
- Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, N.; Dawson, R.; du Bois, J.; Narunsky, K.; Horwith, G.; Phipps, A.J.; Nacy, C.A.; Aarnoutse, R.E.; Boeree, M.J.; Gillespie, S.H.; et al. Early phase evaluation of SQ109 alone and in combination with rifampicin in pulmonary TB patients. J. Antimicrob. Chemother. 2015, 70, 1558–1566. [Google Scholar] [CrossRef] [PubMed]
- Boeree, M.J.; Heinrich, N.; Aarnoutse, R.; Diacon, A.H.; Dawson, R.; Rehal, S.; Kibiki, G.S.; Churchyard, G.; Sanne, I.; Ntinginya, N.E.; et al. High-dose rifampicin, moxifloxacin, and SQ109 for treating tuberculosis: A multi-arm, multi-stage randomised controlled trial. Lancet Infect. Dis. 2017, 17, 39–49. [Google Scholar] [CrossRef]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E., 3rd; et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzewicz, A.E.; Pham, H.; Gundi, V.A.; Scherman, M.S.; North, E.J.; Hess, T.; Jones, V.; Gruppo, V.; Born, S.E.; Kordulakova, J.; et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 2012, 8, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Makobongo, M.O.; Einck, L.; Peek, R.M., Jr.; Merrell, D.S. In vitro characterization of the anti-bacterial activity of SQ109 against Helicobacter pylori. PLoS ONE 2013, 8, e68917. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Santos, P.; Li, K.; Lameira, L.; de Carvalho, T.M.; Huang, G.; Galizzi, M.; Shang, N.; Li, Q.; Gonzalez-Pacanowska, D.; Hernandez-Rodriguez, V.; et al. SQ109, a new drug lead for Chagas disease. Antimicrob. Agents Chemother. 2015, 59, 1950–1961. [Google Scholar] [CrossRef] [PubMed]
