

Single-Cell Transcriptome Sequencing Reveals Molecular Expression Differences and Marker Genes in Testes during the Sexual Maturation of Mongolian Horses

 , , , and
, , , and 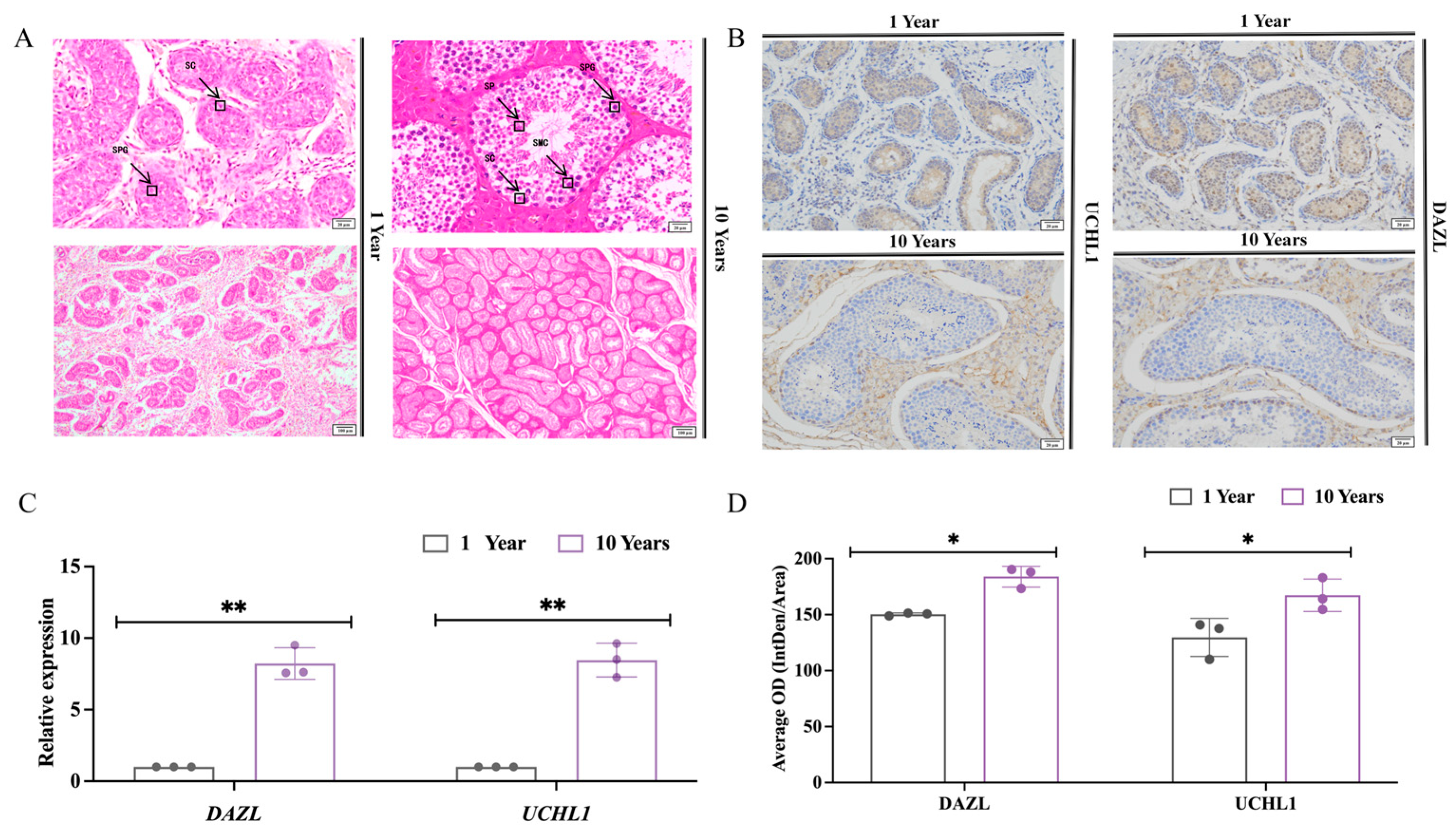{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Paraffin Sectioning of Testicular Tissue
2.2. Hematoxylin and Eosin (H.E.) Staining
2.3. Single-Cell Transcriptome Sequencing
2.4. Bioinformatics Analysis
2.5. RNA Extraction and Reverse Transcription
2.6. Real-Time Quantitative PCR
2.7. Immunohistochemical Staining
2.8. Immunofluorescence
2.9. Western Blot
2.10. Statistical Analysis of Data
3. Results
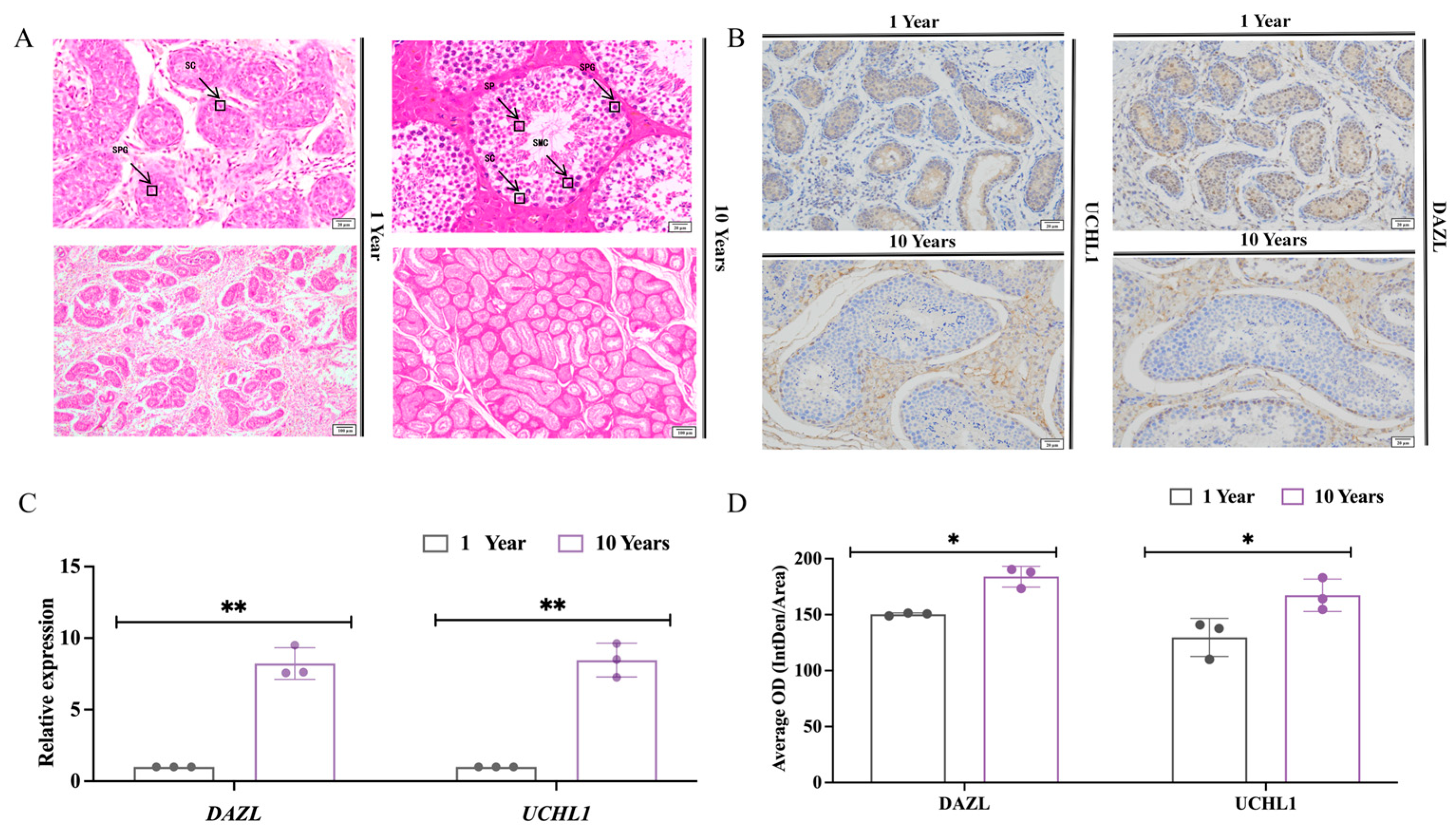
3.1. Histomorphometric Analysis of Testes of Sexually Immature and Sexually Mature Mongolian Horses
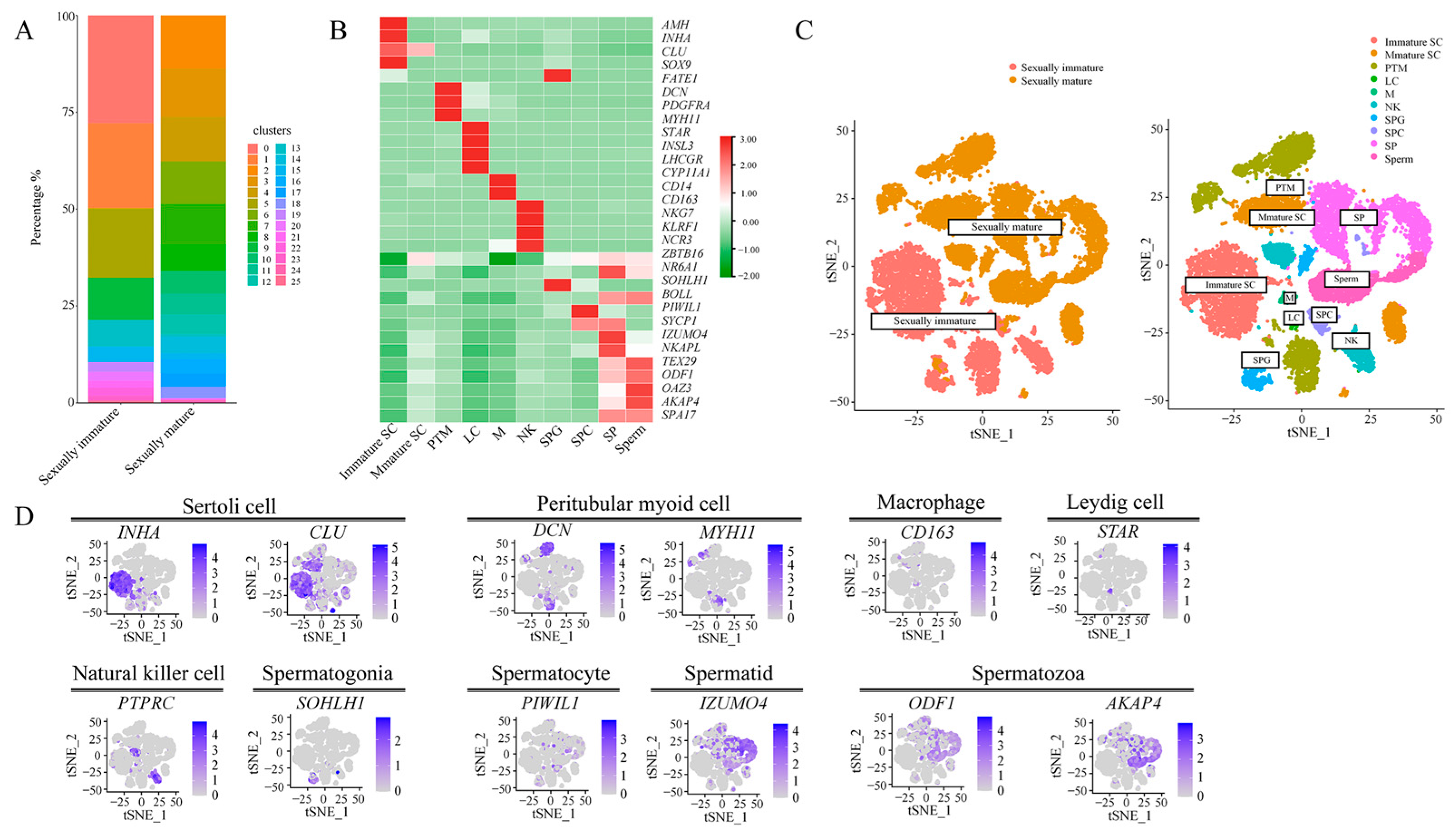
3.2. Classification of Single-Cell Subpopulations and Their Visualization
3.3. Pseudotime Analysis and Visualization of Spermatogenic Cells
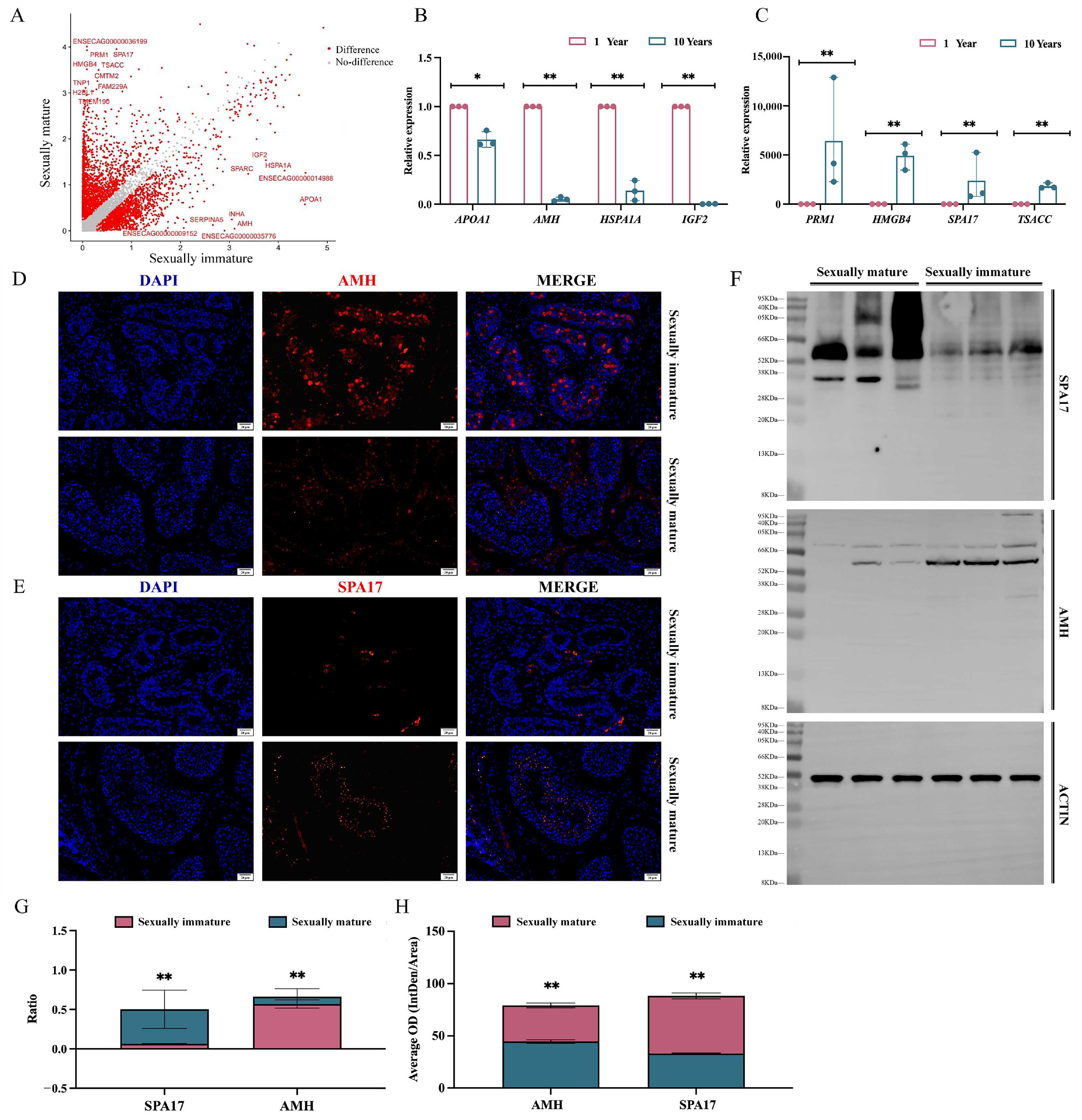
3.4. Analysis of Testicular Differential Genes between Sexually Immature and Sexually Mature Mongolian Horses
3.5. Analysis of Differential Gene Enrichment in the Testes of Sexually Immature and Sexually Mature Mongolian Horses
3.6. Screening for Marker Genes in Sexually Immature and Sexually Mature Mongolian Horses
4. Discussion
4.1. Histomorphologic Differences in Testicular Histomorphology between Sexually Immature and Sexually Mature Mongolian Horses
4.2. Single-Cell Mapping of Mongolian Horse Testes
4.3. Pseudotime Analysis and Visualization of Spermatogenic Cells
4.4. Analysis of Testicular Differential Genes and Enrichment between Sexually Immature and Sexually Mature Mongolian Horses
4.5. Screening of Marker Genes in Sexually Immature and Sexually Mature Mongolian Horses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Neto, F.T.L.; Bach, P.V.; Najari, B.B.; Li, P.S.; Goldstein, M. Spermatogenesis in humans and its affecting factors. Semin. Cell Dev. Biol. 2016, 59, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Staub, C.; Johnson, L. Review: Spermatogenesis in the bull. Animal 2018, 12, s27–s35. [Google Scholar] [CrossRef] [PubMed]
