Messing with the Sentinels—The Interaction of Staphylococcus aureus with Dendritic Cells


{kind=link}
Abstract
:1. Introduction
2. Dendritic Cells
3. Recognition and Uptake of S. aureus by DCs
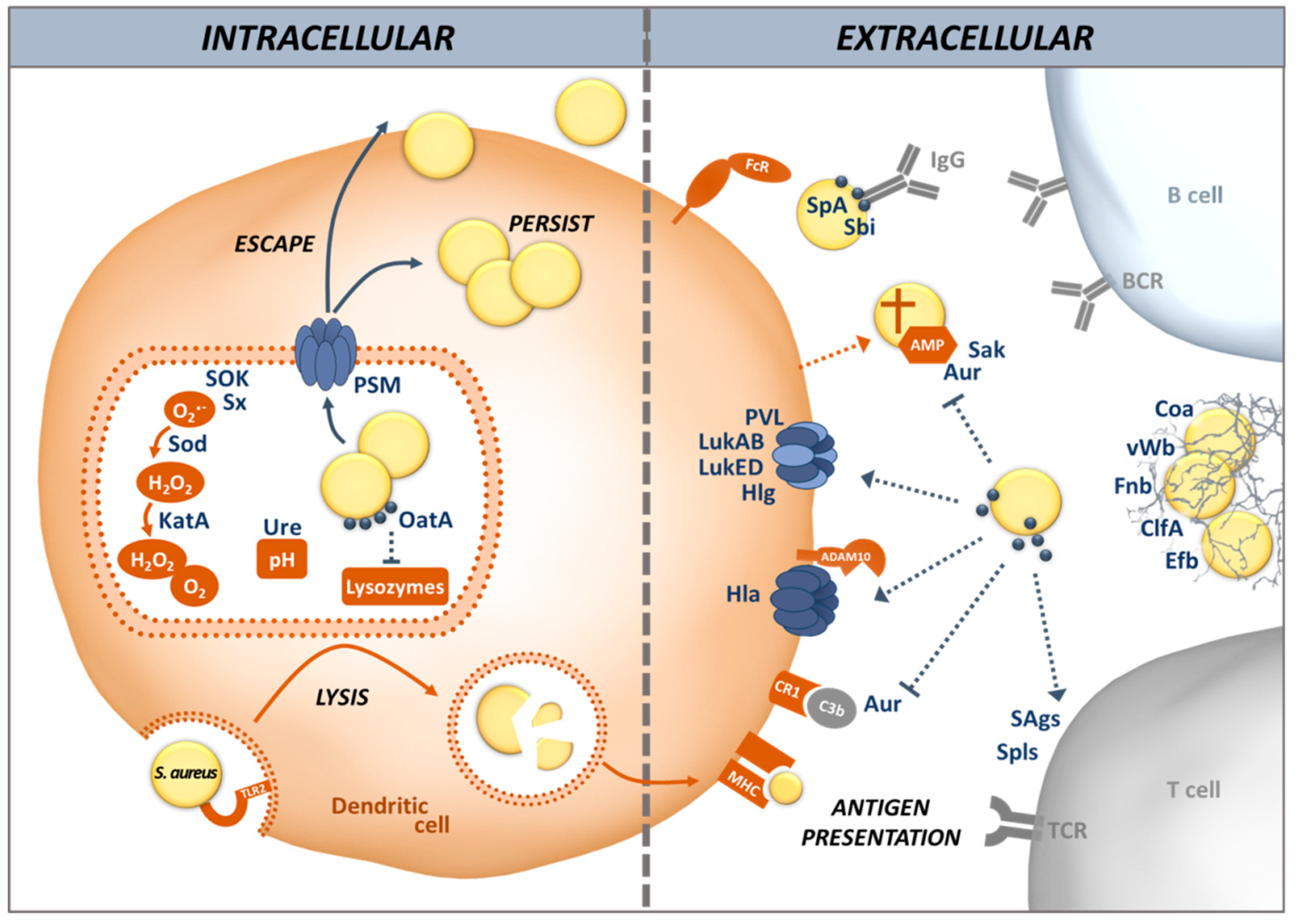
4. S. aureus Evades Killing by DCs as well as Antigen Processing and Presentation
5. Interaction of DCs with S. aureus in the Respiratory Tract
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thammavongsa, V.; Kim, H.K.; Missiakas, D.; Schneewind, O. Staphylococcal manipulation of host immune responses. Nat. Rev. Microbiol. 2015, 13, 529–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, C.H.; Miller, M.D.; Simon, R.A. The united allergic airway: Connections between allergic rhinitis, asthma, and chronic sinusitis. Am. J. Rhinol. Allergy 2012, 26, 187–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Humaidan, A.H.; Elvén, M.; Sonesson, A.; Garred, P.; Sørensen, O.E. Persistent Intracellular Staphylococcus aureus in Keratinocytes Lead to Activation of the Complement System with Subsequent Reduction in the Intracellular Bacterial Load. Front. Immunol. 2018, 9, 396. [Google Scholar] [CrossRef] [PubMed]
- Nagl, M.; Kacani, L.; Müllauer, B.; Lemberger, E.-M.; Stoiber, H.; Sprinzl, G.M.; Schennach, H.; Dierich, M.P. Phagocytosis and Killing of Bacteria by Professional Phagocytes and Dendritic Cells. Clin. Diagn. Lab. Immunol. 2002, 9, 1165–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schindler, D.; Gutierrez, M.G.; Beineke, A.; Rauter, Y.; Rohde, M.; Foster, S.; Goldmann, O.; Medina, E. Dendritic cells are central coordinators of the host immune response to Staphylococcus aureus bloodstream infection. Am. J. Pathol. 2012, 181, 1327–1337. [Google Scholar] [CrossRef] [PubMed]
- Van Kessel, K.P.M.; Bestebroer, J.; van Strijp, J.A.G. Neutrophil-Mediated Phagocytosis of Staphylococcus aureus. Front. Immunol. 2014, 5, 467. [Google Scholar] [CrossRef] [PubMed]
- Berends, E.T.M.; Horswill, A.R.; Haste, N.M.; Monestier, M.; Nizet, V.; Köckritz-Blickwede, M. von. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J. Innate Immun. 2010, 2, 576–586. [Google Scholar] [CrossRef] [PubMed]
- Laarman, A.; Milder, F.; van Strijp, J.; Rooijakkers, S. Complement inhibition by gram-positive pathogens: Molecular mechanisms and therapeutic implications. J. Mol. Med. 2010, 88, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.M.; van Kessel, K.P.M.; van Strijp, J.A.G. Staphylococcal innate immune evasion. Trends Microbiol. 2005, 13, 596–601. [Google Scholar] [CrossRef] [PubMed]
- Von Köckritz-Blickwede, M.; Konrad, S.; Foster, S.; Gessner, J.E.; Medina, E. Protective role of complement C5a in an experimental model of Staphylococcus aureus bacteremia. J. Innate Immun. 2010, 2, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Na, M.; Jarneborn, A.; Ali, A.; Welin, A.; Magnusson, M.; Stokowska, A.; Pekna, M.; Jin, T. Deficiency of the Complement Component 3 but Not Factor B Aggravates Staphylococcus aureus Septic Arthritis in Mice. Infect. Immun. 2016, 84, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Bröker, B.M.; Holtfreter, S.; Bekeredjian-Ding, I. Immune control of Staphylococcus aureus—Regulation and counter-regulation of the adaptive immune response. Int J. Med. Microbiol. 2014, 304, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Holtfreter, S.; Kolata, J.; Bröker, B.M. Towards the immune proteome of Staphylococcus aureus—The anti-S. aureus antibody response. Int. J. Med. Microbiol. 2010, 300, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Stentzel, S.; Sundaramoorthy, N.; Michalik, S.; Nordengrün, M.; Schulz, S.; Kolata, J.; Kloppot, P.; Engelmann, S.; Steil, L.; Hecker, M.; et al. Specific serum IgG at diagnosis of Staphylococcus aureus bloodstream invasion is correlated with disease progression. J. Proteom. 2015, 128, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Stentzel, S.; Hagl, B.; Abel, F.; Kahl, B.C.; Rack-Hoch, A.; Bröker, B.M.; Renner, E.D. Reduced Immunoglobulin (Ig) G Response to Staphylococcus aureus in STAT3 Hyper-IgE Syndrome. Clin. Infect. Dis. 2017, 64, 1279–1282. [Google Scholar] [CrossRef] [PubMed]
- Farmand, S.; Sundin, M. Hyper-IgE syndromes: Recent advances in pathogenesis, diagnostics and clinical care. Curr. Opin. Hematol. 2015, 22, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Guidos, R.; Gilbert, D.; Bradley, J.; Boucher, H.W.; Scheld, W.M.; Bartlett, J.G.; Edwards, J. The epidemic of antibiotic-resistant infections: A call to action for the medical community from the Infectious Diseases Society of America. Clin. Infect. Dis. 2008, 46, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Archer, N.K.; Harro, J.M.; Shirtliff, M.E. Clearance of Staphylococcus aureus nasal carriage is T cell dependent and mediated through interleukin-17A expression and neutrophil influx. Infect. Immun. 2013, 81, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Minegishi, Y.; Saito, M.; Nagasawa, M.; Takada, H.; Hara, T.; Tsuchiya, S.; Agematsu, K.; Yamada, M.; Kawamura, N.; Ariga, T.; et al. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J. Exp. Med. 2009, 206, 1291–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uebele, J.; Stein, C.; Nguyen, M.-T.; Schneider, A.; Kleinert, F.; Tichá, O.; Bierbaum, G.; Götz, F.; Bekeredjian-Ding, I. Antigen delivery to dendritic cells shapes human CD4+ and CD8+ T cell memory responses to Staphylococcus aureus. PLoS Pathog. 2017, 13, e1006387. [Google Scholar] [CrossRef] [PubMed]