- Sacksteder, K.A.; Protopopova, M.; Barry, C.E.; Andries, K.; Nacy, C.A. Discovery and development of sq109: A new antitubercular drug with a novel mechanism of action. Future Microbiol. 2012, 7, 823–837. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Mycobacterial menaquinone biosynthesis pathway and its inhibitors. (A–H) represent chorismate, isochorismate, 2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate, o-succinylbenzoate, o-succinylbenzoyl-CoA, 1,4-dihydroxy-2-naphthoyl CoA, demethylmenaquinone, and menaquinone respectively. Drugs that target the menaquinone biosynthesis enzymes are shown by red flathead arrows.
Figure 1.
Mycobacterial menaquinone biosynthesis pathway and its inhibitors. (A–H) represent chorismate, isochorismate, 2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate, o-succinylbenzoate, o-succinylbenzoyl-CoA, 1,4-dihydroxy-2-naphthoyl CoA, demethylmenaquinone, and menaquinone respectively. Drugs that target the menaquinone biosynthesis enzymes are shown by red flathead arrows.
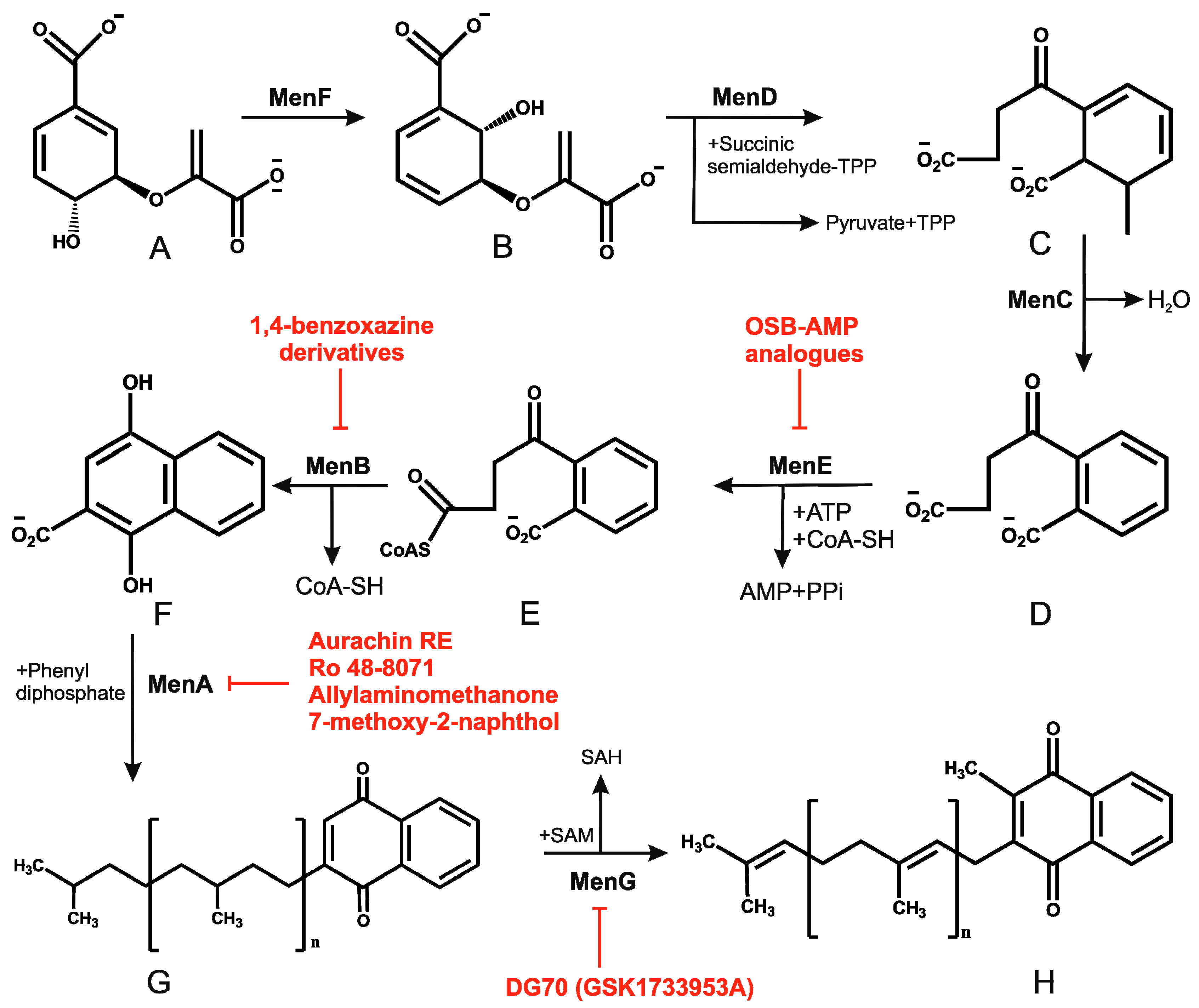
Figure 2.
Inhibition of cytochrome bc1 by LPZ and Q203. The sulfoxide reduction of LPZ converts it to active LPZS, which can bind QcrB of cytochrome bc1 complex. Q203, an imidazopyridine amide, also targets the QcrB subunit of cytochrome bc1 complex. Inhibition of QcrB forces the mycobacteria to use energetically less efficient cytochrome bd oxidase, a decrease in proton motive force (PMF) and ATP levels. Red flathead arrows indicate binding with subunit and inhibition of cytochrome bc1 complex.
Figure 2.
Inhibition of cytochrome bc1 by LPZ and Q203. The sulfoxide reduction of LPZ converts it to active LPZS, which can bind QcrB of cytochrome bc1 complex. Q203, an imidazopyridine amide, also targets the QcrB subunit of cytochrome bc1 complex. Inhibition of QcrB forces the mycobacteria to use energetically less efficient cytochrome bd oxidase, a decrease in proton motive force (PMF) and ATP levels. Red flathead arrows indicate binding with subunit and inhibition of cytochrome bc1 complex.