- Indriastuti, R.; Pardede, B.P.; Gunawan, A.; Ulum, M.F.; Arifiantini, R.I.; Purwantara, B. Sperm Transcriptome Analysis Accurately Reveals Male Fertility Potential in Livestock. Animals 2022, 12, 2955. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.A.; Reuter, M.; Sumner, S. Patterns of reproductive differentiation and reproductive plasticity in the major evolutionary transition to superorganismality. Curr. Opin. Insect Sci. 2019, 34, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Nonacs, P.; Hager, R. The past, present and future of reproductive skew theory and experiments. Biol. Rev. Camb. Philos. Soc. 2011, 86, 271–298. [Google Scholar] [CrossRef] [PubMed]
- Christin-Maitre, S.; Young, J. Androgens and spermatogenesis. Ann. Endocrinol. 2022, 83, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Ramsoomair, C.K.; Alver, C.G.; Flannigan, R.; Ramasamy, R.; Agarwal, A. Spermatogonial Stem Cells and In Vitro Spermatogenesis: How Far Are We from a Human Testis on a Chip? Eur. Urol. Focus 2023, 9, 46–48. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, M.; Zheng, X.; Manske, G.L.; Vargo, A.; Shami, A.N.; Li, J.Z.; Hammoud, S.S. Decoding the Spermatogenesis Program: New Insights from Transcriptomic Analyses. Annu. Rev. Genet. 2022, 56, 339–368. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lu, H.; Li, H.; Yan, C.; Wang, X.; Zang, M.; Rooij, D.G.; Madabhushi, A.; Xu, E.Y. Computerized spermatogenesis staging (CSS) of mouse testis sections via quantitative histomorphological analysis. Med. Image Anal. 2021, 70, 101835. [Google Scholar] [CrossRef]
- Zhang, L.; Guo, M.; Liu, Z.; Liu, R.; Zheng, Y.; Yu, T.; Lv, Y.; Lu, H.; Zeng, W.; Zhang, T.; et al. Single-cell RNA-seq analysis of testicular somatic cell development in pigs. J. Genet. Genom. 2022, 49, 1016–1028. [Google Scholar] [CrossRef]
- Bashawat, M.; Braun, B.C.; Müller, K.; Hermann, B.P. Molecular phenotyping of domestic cat (Felis catus) testicular cells across postnatal development—A model for wild felids. Theriogenol. Wild 2023, 2, 100031. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Wang, H.; Ma, K.; Wu, Y.; Qi, X.; Liu, Z.; Li, Q.; Zhang, Y.; Ma, Y. Identification and functional characterization of developmental-stage-dependent piRNAs in Tibetan sheep testes. J. Anim. Sci. 2023, 101, skad189. [Google Scholar] [CrossRef]
- Zhou, X.; Cui, J.; Meng, J.; Luan, Y. Interactions and links among the noncoding RNAs in plants under stresses. Theor. Appl. Genet. 2020, 133, 3235–3248. [Google Scholar] [CrossRef]