- Kolata, J.B.; Kühbandner, I.; Link, C.; Normann, N.; Vu, C.H.; Steil, L.; Weidenmaier, C.; Bröker, B.M. The Fall of a Dogma? Unexpected High T-Cell Memory Response to Staphylococcus aureus in Humans. J. Infect. Dis. 2015, 212, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.F.; Murphy, A.G.; Lalor, S.J.; Leech, J.M.; O’Keeffe, K.M.; Mac Aogáin, M.; O’Halloran, D.P.; Lacey, K.A.; Tavakol, M.; Hearnden, C.H.; et al. Memory Th1 Cells Are Protective in Invasive Staphylococcus aureus Infection. PLoS Pathog. 2015, 11, e1005226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langerhans, P. Ueber die Nerven der menschlichen Haut. Arch. Pathol. Anat. 1868, 44, 325–337. [Google Scholar] [CrossRef]
- Steinman, R.M. Identification of a novel cell type in peripheral lymphoid organs of mice: I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef] [PubMed]
- Janeway, C.A.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Hawiger, D.; Nussenzweig, M.C. Tolerogenic dendritic cells. Annu. Rev. Immunol. 2003, 21, 685–711. [Google Scholar] [CrossRef] [PubMed]
- Kelly, B.; O’Neill, L.A.J. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savina, A.; Amigorena, S. Phagocytosis and antigen presentation in dendritic cells. Immunol. Rev. 2007, 219, 143–156. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, K.M.; Wilk, M.M.; Leech, J.M.; Murphy, A.G.; Laabei, M.; Monk, I.R.; Massey, R.C.; Lindsay, J.A.; Foster, T.J.; Geoghegan, J.A.; et al. Manipulation of Autophagy in Phagocytes Facilitates Staphylococcus aureus Bloodstream Infection. Infect. Immun. 2015, 83, 3445–3457. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.-Y.; Roberts, L.L.; Robinson, C.M. The presence of interleukin-27 during monocyte-derived dendritic cell differentiation promotes improved antigen processing and stimulation of T cells. Immunology 2015, 144, 649–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishat, S.; Wuescher, L.M.; Worth, R.G. Platelets enhance dendritic cell responses against S. aureus through CD40-CD40L interactions. Infect. Immun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schraml, B.U.; Reis e Sousa, C. Defining dendritic cells. Curr. Opin. Immunol. 2015, 32, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Hart, D.N. Dendritic cells: Unique leukocyte populations which control the primary immune response. Blood 1997, 90, 3245–3287. [Google Scholar] [PubMed]
- Ueno, H.; Klechevsky, E.; Morita, R.; Aspord, C.; Cao, T.; Matsui, T.; Di Pucchio, T.; Connolly, J.; Fay, J.W.; Pascual, V.; et al. Dendritic cell subsets in health and disease. Immunol. Rev. 2007, 219, 118–142. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice: II. Functional properties in vitro. J. Exp. Med. 1974, 139, 380–397. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Lustig, D.S.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice: III. Functional properties in vivo. J. Exp. Med. 1974, 139, 1431–1445. [Google Scholar] [CrossRef] [PubMed]
- Lennert, K.; Remmele, W. Karyometrische Untersuchungen an Lymphknotenzellen des Menschen. Acta Haematol. 2004, 19, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Victora, G.D.; Schwickert, T.A.; Guermonprez, P.; Meredith, M.M.; Yao, K.; Chu, F.-F.; Randolph, G.J.; Rudensky, A.Y.; Nussenzweig, M. In Vivo Analysis of Dendritic Cell Development and Homeostasis. Science 2009, 324, 392–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamazaki, C.; Sugiyama, M.; Ohta, T.; Hemmi, H.; Hamada, E.; Sasaki, I.; Fukuda, Y.; Yano, T.; Nobuoka, M.; Hirashima, T.; et al. Critical roles of a dendritic cell subset expressing a chemokine receptor, XCR1. J. Immunol. 2013, 190, 6071–6082. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef] [PubMed]
- Dress, R.J.; Wong, A.Y.W.; Ginhoux, F. Homeostatic control of dendritic cell numbers and differentiation. Immunol. Cell Boil. 2018, 96, 463–476. [Google Scholar] [CrossRef] [PubMed]
- McGovern, N.; Schlitzer, A.; Gunawan, M.; Jardine, L.; Shin, A.; Poyner, E.; Green, K.; Dickinson, R.; Wang, X.-N.; Low, D.; et al. Human Dermal CD14 + Cells Are a Transient Population of Monocyte-Derived Macrophages. Immunity 2014, 41, 465–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bandola-Simon, J.; Roche, P.A. Dysfunction of antigen processing and presentation by dendritic cells in cancer. Mol. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Collin, M.; Bigley, V. Human dendritic cell subsets: An update. Immunology 2018, 154, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Grouard, G.; Rissoan, M.-C.; Filgueira, L.; Durand, I.; Banchereau, J.; Liu, Y.