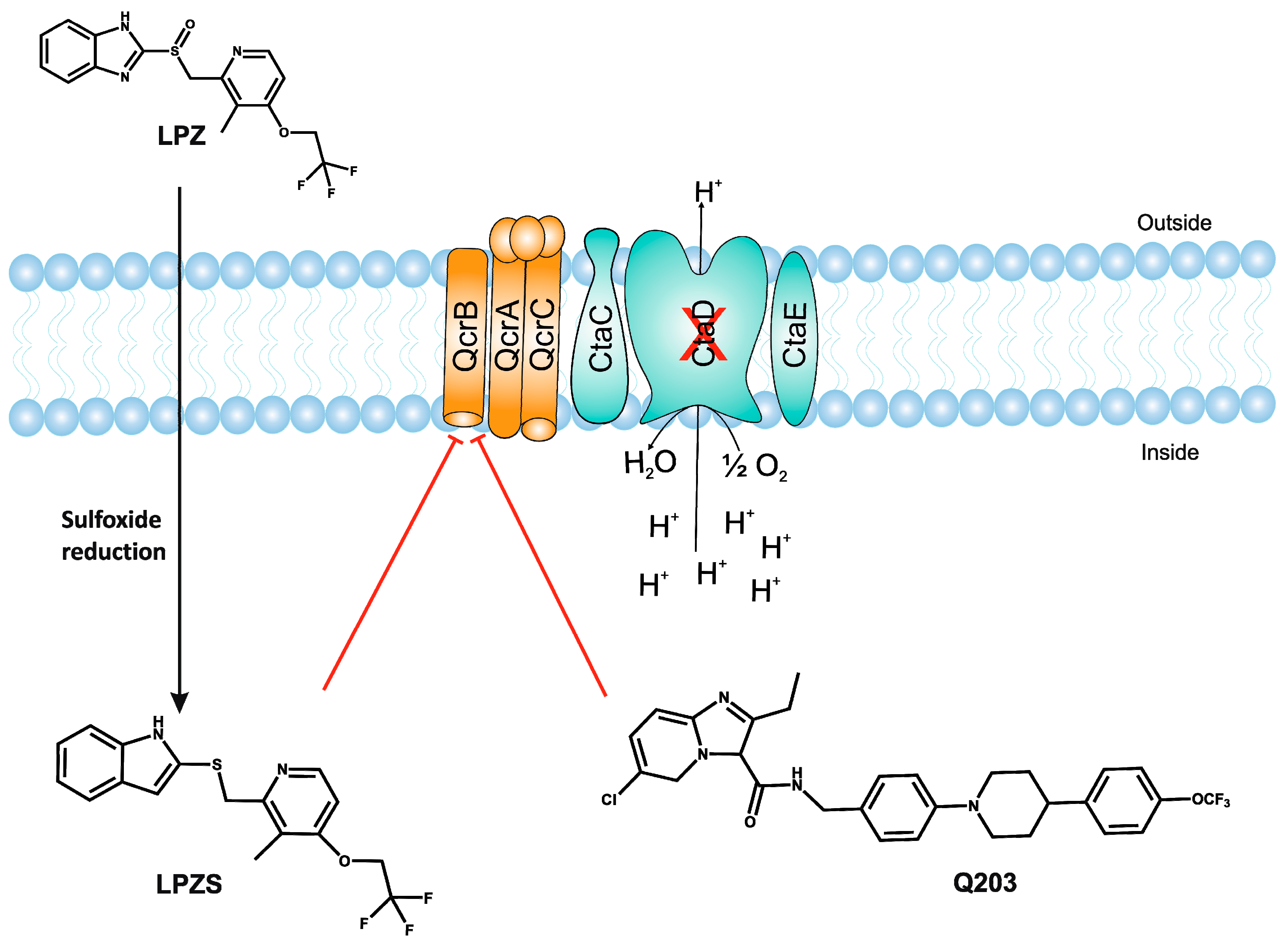
Figure 3.
Schematic view of ATP synthase and its interaction with bedaquiline (BDQ). (A) Depicts the top view of C-ring of ATP synthase Fo complex and comparison of C-rings of mitochondria (C10), E. coli (C8) and mycobacteria (C9). (B) BDQ binds between the two c subunits of the C-ring. The interaction of BDQ with C ring is illustrated in the zoomed region. BDQ specifically interacts with Glu65 (E65), Phe69 (F69), Leu63 (L63), Asp32 (D32), and Ile70 (I70) of adjacent c subunits.
Figure 3.
Schematic view of ATP synthase and its interaction with bedaquiline (BDQ). (A) Depicts the top view of C-ring of ATP synthase Fo complex and comparison of C-rings of mitochondria (C10), E. coli (C8) and mycobacteria (C9). (B) BDQ binds between the two c subunits of the C-ring. The interaction of BDQ with C ring is illustrated in the zoomed region. BDQ specifically interacts with Glu65 (E65), Phe69 (F69), Leu63 (L63), Asp32 (D32), and Ile70 (I70) of adjacent c subunits.

Figure 4.