- Banfalvi, G. Origin of Coding RNA from Random-Sequence RNA. DNA Cell Biol. 2019, 38, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Varma, E.; Luo, X.; Muthukumar, T. Dissecting the human kidney allograft transcriptome: Single-cell RNA sequencing. Curr. Opin. Organ. Transplant. 2021, 26, 43–51. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhou, X.; Li, R.; Michal, J.J.; Zhang, S.; Dodson, M.V.; Zhang, Z.; Harland, R.M. Whole transcriptome analysis with sequencing: Methods, challenges and potential solutions. Cell Mol. Life Sci. 2015, 72, 3425–3439. [Google Scholar] [CrossRef] [PubMed]
- Zormpas, E.; Queen, R.; Comber, A.; Cockell, S.J. Mapping the transcriptome: Realizing the full potential of spatial data analysis. Cell 2023, 186, 5677–5689. [Google Scholar] [CrossRef]
- Bawa, G.; Liu, Z.; Yu, X.; Tran, L.P.; Sun, X. Introducing single cell stereo-sequencing technology to transform the plant transcriptome landscape. Trends Plant Sci. 2024, 29, 249–265. [Google Scholar] [CrossRef]
- Chen, Y.; Song, J.; Ruan, Q.; Zeng, X.; Wu, L.; Cai, L.; Wang, X.; Yang, C. Single-Cell Sequencing Methodologies: From Transcriptome to Multi-Dimensional Measurement. Small Methods 2021, 5, e2100111. [Google Scholar] [CrossRef]
- Tang, F.; Barbacioru, C.; Wang, Y.; Nordman, E.; Lee, C.; Xu, N.; Wang, X.; Bodeau, J.; Tuch, B.B.; Siddiqui, A.; et al. mRNA-Seq whole-transcriptome analysis of a single cell. Nat. Methods 2009, 6, 377–382. [Google Scholar] [CrossRef]
- Islam, S.; Kjällquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lönnerberg, P.; Linnarsson, S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Butler, A.; Hoffman, P.; Hafemeister, C.; Papalexi, E.; Mauck, W.M.; Hao, Y.; Stoeckius, M.; Smibert, P.; Satija, R. Comprehensive Integration of Single-Cell Data. Cell 2019, 177, 1888–1902.e21. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; He, X.; Zhao, Y.; Bai, D.; Du, M.; Song, L.; Liu, Z.; Yin, Z.; Manglai, D. Transcriptome profiling of developing testes and spermatogenesis in the Mongolian horse. BMC Genet. 2020, 21, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.L.; Zhang, X.Y.; Zhao, J.X.; Zhu, K.X.; Liu, S.Q.; Zhang, T.; Sun, Y.J.; Wang, J.J.; Shen, W. Multispecies comparative analysis reveals transcriptional specificity during Mongolian horse testicular development. Reprod. Domest. Anim. 2022, 57, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.D.; Lane, J.G.; Waldron, K.R. Is dentition an accurate indication of the age of a horse? Vet. Rec. 1994, 135, 31–34. [Google Scholar] [CrossRef]