-J. The Enigmatic Plasmacytoid T Cells Develop into Dendritic Cells with Interleukin (IL)-3 and CD40-Ligand. J. Exp. Med. 1997, 185, 1101–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rissoan, M.C.; Soumelis, V.; Kadowaki, N.; Grouard, G.; Briere, F.; de Waal Malefyt, R.; Liu, Y.J. Reciprocal control of T helper cell and dendritic cell differentiation. Science 1999, 283, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Wollenberg, A.; Günther, S.; Moderer, M.; Wetzel, S.; Wagner, M.; Towarowski, A.; Tuma, E.; Rothenfusser, S.; Endres, S.; Hartmann, G. Plasmacytoid Dendritic Cells: A New Cutaneous Dendritic Cell Subset with Distinct Role in Inflammatory Skin Diseases. J. Investig. Dermatol. 2002, 119, 1096–1102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, K.P.A. Characterization of human blood dendritic cell subsets. Blood 2002, 100, 4512–4520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onai, N.; Kurabayashi, K.; Hosoi-Amaike, M.; Toyama-Sorimachi, N.; Matsushima, K.; Inaba, K.; Ohteki, T. A Clonogenic Progenitor with Prominent Plasmacytoid Dendritic Cell Developmental Potential. Immunity 2013, 38, 943–957. [Google Scholar] [CrossRef] [PubMed]
- Kamath, A.T.; Pooley, J.; O’Keeffe, M.A.; Vremec, D.; Zhan, Y.; Lew, A.M.; D’Amico, A.; Wu, L.; Tough, D.F.; Shortman, K. The development, maturation, and turnover rate of mouse spleen dendritic cell populations. J. Immunol. 2000, 165, 6762–6770. [Google Scholar] [CrossRef] [PubMed]
- Fonteneau, J.-F. Activation of influenza virus-specific CD4+ and CD8+ T cells: A new role for plasmacytoid dendritic cells in adaptive immunity. Blood 2003, 101, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; van Nimwegen, M.; Willart, M.A.M.; Muskens, F.; Boon, L.; Smit, J.J.; Coyle, A.; Clausen, B.E.; Hoogsteden, H.C.; Lambrecht, B.N.; et al. An anti-inflammatory role for plasmacytoid dendritic cells in allergic airway inflammation. J. Immunol. 2009, 183, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanzelmann, D.; Joo, H.-S.; Franz-Wachtel, M.; Hertlein, T.; Stevanovic, S.; Macek, B.; Wolz, C.; Götz, F.; Otto, M.; Kretschmer, D.; et al. Toll-like receptor 2 activation depends on lipopeptide shedding by bacterial surfactants. Nat. Commun. 2016, 7, 12304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fournier, B.; Philpott, D.J. Recognition of Staphylococcus aureus by the innate immune system. Clin. Microbiol. Rev. 2005, 18, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B. The function of TLR2 during staphylococcal diseases. Front. Cell. Infect. Microbiol. 2012, 2, 167. [Google Scholar] [CrossRef] [PubMed]
- Koymans, K.J.; Feitsma, L.J.; Brondijk, T.H.C.; Aerts, P.C.; Lukkien, E.; Lössl, P.; van Kessel, K.P.M.; de Haas, C.J.C.; van Strijp, J.A.G.; Huizinga, E.G. Structural basis for inhibition of TLR2 by staphylococcal superantigen-like protein 3 (SSL3). Proc. Natl. Acad. Sci. USA 2015, 112, 11018–11023. [Google Scholar] [CrossRef] [PubMed]
- Koymans, K.J.; Goldmann, O.; Karlsson, C.A.Q.; Sital, W.; Thänert, R.; Bisschop, A.; Vrieling, M.; Malmström, J.; van Kessel, K.P.M.; de Haas, C.J.C.; et al. The TLR2 Antagonist Staphylococcal Superantigen-Like Protein 3 Acts as a Virulence Factor to Promote Bacterial Pathogenicity in vivo. J. Innate Immun. 2017, 9, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, K.; Stroisch, T.J.; Koch, S.; Herrmann, N.; Leib, N.; Bieber, T. Langerhans and inflammatory dendritic epidermal cells in atopic dermatitis are tolerized towards TLR2 activation. Allergy 2018. [Google Scholar] [CrossRef] [PubMed]
- Aoyagi, S.; Oda, T.; Wada, K.; Nakamura, E.; Kosuga, T.; Yasunaga, H. Infective Endocarditis Associated with Atopic Dermatitis. Int. Heart J. 2018, 59, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Bergstrøm, B.; Aune, M.H.; Awuh, J.A.; Kojen, J.F.; Blix, K.J.; Ryan, L.; Flo, T.H.; Mollnes, T.E.; Espevik, T.; Stenvik, J. TLR8 Senses Staphylococcus aureus RNA in Human Primary Monocytes and Macrophages and Induces IFN-β Production via a TAK1-IKKβ-IRF5 Signaling Pathway. J. Immunol. 2015, 195, 1100–1111. [Google Scholar] [CrossRef] [PubMed]
- Ugolini, M.; Gerhard, J.; Burkert, S.; Jensen, K.J.; Georg, P.; Ebner, F.; Volkers, S.M.; Thada, S.; Dietert, K.; Bauer, L.; et al. Recognition of microbial viability via TLR8 drives TFH cell differentiation and vaccine responses. Nat. Immunol. 2018, 19, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Parker, D.; Prince, A. Staphylococcus aureus induces type I IFN signaling in dendritic cells via TLR9. J. Immunol. 2012, 189, 4040–4046. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, W.; Domann, E.; Chakraborty, T.; Mannala, G.; Lips, K.S.; Heiss, C.; Schnettler, R.; Alt, V. TLR9 mediates S. aureus killing inside osteoblasts via induction of oxidative stress. BMC Microbiol. 2016, 16, 230. [Google Scholar] [CrossRef] [PubMed]
- Mansur, N.; Hazzan, R.; Paul, M.; Bishara, J.; Leibovici, L. Does sex affect 30-day mortality in Staphylococcus aureus bacteremia? Gend. Med. 2012, 9, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Nurjadi, D.; Heeg, K.; Weber, A.N.R.; Zanger, P. Toll-like receptor 9 (TLR-9) promotor polymorphisms and gene expression are associated with persistent Staphylococcus aureus nasal carriage. Clin. Microbiol. Infect. 2018. [Google Scholar] [CrossRef] [PubMed]