Schematic representation of the mycobacterial electron transport chain and its inhibitors. NADH derived via glycolysis and tricarboxylic acid (TCA) cycle feds electrons into the electron transport chain by NADH dehydrogenase. The menaquinone (MK) pool can be reduced by primary dehydrogenases such as NADH dehydrogenases (NDH1 and NDH2) and via succinate dehydrogenase (SDH). Electrons from the menaquinone pool are accepted directly by cytochrome bd-type terminal oxidase or via bc1-aa3 supercomplex. A proton motive force (PMF) is generated during electron transport chain because of pumping of protons across the membrane. This PMF is used by the ATP synthase to generate ATP. Drugs that target the oxidative phosphorylation are shown by red flathead arrows. Abbreviations: CFZ, clofazimine; TPZ, trifluoperazine; QPs, quinolinyl pyrimidines; Q203, imidazopyridine amide; LPZ, lansoprazole; Ro 48-8071, (4-bromophenyl)[2-fluoro-4-[[6-(methyl-2-propenylamino)hexyl]oxy]phenyl]-methanone; DG70,biphenyl amide; BDQ, bedaquiline; SQR, squaramide; SQ109, N-adamantan-2-yl-N-((E)-3,7-dimethyl-octa-2,6-dienyl)-ethane-1,2-diamine; PYZ, pyrazinamide. Red arrows in the TCA cycle depict the glyoxylate shunt.