- Carmalt, J.L.; Allen, A.L. Morphology of the occlusal surfaces of premolar and molar teeth as an indicator of age in the horse. J. Vet. Dent. 2008, 25, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Dard-Dascot, C.; Naquin, D.; d’Aubenton-Carafa, Y.; Alix, K.; Thermes, C.; van Dijk, E. Systematic comparison of small RNA library preparation protocols for next-generation sequencing. BMC Genom. 2018, 19, 118. [Google Scholar] [CrossRef] [PubMed]
- Dunning, M.J.; Barbosa-Morais, N.L.; Lynch, A.G.; Tavaré, S.; Ritchie, M.E. Statistical issues in the analysis of Illumina data. BMC Bioinform. 2008, 9, 85. [Google Scholar] [CrossRef]
- Benitez, J.A.; Cheng, S.; Deng, Q. Revealing allele-specific gene expression by single-cell transcriptomics. Int. J. Biochem. Cell Biol. 2017, 90, 155–160. [Google Scholar] [CrossRef] [PubMed]
- You, Y.; Tian, L.; Su, S.; Dong, X.; Jabbari, J.S.; Hickey, P.F.; Ritchie, M.E. Benchmarking UMI-based single-cell RNA-seq preprocessing workflows. Genome Biol. 2021, 22, 339. [Google Scholar] [CrossRef]
- Giannone, A.A.; Sellitto, C.; Rosati, B.; McKinnon, D.; White, T.W. Single-Cell RNA Sequencing Analysis of the Early Postnatal Mouse Lens Epithelium. Investig. Ophthalmol. Vis. Sci. 2023, 64, 37. [Google Scholar] [CrossRef] [PubMed]
- Niu, W.; Zhang, Y.; Liu, H.; Liang, N.; Xu, L.; Li, Y.; Yao, W.; Shi, W.; Liu, Z. Single-Cell Profiling Uncovers the Roles of Endometrial Fibrosis and Microenvironmental Changes in Adenomyosis. J. Inflamm. Res. 2023, 16, 1949–1965. [Google Scholar] [CrossRef]
- Li, B.; He, X.; Zhao, Y.; Bai, D.; Li, D.; Zhou, Z.; Manglai, D. Analysis of the miRNA transcriptome during testicular development and spermatogenesis of the Mongolian horse. Reprod. Fertil. Dev. 2020, 32, 582–593. [Google Scholar] [CrossRef] [PubMed]
- Bao, T.; Han, H.; Li, B.; Zhao, Y.; Bou, G.; Zhang, X.; Du, M.; Zhao, R.; Mongke, T.; Laxima; et al. The distinct transcriptomes of fast-twitch and slow-twitch muscles in Mongolian horses. Comp. Biochem. Physiol. Part D Genom. Proteom. 2020, 33, 100649. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Bou, G.; Zhang, X.; Tao, L.; Shen, Y.; Na, R.; Liu, G.; Ren, H.; Ren, X.; Song, L.; et al. A Fast PCR Test for the Simultaneous Identification of Species and Gender in Horses, Donkeys, Mules and Hinnies. J. Equine Vet. Sci. 2021, 102, 103458. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; McGivney, B.A.; Allen, L.; Bai, D.; Corduff, L.R.; Davaakhuu, G.; Davaasambuu, J.; Dorjgotov, D.; Hall, T.J.; Hemmings, A.J.; et al. Common protein-coding variants influence the racing phenotype in galloping racehorse breeds. Commun. Biol. 2022, 5, 1320. [Google Scholar] [CrossRef] [PubMed]