- Fraunholz, M.; Sinha, B. Intracellular Staphylococcus aureus: Live-in and let die. Front. Cell. Infect. Microbiol. 2012, 2, 43. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Strobel, M.; Pförtner, H.; Tuchscherr, L.; Völker, U.; Schmidt, F.; Kramko, N.; Schnittler, H.-J.; Fraunholz, M.J.; Löffler, B.; Peters, G.; et al. Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin. Microbiol. Infect. 2016, 22, 799–809. [Google Scholar] [CrossRef] [PubMed]
- Patti, J.M.; Allen, B.L.; McGavin, M.J.; Höök, M. MSCRAMM-mediated adherence of microorganisms to host tissues. Annu. Rev. Microbiol. 1994, 48, 585–617. [Google Scholar] [CrossRef] [PubMed]
- Sinha, B.; François, P.P.; Nüsse, O.; Foti, M.; Hartford, O.M.; Vaudaux, P.; Foster, T.J.; Lew, D.P.; Herrmann, M.; Krause, K.H. Fibronectin-binding protein acts as Staphylococcus aureus invasin via fibronectin bridging to integrin alpha5beta1. Cell. Microbiol. 1999, 1, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Schwarz-Linek, U.; Werner, J.M.; Pickford, A.R.; Gurusiddappa, S.; Kim, J.H.; Pilka, E.S.; Briggs, J.A.G.; Gough, T.S.; Höök, M.; Campbell, I.D.; et al. Pathogenic bacteria attach to human fibronectin through a tandem beta-zipper. Nature 2003, 423, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Porayath, C.; Suresh, M.K.; Biswas, R.; Nair, B.G.; Mishra, N.; Pal, S. Autolysin mediated adherence of Staphylococcus aureus with Fibronectin, Gelatin and Heparin. Int. J. Biol. Macromol. 2018, 110, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Chavakis, E.; Aicher, A.; Heeschen, C.; Sasaki, K.-i.; Kaiser, R.; El Makhfi, N.; Urbich, C.; Peters, T.; Scharffetter-Kochanek, K.; Zeiher, A.M.; et al. Role of beta2-integrins for homing and neovascularization capacity of endothelial progenitor cells. J. Exp. Med. 2005, 201, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; von Eiff, C.; Sinha, B.; Joost, I.; Herrmann, M.; Peters, G.; Becker, K. Eap Gene as novel target for specific identification of Staphylococcus aureus. J. Clin. Microbiol. 2008, 46, 470–476. [Google Scholar] [CrossRef] [PubMed]
- McAdow, M.; Kim, H.K.; Dedent, A.C.; Hendrickx, A.P.A.; Schneewind, O.; Missiakas, D.M. Preventing sepsis through the inhibition of its agglutination in blood. PLoS Pathog. 2011, 7, e1002307. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bruhns, P.; Saeys, Y.; Hammad, H.; Lambrecht, B.N. The function of Fcγ receptors in dendritic cells and macrophages. Nat. Rev. Immunol. 2014, 14, 94–108. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, A.; Stapels, D.A.C.; Weerwind, L.T.; Ko, Y.-P.; Ruyken, M.; Lee, J.C.; van Kessel, K.P.M.; Rooijakkers, S.H.M. The Staphylococcus aureus polysaccharide capsule and Efb-dependent fibrinogen shield act in concert to protect against phagocytosis. Microbiology 2016, 162, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- O’Riordan, K.; Lee, J.C. Staphylococcus aureus Capsular Polysaccharides. Clin. Microbiol. Rev. 2004, 17, 218–234. [Google Scholar] [CrossRef] [PubMed]
- Becker, S.; Frankel, M.B.; Schneewind, O.; Missiakas, D. Release of protein A from the cell wall of Staphylococcus aureus. Proc. Natl. Acad. Sci. USA 2014, 111, 1574–1579. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, C.S.; Silverman, G.J. Staphylococcal toxin induced preferential and prolonged in vivo deletion of innate-like B lymphocytes. Proc. Natl. Acad. Sci. USA 2004, 101, 11392–11397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukácsi, S.; Nagy-Baló, Z.; Erdei, A.; Sándor, N.; Bajtay, Z. The role of CR3 (CD11b/CD18) and CR4 (CD11c/CD18) in complement-mediated phagocytosis and podosome formation by human phagocytes. Immunol. Lett. 2017, 189, 64–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietrocola, G.; Nobile, G.; Rindi, S.; Speziale, P. Staphylococcus aureus Manipulates Innate Immunity through Own and Host-Expressed Proteases. Front. Cell. Infect. Microbiol. 2017, 7, 787. [Google Scholar] [CrossRef] [PubMed]
- Laarman, A.J.; Ruyken, M.; Malone, C.L.; van Strijp, J.A.G.; Horswill, A.R.; Rooijakkers, S.H.M. Staphylococcus aureus metalloprotease aureolysin cleaves complement C3 to mediate immune evasion. J. Immunol. 2011, 186, 6445–6453. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Bokarewa, M.; Foster, T.; Mitchell, J.; Higgins, J.; Tarkowski, A. Staphylococcus aureus Resists Human Defensins by Production of Staphylokinase, a Novel Bacterial Evasion Mechanism. J. Immunol. 2004, 172, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Sieprawska-Lupa, M.; Mydel, P.; Krawczyk, K.; Wojcik, K.; Puklo, M.; Lupa, B.; Suder, P.; Silberring, J.; Reed, M.; Pohl, J.; et al. Degradation of Human Antimicrobial Peptide LL-37 by Staphylococcus aureus-Derived Proteinases. Antimicrob. Agents Chemother. 2004, 48, 4673–4679. [Google Scholar] [CrossRef] [PubMed]