Figure 4.
Schematic representation of the mycobacterial electron transport chain and its inhibitors. NADH derived via glycolysis and tricarboxylic acid (TCA) cycle feds electrons into the electron transport chain by NADH dehydrogenase. The menaquinone (MK) pool can be reduced by primary dehydrogenases such as NADH dehydrogenases (NDH1 and NDH2) and via succinate dehydrogenase (SDH). Electrons from the menaquinone pool are accepted directly by cytochrome bd-type terminal oxidase or via bc1-aa3 supercomplex. A proton motive force (PMF) is generated during electron transport chain because of pumping of protons across the membrane. This PMF is used by the ATP synthase to generate ATP. Drugs that target the oxidative phosphorylation are shown by red flathead arrows. Abbreviations: CFZ, clofazimine; TPZ, trifluoperazine; QPs, quinolinyl pyrimidines; Q203, imidazopyridine amide; LPZ, lansoprazole; Ro 48-8071, (4-bromophenyl)[2-fluoro-4-[[6-(methyl-2-propenylamino)hexyl]oxy]phenyl]-methanone; DG70,biphenyl amide; BDQ, bedaquiline; SQR, squaramide; SQ109, N-adamantan-2-yl-N-((E)-3,7-dimethyl-octa-2,6-dienyl)-ethane-1,2-diamine; PYZ, pyrazinamide. Red arrows in the TCA cycle depict the glyoxylate shunt.