- Bou, T.; Han, H.; Mongke, T.; Zhao, R.; La, X.; Ding, W.; Jia, Z.; Liu, H.; Tiemuqier, A.; An, T.; et al. Fast and slow myofiber-specific expression profiles are affected by noncoding RNAs in Mongolian horses. Comp. Biochem. Physiol. Part D Genom. Proteom. 2022, 41, 100942. [Google Scholar] [CrossRef] [PubMed]
- Bou, T.; Ding, W.; Liu, H.; Gong, W.; Jia, Z.; Dugarjaviin, M.; Bai, D. A genome-wide landscape of mRNAs, miRNAs, lncRNAs, and circRNAs of skeletal muscles during dietary restriction in Mongolian horses. Comp. Biochem. Physiol. Part D Genom. Proteom. 2023, 46, 101084. [Google Scholar] [CrossRef]
- Xing, J.; Xie, L.; Qi, X.; Liu, G.; Akhtar, M.F.; Li, X.; Bou, G.; Bai, D.; Zhao, Y.; Dugarjaviin, M.; et al. Integrated analysis of transcriptome and proteome for exploring mechanism of promoting proliferation of equine satellite cells associated with leucine. Comp. Biochem. Physiol. Part D Genom. Proteom. 2023, 48, 101118. [Google Scholar] [CrossRef]
- Xing, J.; Qi, X.; Liu, G.; Li, X.; Gao, X.; Bou, G.; Bai, D.; Zhao, Y.; Du, M.; Dugarjaviin, M.; et al. A Transcriptomic Regulatory Network among miRNAs, lncRNAs, circRNAs, and mRNAs Associated with L-leucine-induced Proliferation of Equine Satellite Cells. Animals 2023, 13, 208. [Google Scholar] [CrossRef]
- Ren, X.; Liu, Y.; Zhao, Y.; Li, B.; Bai, D.; Bou, G.; Zhang, X.; Du, M.; Wang, X.; Bou, T.; et al. Analysis of the Whole-Genome Sequences from an Equus Parent-Offspring Trio Provides Insight into the Genomic Incompatibilities in the Hybrid Mule. Genes 2022, 13, 2188. [Google Scholar] [CrossRef] [PubMed]
- Kruppa, J.; Hothorn, L. A comparison study on modeling of clustered and overdispersed count data for multiple comparisons. J. Appl. Stat. 2020, 48, 3220–3232. [Google Scholar] [CrossRef] [PubMed]
- Lowry, S.R. Use and misuse of multiple comparisons in animal experiments. J. Anim. Sci. 1992, 70, 1971–1977. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, M.G.; Gagliano-Jucá, T.; Basaria, S. Male Reproduction and Aging. Endocrinol. Metab. Clin. N. Am. 2023, 52, 211–228. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Guo, J.; Zhou, B.; Li, C.; Qiao, T.; Hua, L.; Jiang, Y.; Mai, Z.; Yu, S.; Tian, Y.; et al. Chestnut polysaccharide rescues the damaged spermatogenesis process of asthenozoospermia-model mice by upregulating the level of palmitic acid. Front. Endocrinol. 2023, 14, 1222635. [Google Scholar] [CrossRef] [PubMed]