- Noore, J.; Noore, A.; Li, B. Cationic antimicrobial peptide LL-37 is effective against both extra- and intracellular Staphylococcus aureus. Antimicrob. Agents Chemother. 2013, 57, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Spaan, A.N.; van Strijp, J.A.G.; Torres, V.J. Leukocidins: Staphylococcal bi-component pore-forming toxins find their receptors. Nat. Rev. Microbiol. 2017, 15, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Alonzo, F.; Kozhaya, L.; Rawlings, S.A.; Reyes-Robles, T.; DuMont, A.L.; Myszka, D.G.; Landau, N.R.; Unutmaz, D.; Torres, V.J. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 2013, 493, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Reyes-Robles, T.; Alonzo, F.; Kozhaya, L.; Lacy, D.B.; Unutmaz, D.; Torres, V.J. Staphylococcus aureus leukotoxin ED targets the chemokine receptors CXCR1 and CXCR2 to kill leukocytes and promote infection. Cell Host Microbe 2013, 14, 453–459. [Google Scholar] [CrossRef] [PubMed]
- DuMont, A.L.; Yoong, P.; Day, C.J.; Alonzo, F.; McDonald, W.H.; Jennings, M.P.; Torres, V.J. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proc. Natl. Acad. Sci. USA 2013, 110, 10794–10799. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.K.; Springer, T.A. Mac-1 antigen: Quantitative expression in macrophage populations and tissues, and immunofluorescent localization in spleen. J. Immunol. 1982, 128, 2281–2286. [Google Scholar] [PubMed]
- DuMont, A.L.; Yoong, P.; Liu, X.; Day, C.J.; Chumbler, N.M.; James, D.B.A.; Alonzo, F.; Bode, N.J.; Lacy, D.B.; Jennings, M.P.; et al. Identification of a crucial residue required for Staphylococcus aureus LukAB cytotoxicity and receptor recognition. Infect. Immun. 2014, 82, 1268–1276. [Google Scholar] [CrossRef] [PubMed]
- Vrieling, M.; Koymans, K.J.; Heesterbeek, D.A.C.; Aerts, P.C.; Rutten, V.P.M.G.; de Haas, C.J.C.; van Kessel, K.P.M.; Koets, A.P.; Nijland, R.; van Strijp, J.A.G. Bovine Staphylococcus aureus Secretes the Leukocidin LukMF’ To Kill Migrating Neutrophils through CCR1. mBio 2015, 6, e00335. [Google Scholar] [CrossRef] [PubMed]
- Sozzani, S.; Luini, W.; Borsatti, A.; Polentarutti, N.; Zhou, D.; Piemonti, L.; D’Amico, G.; Power, C.A.; Wells, T.N.; Gobbi, M.; et al. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J. Immunol. 1997, 159, 1993–2000. [Google Scholar] [PubMed]
- Spaan, A.N.; Reyes-Robles, T.; Badiou, C.; Cochet, S.; Boguslawski, K.M.; Yoong, P.; Day, C.J.; de Haas, C.J.C.; van Kessel, K.P.M.; Vandenesch, F.; et al. Staphylococcus aureus Targets the Duffy Antigen Receptor for Chemokines (DARC) to Lyse Erythrocytes. Cell Host Microbe 2015, 18, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Spaan, A.N.; Vrieling, M.; Wallet, P.; Badiou, C.; Reyes-Robles, T.; Ohneck, E.A.; Benito, Y.; de Haas, C.J.C.; Day, C.J.; Jennings, M.P.; et al. The staphylococcal toxins γ-haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nat. Commun. 2014, 5, 5438. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, F.; Girardot, R.; Piémont, Y.; Prévost, G.; Colin, D.A. Analysis of the specificity of Panton-Valentine leucocidin and gamma-hemolysin F component binding. Infect. Immun. 2009, 77, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Dalla Serra, M.; Coraiola, M.; Viero, G.; Comai, M.; Potrich, C.; Ferreras, M.; Baba-Moussa, L.; Colin, D.A.; Menestrina, G.; Bhakdi, S.; et al. Staphylococcus aureus bicomponent gamma-hemolysins, HlgA, HlgB, and HlgC, can form mixed pores containing all components. J. Chem. Inf. Model. 2005, 45, 1539–1545. [Google Scholar] [CrossRef] [PubMed]
- Rybicka, J.M.; Balce, D.R.; Chaudhuri, S.; Allan, E.R.O.; Yates, R.M. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J. 2012, 31, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Ip, W.K.E.; Sokolovska, A.; Charriere, G.M.; Boyer, L.; Dejardin, S.; Cappillino, M.P.; Yantosca, L.M.; Takahashi, K.; Moore, K.J.; Lacy-Hulbert, A.; et al. Phagocytosis and phagosome acidification are required for pathogen processing and MyD88-dependent responses to Staphylococcus aureus. J. Immunol. 2010, 184, 7071–7081. [Google Scholar] [CrossRef] [PubMed]
- Horn, J.; Stelzner, K.; Rudel, T.; Fraunholz, M. Inside job: Staphylococcus aureus host-pathogen interactions. Int. J. Med. Microbiol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bore, E.; Langsrud, S.; Langsrud, O.; Rode, T.M.; Holck, A. Acid-shock responses in Staphylococcus aureus investigated by global gene expression analysis. Microbiology 2007, 153, 2289–2303. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Essex, A.; Buchanan, J.T.; Datta, V.; Hoffman, H.M.; Bastian, J.F.; Fierer, J.; Nizet, V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J. Exp. Med. 2005, 202, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; Kohler, P.L.; Schlievert, P.M.; Chuang, O.N.; Dunny, G.M.; Kobayashi, S.D.; Miedzobrodzki, J.; Bohach, G.A.; Seo, K.S. Characterization of a Staphylococcus aureus surface virulence factor that promotes resistance to oxidative killing and infectious endocarditis. Infect. Immun. 2011, 79, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Ballal, A.; Manna, A.C. Regulation of superoxide dismutase (sod) genes by SarA in Staphylococcus aureus. J. Bacteriol. 2009, 191, 3301–3310. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, K.; Coutts, G.; Jonsson, I.-M.; Tarkowski, A.; Kokai-Kun, J.F.; Mond, J.J.; Foster, S.J. Catalase (KatA) and alkyl hydroperoxide reductase (AhpC) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J. Bacteriol. 2007, 189, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- De Jong, N.W.M.; Ramyar, K.X.; Guerra, F.E.; Nijland, R.; Fevre, C.; Voyich, J.M.; McCarthy, A.J.; Garcia, B.L.; van Kessel, K.P.M.; van Strijp, J.A.G.; et al. Immune evasion by a staphylococcal inhibitor of myeloperoxidase. Proc. Natl. Acad. Sci. USA 2017, 114, 9439–9444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odobasic, D.; Kitching, A.R.; Yang, Y.; O’Sullivan, K.M.; Muljadi, R.C.M.; Edgtton, K.L.; Tan, D.S.Y.; Summers, S.A.; Morand, E.F.; Holdsworth, S.R. Neutrophil myeloperoxidase regulates T-cell-driven tissue inflammation in mice by inhibiting dendritic cell function. Blood 2013, 121, 4195–4204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz, W.; Platzer, B.; Schumich, A.; Höcher, B.; Fritsch, G.; Knapp, W.; Strobl, H. Initial human myeloid/dendritic cell progenitors identified by absence of myeloperoxidase protein expression. Exp. Hematol. 2004, 32, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Herbert, S.; Jakob, A.; Vollmer, W.; Götz, F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan O-acetyltransferase OatA is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 2005, 55, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Vu, C.H.; Kolata, J.; Stentzel, S.; Beyer, A.; Gesell Salazar, M.; Steil, L.; Pané-Farré, J.; Rühmling, V.; Engelmann, S.; Götz, F.; et al. Adaptive immune response to lipoproteins of Staphylococcus aureus in healthy subjects. Proteomics 2016, 16, 2667–2677. [Google Scholar] [CrossRef] [PubMed]
- Giese, B.; Glowinski, F.; Paprotka, K.; Dittmann, S.; Steiner, T.; Sinha, B.; Fraunholz, M.J. Expression of δ-toxin by Staphylococcus aureus mediates escape from phago-endosomes of human epithelial and endothelial cells in the presence of β-toxin. Cell. Microbiol. 2011, 13, 316–329. [Google Scholar] [CrossRef] [PubMed]
- Grosz, M.; Kolter, J.; Paprotka, K.; Winkler, A.-C.; Schäfer, D.; Chatterjee, S.S.; Geiger, T.; Wolz, C.; Ohlsen, K.; Otto, M.; et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin α. Cell. Microbiol. 2014, 16, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Surewaard, B.G.J.; de Haas, C.J.C.; Vervoort, F.; Rigby, K.M.; DeLeo, F.R.; Otto, M.; van Strijp, J.A.G.; Nijland, R. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell. Microbiol. 2013, 15, 1427–1437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armbruster, N.S.; Richardson, J.R.; Schreiner, J.; Klenk, J.; Günter, M.; Autenrieth, S.E. Staphylococcus aureus PSM peptides induce tolerogenic dendritic cells upon treatment with ligands of extracellular and intracellular TLRs. Int. J. Med. Microbiol. 2016, 306, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Armbruster, N.S.; Richardson, J.R.; Schreiner, J.; Klenk, J.; Günter, M.; Kretschmer, D.; Pöschel, S.; Schenke-Layland, K.; Kalbacher, H.; Clark, K.; et al. PSM Peptides of Staphylococcus aureus Activate the p38-CREB Pathway in Dendritic Cells, Thereby Modulating Cytokine Production and T Cell Priming. J. Immunol. 2016, 196, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Deplanche, M.; Alekseeva, L.; Semenovskaya, K.; Fu, C.-L.; Dessauge, F.; Finot, L.; Petzl, W.; Zerbe, H.; Le Loir, Y.; Rainard, P.; et al. Staphylococcus aureus Phenol-Soluble Modulins Impair Interleukin Expression in Bovine Mammary Epithelial Cells. Infect. Immun. 2016, 84, 1682–1692. [Google Scholar] [CrossRef] [PubMed]
- Dey, S.; Bishayi, B. Killing of Staphylococcus aureus in murine macrophages by chloroquine used alone and in combination with ciprofloxacin or azithromycin. J. Inflamm. Res. 2015, 8, 29–47. [Google Scholar] [CrossRef] [PubMed]
- Leimer, N.; Rachmühl, C.; Palheiros Marques, M.; Bahlmann, A.S.; Furrer, A.; Eichenseher, F.; Seidl, K.; Matt, U.; Loessner, M.J.; Schuepbach, R.A.; et al. Nonstable Staphylococcus aureus Small-Colony Variants Are Induced by Low pH and Sensitized to Antimicrobial Therapy by Phagolysosomal Alkalinization. J. Infect. Dis. 2016, 213, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Neumann, Y.; Bruns, S.A.; Rohde, M.; Prajsnar, T.K.; Foster, S.J.; Schmitz, I. Intracellular Staphylococcus aureus eludes selective autophagy by activating a host cell kinase. Autophagy 2016, 12, 2069–2084. [Google Scholar] [CrossRef] [PubMed]
- Maurer, K.; Reyes-Robles, T.; Alonzo, F.; Durbin, J.; Torres, V.J.; Cadwell, K. Autophagy mediates tolerance to Staphylococcus aureus alpha-toxin. Cell Host Microbe 2015, 17, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Mestre, M.B.; Fader, C.M.; Sola, C.; Colombo, M.I. α-hemolysin is required for the activation of the autophagic pathway in Staphylococcus aureus infected cells. Autophagy 2010, 6, 110–125. [Google Scholar] [CrossRef] [PubMed]