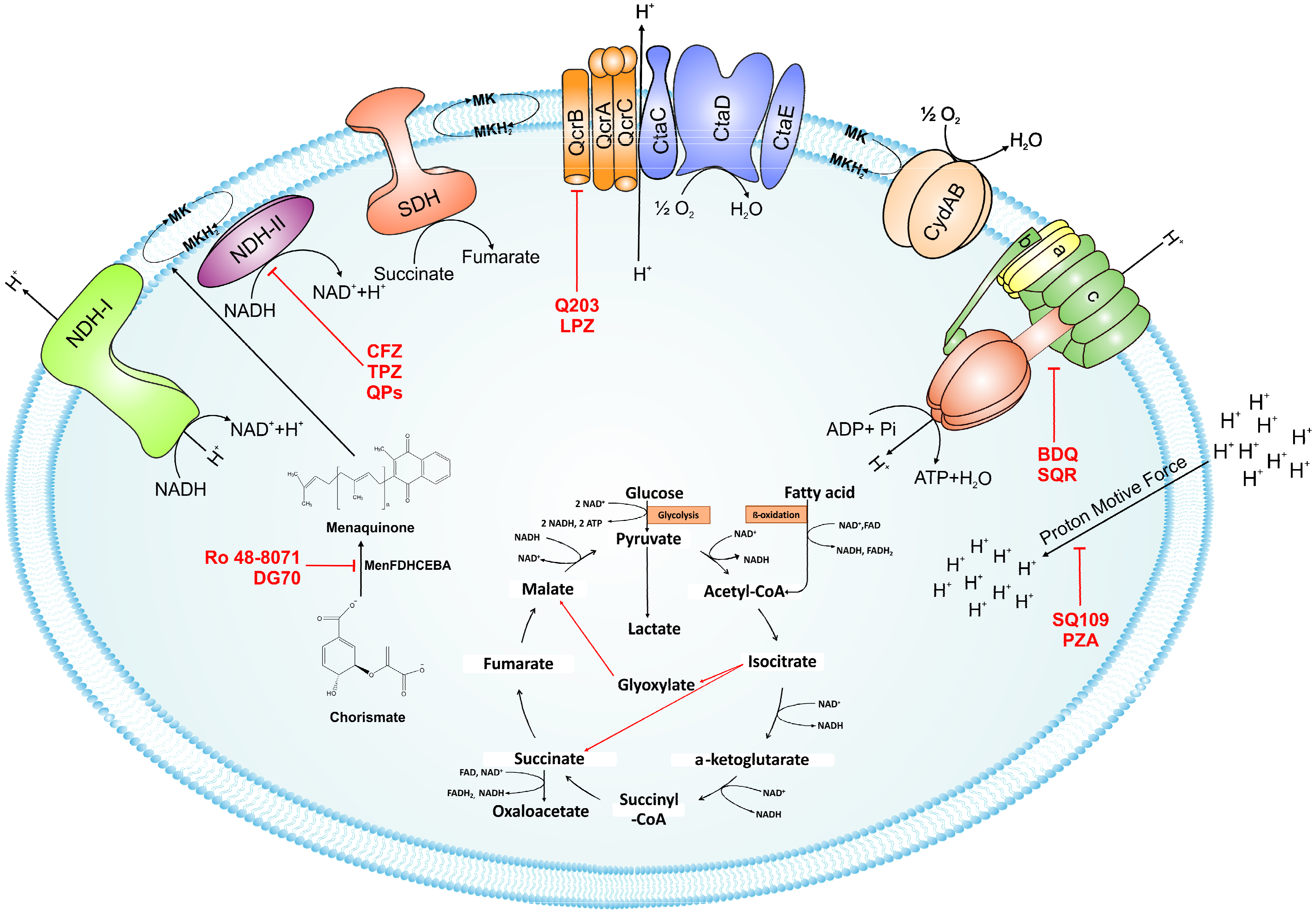

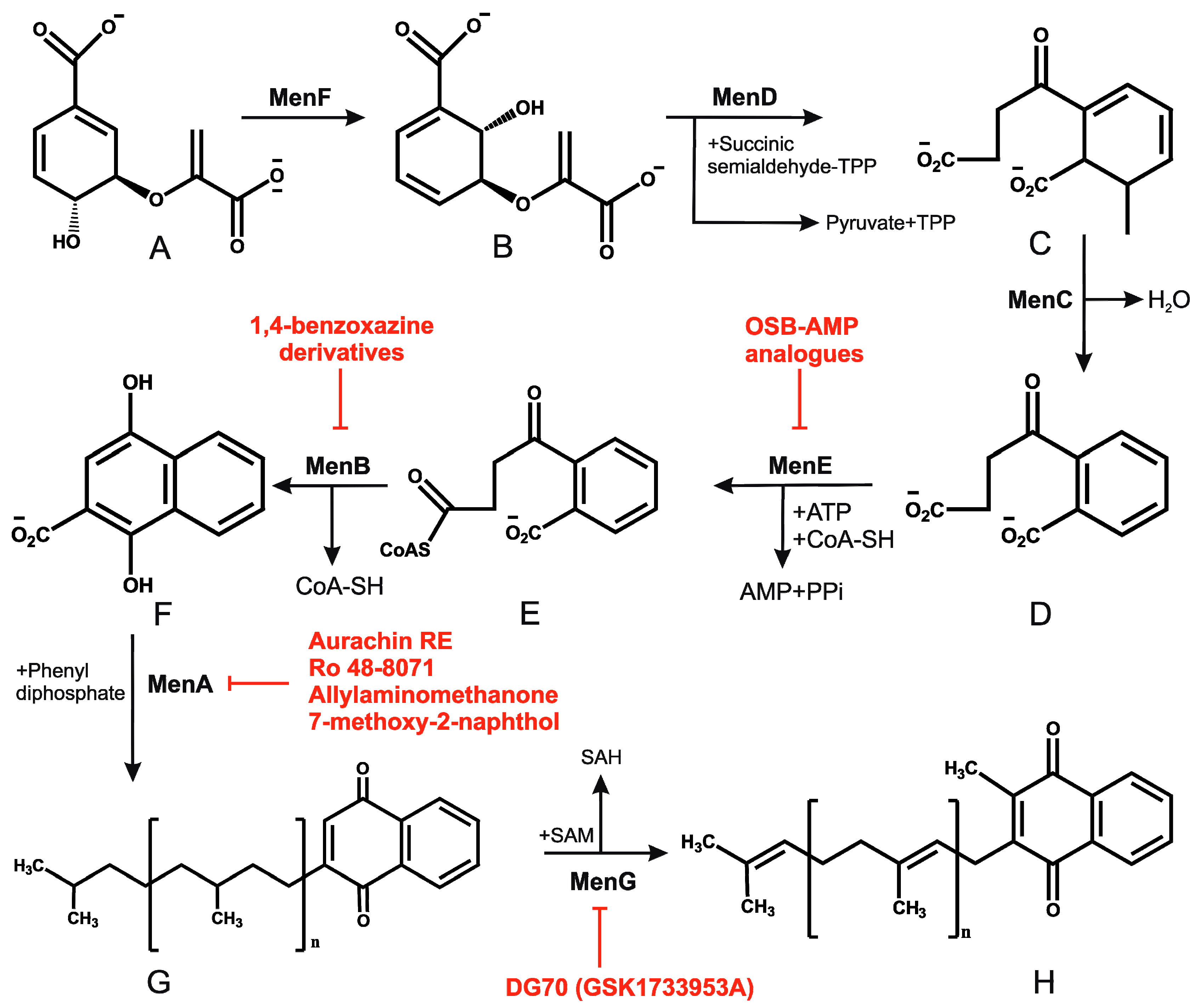{kind=link}
{kind=link}
{kind=link}
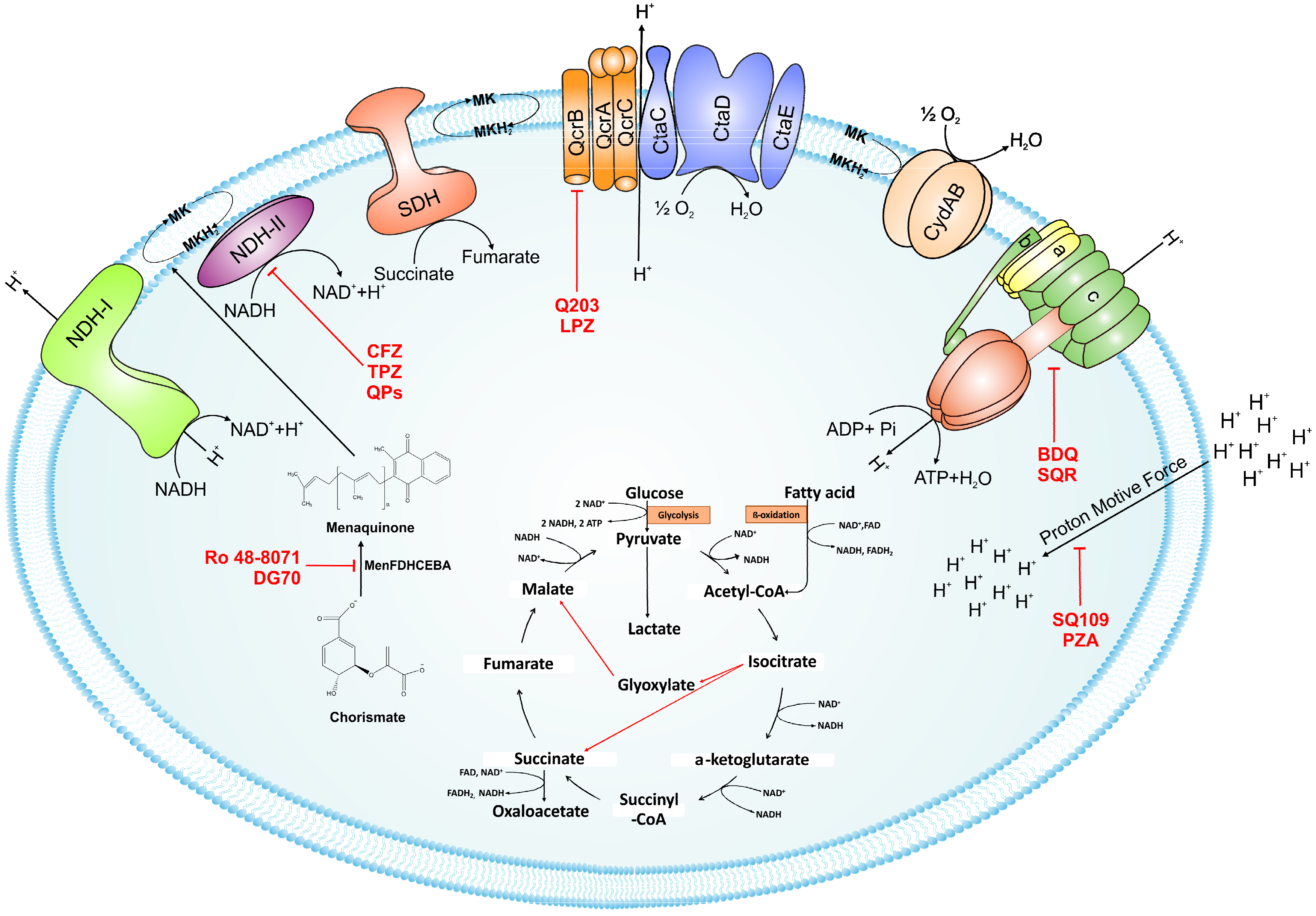{kind=link}
Table 1.
Summary of potential drug molecules with their respective targets, bactericidal properties, and structures.
Table 1.
Summary of potential drug molecules with their respective targets, bactericidal properties, and structures.
| Drug | Target | Properties | Structure |
|---|---|---|---|
| Thioridazine | NDH-2 | MIC90-8–15 µg/mL Approved |  |
| Trifluoperazine | NDH-2 | MIC90-19.2 µg/mL |  |
| Clofazimine | NDH-2 | MIC90-0.25 µg/mL Phase III clinical trials |  |
| Quinolinyl pyrimidine | NDH-2 | MIC90-8-32 µg/mL |  |
| 3-Nitropropionate (3NP) | SDH | MIC90-3.3 µM Active in vivo |  |
| DG70 | MenG | MIC90-4.8 µg/mL |  |
| Aurachin RE | MenA | MIC90 < 12.5 µg/mL |  |
| Ro 48-8071 | MenA | MIC90-3 µg/mL |  |
| 7-Methoxy-2-naphthol | MenA | MIC90-3 µg/mL |  |
| Allylaminomethanone | MenA | MIC90-1 µg/mL |  |
| Aurachin D | Cytochrome bd oxidase | MIC90-85 µM |  |
| PABS (Phenoxyalkylbenzimidazoles) | QcrB | MIC90-0.056 µM |  |