- Valentini, L.; Zupa, R.; Pousis, C.; Cuko, R.; Corriero, A. Proliferation and Apoptosis of Cat (Felis catus) Male Germ Cells during Breeding and Non-Breeding Seasons. Vet. Sci. 2022, 9, 447. [Google Scholar] [CrossRef] [PubMed]
- Lesch, B.J.; Silber, S.J.; McCarrey, J.R.; Page, D.C. Parallel evolution of male germline epigenetic poising and somatic development in animals. Nat. Genet. 2016, 48, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Kleemann, D.O.; Kelly, J.M.; Arney, L.J.; Len, J.; Tilbrook, A.J.; Walker, S.K. Sexual behaviour, semen quality and fertility of young Border Leicester rams administered melatonin during spring. Anim. Reprod. Sci. 2021, 231, 106804. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; He, X.; Zhao, Y.; Bai, D.; Bou, G.; Zhang, X.; Su, S.; Dao, L.; Liu, R.; Wang, Y.; et al. Identification of piRNAs and piRNA clusters in the testes of the Mongolian horse. Sci. Rep. 2019, 9, 5022. [Google Scholar] [CrossRef]
- Cheung, S.; Xie, P.; Rosenwaks, Z.; Palermo, G.D. Profiling the male germline genome to unravel its reproductive potential. Fertil. Steril. 2023, 119, 196–206. [Google Scholar] [CrossRef]
- Griswold, M.D. The central role of Sertoli cells in spermatogenesis. Semin. Cell Dev. Biol. 1998, 9, 411–416. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liang, Z.; Yang, J.; Wang, D.; Wang, H.; Zhu, M.; Geng, B.; Xu, E.Y. DAZL is a master translational regulator of murine spermatogenesis. Natl. Sci. Rev. 2019, 6, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Grow, E.J.; Mlcochova, H.; Maher, G.J.; Lindskog, C.; Nie, X.; Guo, Y.; Takei, Y.; Yun, J.; Cai, L.; et al. The adult human testis transcriptional cell atlas. Cell Res. 2018, 28, 1141–1157. [Google Scholar] [CrossRef] [PubMed]
- Green, C.D.; Ma, Q.; Manske, G.L.; Shami, A.N.; Zheng, X.; Marini, S.; Moritz, L.; Sultan, C.; Gurczynski, S.J.; Moore, B.B.; et al. A Comprehensive Roadmap of Murine Spermatogenesis Defined by Single-Cell RNA-Seq. Dev. Cell 2018, 46, 651–667.e10. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Zhang, J.; Zhang, P.; Huang, X.; Yang, W.; Liu, R.; Sun, Q.; Lu, Y.; Zhang, M.; Fu, Q. Single-cell RNA sequencing uncovers dynamic roadmap and cell-cell communication during buffalo spermatogenesis. iScience 2022, 26, 105733. [Google Scholar] [CrossRef]
- Hess, R.A.; Renato de Franca, L. Spermatogenesis and cycle of the seminiferous epithelium. Adv. Exp. Med. Biol. 2008, 636, 1–15. [Google Scholar]