- Korea, C.G.; Balsamo, G.; Pezzicoli, A.; Merakou, C.; Tavarini, S.; Bagnoli, F.; Serruto, D.; Unnikrishnan, M. Staphylococcal Esx proteins modulate apoptosis and release of intracellular Staphylococcus aureus during infection in epithelial cells. Infect. Immun. 2014, 82, 4144–4153. [Google Scholar] [CrossRef] [PubMed]
- Cruciani, M.; Etna, M.P.; Camilli, R.; Giacomini, E.; Percario, Z.A.; Severa, M.; Sandini, S.; Rizzo, F.; Brandi, V.; Balsamo, G.; et al. Staphylococcus aureus Esx Factors Control Human Dendritic Cell Functions Conditioning Th1/Th17 Response. Front. Cell. Infect. Microbiol. 2017, 7, 330. [Google Scholar] [CrossRef] [PubMed]
- Koymans, K.J.; Vrieling, M.; Gorham, R.D.; van Strijp, J.A.G. Staphylococcal Immune Evasion Proteins: Structure, Function, and Host Adaptation. Curr. Top. Microbiol. Immunol. 2017, 409, 441–489. [Google Scholar] [CrossRef] [PubMed]
- Argudín, M.Á.; Mendoza, M.C.; Rodicio, M.R. Food poisoning and Staphylococcus aureus enterotoxins. Toxins 2010, 2, 1751–1773. [Google Scholar] [CrossRef] [PubMed]
- Grumann, D.; Nübel, U.; Bröker, B.M. Staphylococcus aureus toxins—Their functions and genetics. Infect. Genet. Evol. 2014, 21, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Llewelyn, M.; Cohen, J. Superantigens: Microbial agents that corrupt immunity. Lancet Infect. Dis. 2002, 2, 156–162. [Google Scholar] [CrossRef]
- Sollid, J.U.E.; Furberg, A.S.; Hanssen, A.M.; Johannessen, M. Staphylococcus aureus: Determinants of human carriage. Infect. Genet. Evol. 2014, 21, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Frazee, B.W. Severe methicillin-resistant Staphylococcus aureus community-acquired pneumonia associated with influenza—Louisiana and Georgia, December 2006-January 2007. MMWR Morb. Mortal. Wkly. Rep. 2007, 56, 325–329. [Google Scholar]
- Barnes, P.J. Intrinsic asthma: Not so different from allergic asthma but driven by superantigens? Clin. Exp. Allergy 2009, 39, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; Zhang, N. Chronic rhinosinusitis and asthma: Novel understanding of the role of IgE ‘above atopy’. J. Int. Med. 2012, 272, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.F.; Peng, R.D.; McCormack, M.C.; Matsui, E.C. Staphylococcus aureus colonization is associated with wheeze and asthma among US children and young adults. J. Allergy Clin. Immunol. 2015, 135, 811–813.e5. [Google Scholar] [CrossRef] [PubMed]
- Van Zele, T.; Gevaert, P.; Watelet, J.-B.; Claeys, G.; Holtappels, G.; Claeys, C.; van Cauwenberge, P.; Bachert, C. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J. Allergy Clin. Immunol. 2004, 114, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Bachert, C.; van Steen, K.; Zhang, N.; Holtappels, G.; Cattaert, T.; Maus, B.; Buhl, R.; Taube, C.; Korn, S.; Kowalski, M.; et al. Specific IgE against Staphylococcus aureus enterotoxins: An independent risk factor for asthma. J. Allergy Clin. Immunol. 2012, 130, 376–381.e8. [Google Scholar] [CrossRef] [PubMed]
- Stentzel, S.; Teufelberger, A.; Nordengrün, M.; Kolata, J.; Schmidt, F.; van Crombruggen, K.; Michalik, S.; Kumpfmüller, J.; Tischer, S.; Schweder, T.; et al. Staphylococcal serine protease–like proteins are pacemakers of allergic airway reactions to Staphylococcus aureus. J. Allergy Clin. Immunol. 2017, 139, 492–500.e8. [Google Scholar] [CrossRef] [PubMed]
- Kapsenberg, M.L. Dendritic-cell control of pathogen-driven T-cell polarization. Nat. Rev. Immunol. 2003, 3, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Condon, T.V.; Sawyer, R.T.; Fenton, M.J.; Riches, D.W.H. Lung dendritic cells at the innate-adaptive immune interface. J. Leukoc. Biol. 2011, 90, 883–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, J.-O.; Zhang, W.; Du, J.-Y.; Yu, Q. BDCA1-positive dendritic cells (DCs) represent a unique human myeloid DC subset that induces innate and adaptive immune responses to Staphylococcus aureus Infection. Infect. Immun. 2014, 82, 4466–4476. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, C.; Goldmann, O.; Hobeika, E.; Geffers, R.; Peters, G.; Medina, E. The dynamics of T cells during persistent Staphylococcus aureus infection: From antigen-reactivity to in vivo anergy. EMBO Mol. Med. 2011, 3, 652–666. [Google Scholar] [CrossRef] [PubMed]
- Holtfreter, S.; Kolata, J.; Stentzel, S.; Bauerfeind, S.; Schmidt, F.; Sundaramoorthy, N.; Bröker, B.M. Omics Approaches for the Study of Adaptive Immunity to Staphylococcus aureus and the Selection of Vaccine Candidates. Proteomes 2016, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.F.; Ludwig, S.; Brigham, E.P.; McCormack, M.C.; Matsui, E.C. Effect of home exposure to Staphylococcus aureus on asthma in adolescents. J. Allergy Clin. Immunol. 2018, 141, 402–405.e10. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Hamid, Q.; Bentley, A.; Ying, S.; Kay, A.B.; Durham, S.R. Activation of CD4+ T cells, increased TH2-type cytokine mRNA expression, and eosinophil recruitment in bronchoalveolar lavage after allergen inhalation challenge in patients with atopic asthma. J. Allergy Clin. Immunol. 1993, 92, 313–324. [Google Scholar] [CrossRef]