| Q203 (imidazopyridine amide) | QcrB | MIC50-0.28 nM ex vivo MIC50-2.7 nM in vitro Phase I clinical trials |  |
| Pyrrolo[3,4-c]pyridine-1,3-dione | QcrB |  | |
| Lansoprazole | QcrB | MIC90-104 µg/mL Active in vivo |  |
| Bedaquiline | ATP synthase | MIC90-0.004-0.13 µg/mL Approved for MDR-TB Phase III clinical trials for DS-TB |  |
| Squaramide | ATP synthase | MIC90-0.5 µM Active in vivo Pre-clinical trials |  |
| SQ109 | PMF | MIC90-0.78 µg/mL Phase II clinical trials |  |
| Pyrazinamide | PMF | MIC90-100 µg/mL Approved |  |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Iqbal, I.K.; Bajeli, S.; Akela, A.K.; Kumar, A. Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery. Pathogens 2018, 7, 24. https://doi.org/10.3390/pathogens7010024
AMA Style
Iqbal IK, Bajeli S, Akela AK, Kumar A. Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery. Pathogens. 2018; 7(1):24. https://doi.org/10.3390/pathogens7010024
Chicago/Turabian StyleIqbal, Iram Khan, Sapna Bajeli, Ajit Kumar Akela, and Ashwani Kumar. 2018. "Bioenergetics of Mycobacterium: An Emerging Landscape for Drug Discovery" Pathogens 7, no. 1: 24. https://doi.org/10.3390/pathogens7010024
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.