- Hilz, S.; Modzelewski, A.J.; Cohen, P.E.; Grimson, A. The roles of microRNAs and siRNAs in mammalian spermatogenesis. Development 2016, 143, 3061–3073. [Google Scholar] [CrossRef]
- Maclean, J.A.; Wilkinson, M.F. Gene regulation in spermatogenesis. Curr. Top. Dev. Biol. 2005, 71, 131–197. [Google Scholar]
- Beckers, A.; Fuhl, F.; Ott, T.; Boldt, K.; Brislinger, M.M.; Walentek, P.; Schuster-Gossler, K.; Hegermann, J.; Alten, L.; Kremmer, E.; et al. The highly conserved FOXJ1 target CFAP161 is dispensable for motile ciliary function in mouse and Xenopus. Sci. Rep. 2021, 11, 13333. [Google Scholar] [CrossRef]
- Raghupathy, R.K.; Zhang, X.; Liu, F.; Alhasani, R.H.; Biswas, L.; Akhtar, S.; Pan, L.; Moens, C.B.; Li, W.; Liu, M.; et al. Rpgrip1 is required for rod outer segment development and ciliary protein trafficking in zebrafish. Sci. Rep. 2017, 7, 16881. [Google Scholar] [CrossRef]
- Ishijima, S.; Mohri, H. A quantitative description of flagellar movement in golden hamster spermatozoa. J. Exp. Biol. 1985, 114, 463–475. [Google Scholar] [CrossRef] [PubMed]
- François-Heude, M.C.; Lebigot, E.; Roze, E.; Warde, M.T.A.; Cances, C.; Damaj, L.; Espil, C.; Fluss, J.; de Lonlay, P.; Kern, I.; et al. Movement disorders in valine métabolism diseases caused by HIBCH and ECHS1 deficiencies. Eur. J. Neurol. 2022, 29, 3229–3242. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Roser, J.F.; Yoon, M. UTF1, a putative marker for spermatogonial stem cells in stallions. PLoS ONE 2014, 9, e108825. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Sun, L.; Wang, L.; Yao, B.; Mo, H.; Yang, W. LncRNA SNHG7 accelerates the proliferation, migration and invasion of hepatocellular carcinoma cells via regulating miR-122-5p and RPL4. Biomed. Pharmacother. 2019, 118, 109386. [Google Scholar] [CrossRef] [PubMed]
- Grasser, K.D. The FACT Histone Chaperone: Tuning Gene Transcription in the Chromatin Context to Modulate Plant Growth and Development. Front. Plant Sci. 2020, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Holleboom, A.G.; Jakulj, L.; Franssen, R.; Decaris, J.; Vergeer, M.; Koetsveld, J.; Luchoomun, J.; Glass, A.; Hellerstein, M.K.; Kastelein, J.J.; et al. In vivo tissue cholesterol efflux is reduced in carriers of a mutation in APOA1. J. Lipid Res. 2013, 54, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Gautam, D.; Vats, A.; Pal, P.; Haldar, A.; De, S. Characterization of Anti-Müllerian Hormone (AMH) Gene in Buffaloes and Goats. Front. Vet. Sci. 2021, 8, 627094. [Google Scholar] [CrossRef]