- Romagnani, S. The role of lymphocytes in allergic disease. J. Allergy Clin. Immunol. 2000, 105, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Lambrecht, B.N.; Salomon, B.; Klatzmann, D.; Pauwels, R.A. Dendritic cells are required for the development of chronic eosinophilic airway inflammation in response to inhaled antigen in sensitized mice. J. Immunol. 1998, 160, 4090–4097. [Google Scholar] [PubMed]
- Andreas, N.; Riemann, M.; Castro, C.N.; Groth, M.; Koliesnik, I.; Engelmann, C.; Sparwasser, T.; Kamradt, T.; Haenold, R.; Weih, F. A new RelB-dependent CD117+ CD172a+ murine DC subset preferentially induces Th2 differentiation and supports airway hyperresponses in vivo. Eur. J. Immunol. 2018, 48, 923–936. [Google Scholar] [CrossRef] [PubMed]
- Georas, S.N.; Rezaee, F. Epithelial barrier function: At the front line of asthma immunology and allergic airway inflammation. J. Allergy Clin. Immunol. 2014, 134, 509–520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berube, B.J.; Bubeck Wardenburg, J. Staphylococcus aureus α-Toxin: Nearly a Century of Intrigue. Toxins 2013, 5, 1140–1166. [Google Scholar] [CrossRef] [PubMed]
- Wilke, G.A.; Bubeck Wardenburg, J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc. Natl. Acad. Sci. USA 2010, 107, 13473–13478. [Google Scholar] [CrossRef] [PubMed]
- Damle, S.R.; Martin, R.K.; Cockburn, C.L.; Lownik, J.C.; Carlyon, J.A.; Smith, A.D.; Conrad, D.H. ADAM10 and Notch1 on murine dendritic cells control the development of type 2 immunity and IgE production. Allergy 2018, 73, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Breuer, K.; Wittmann, M.; Kempe, K.; Kapp, A.; Mai, U.; Dittrich-Breiholz, O.; Kracht, M.; Mrabet-Dahbi, S.; Werfel, T. Alpha-toxin is produced by skin colonizing Staphylococcus aureus and induces a T helper type 1 response in atopic dermatitis. Clin. Exp. Allergy 2005, 35, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Niebuhr, M.; Gathmann, M.; Scharonow, H.; Mamerow, D.; Mommert, S.; Balaji, H.; Werfel, T. Staphylococcal alpha-toxin is a strong inducer of interleukin-17 in humans. Infect. Immun. 2011, 79, 1615–1622. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.M.; Zhou, T.; Moreno-Vinasco, L.; Hollett, B.; Garcia, J.G.N.; Bubeck Wardenburg, J. Host response signature to Staphylococcus aureus alpha-hemolysin implicates pulmonary Th17 response. Infect. Immun. 2012, 80, 3161–3169. [Google Scholar] [CrossRef] [PubMed]
- Inoshima, I.; Inoshima, N.; Wilke, G.A.; Powers, M.E.; Frank, K.M.; Wang, Y.; Bubeck Wardenburg, J. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011, 17, 1310–1314. [Google Scholar] [CrossRef] [PubMed]
- Ezekwe, E.A.D.; Weng, C.; Duncan, J.A. ADAM10 Cell Surface Expression but Not Activity Is Critical for Staphylococcus aureus α-Hemolysin-Mediated Activation of the NLRP3 Inflammasome in Human Monocytes. Toxins 2016, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Hildebrandt, J.-P. Pore-forming virulence factors of Staphylococcus aureus destabilize epithelial barriers-effects of alpha-toxin in the early phases of airway infection. AIMS Microbiol. 2015, 1, 11–36. [Google Scholar] [CrossRef]
- Giersing, B.K.; Dastgheyb, S.S.; Modjarrad, K.; Moorthy, V. Status of vaccine research and development of vaccines for Staphylococcus aureus. Vaccine 2016, 34, 2962–2966. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.M.; Allen, C.; Rich, G.; Dryja, D.; Bina, P.; Reiser, R.; Ballow, M.; Wilding, G.E. Further observations on the role of Staphylococcus aureus exotoxins and IgE in the pathogenesis of nasal polyposis. Laryngoscope 2011, 121, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, M.; Klingenberg, C.; Wickman, M.; Sollid, J.U.E.; Furberg, A.-S.; Bachert, C.; Bousquet, J. Staphylococcus aureus enterotoxin sensitization is associated with allergic poly-sensitization and allergic multimorbidity in adolescents. Allergy 2017, 72, 1548–1555. [Google Scholar] [CrossRef] [PubMed]
- Bacher, P.; Heinrich, F.; Stervbo, U.; Nienen, M.; Vahldieck, M.; Iwert, C.; Vogt, K.; Kollet, J.; Babel, N.; Sawitzki, B.; et al. Regulatory T Cell Specificity Directs Tolerance versus Allergy against Aeroantigens in Humans. Cell 2016, 167, 1067–1078. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Darisipudi, M.N.; Nordengrün, M.; Bröker, B.M.; Péton, V. Messing with the Sentinels—The Interaction of Staphylococcus aureus with Dendritic Cells. Microorganisms 2018, 6, 87. https://doi.org/10.3390/microorganisms6030087
Darisipudi MN, Nordengrün M, Bröker BM, Péton V. Messing with the Sentinels—The Interaction of Staphylococcus aureus with Dendritic Cells. Microorganisms. 2018; 6(3):87. https://doi.org/10.3390/microorganisms6030087
Chicago/Turabian StyleDarisipudi, Murthy N., Maria Nordengrün, Barbara M. Bröker, and Vincent Péton. 2018. "Messing with the Sentinels—The Interaction of Staphylococcus aureus with Dendritic Cells" Microorganisms 6, no. 3: 87. https://doi.org/10.3390/microorganisms6030087
APA StyleDarisipudi, M. N., Nordengrün, M., Bröker, B. M., & Péton, V. (2018). Messing with the Sentinels—The Interaction of Staphylococcus aureus with Dendritic Cells. Microorganisms, 6(3), 87. https://doi.org/10.3390/microorganisms6030087