- Vandova, V.; Vankova, P.; Durech, M.; Houser, J.; Kavan, D.; Man, P.; Muller, P.; Trcka, F. HSPA1A conformational mutants reveal a conserved structural unit in Hsp70 proteins. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129458. [Google Scholar] [CrossRef] [PubMed]
- Cannarella, R.; Mancuso, F.; Arato, I.; Lilli, C.; Bellucci, C.; Gargaro, M.; Curto, R.; Aglietti, M.C.; La Vignera, S.; Condorelli, R.A.; et al. Sperm-carried IGF2 downregulated the expression of mitogens produced by Sertoli cells: A paracrine mechanism for regulating spermatogenesis. Front. Endocrinol. 2022, 13, 1010796. [Google Scholar] [CrossRef]
- Intasqui, P.; Agarwal, A.; Sharma, R.; Samanta, L.; Bertolla, R.P. Towards the identification of reliable sperm biomarkers for male infertility: A sperm proteomic approach. Andrologia 2018, 50, e12919. [Google Scholar] [CrossRef]
- Nasirshalal, M.; Tahmasebi-Birgani, M.; Dadfar, M.; Nikbakht, R.; Saberi, A.; Ghandil, P. Identification of the PRM1 gene mutations in oligoasthenoteratozoospermic men. Andrologia 2020, 52, e13872. [Google Scholar] [CrossRef]
- Siebert-Kuss, L.M.; Krenz, H.; Tekath, T.; Wöste, M.; Di Persio, S.; Terwort, N.; Wyrwoll, M.J.; Cremers, J.F.; Wistuba, J.; Dugas, M.; et al. Transcriptome analyses in infertile men reveal germ cell-specific expression and splicing patterns. Life Sci. Alliance 2022, 6, e202201633. [Google Scholar] [CrossRef] [PubMed]
- Meistrich, M.L.; Hess, R.A. Assessment of spermatogenesis through staging of seminiferous tubules. Methods Mol. Biol. 2013, 927, 299–307. [Google Scholar] [PubMed]
- Zirkin, B.R. Spermatogenesis: Its regulation by testosterone and FSH. Semin. Cell Dev. Biol. 1998, 9, 417–421. [Google Scholar] [CrossRef] [PubMed]
- Saewu, A.; Kongmanas, K.; Raghupathy, R.; Netherton, J.; Kadunganattil, S.; Linton, J.J.; Chaisuriyong, W.; Faull, K.F.; Baker, M.A.; Tanphaichitr, N. Primary Sertoli Cell Cultures from Adult Mice Have Different Properties Compared with Those Derived from 20-Day-Old Animals. Endocrinology 2020, 161, bqz020. [Google Scholar] [CrossRef] [PubMed]
- Kasimanickam, V.R.; Kasimanickam, R.K. Sertoli, Leydig, and Spermatogonial Cells’ Specific Gene and Protein Expressions as Dog Testes Evolve from Immature into Mature States. Animals 2022, 12, 271. [Google Scholar] [CrossRef]
- Fayomi, A.P.; Orwig, K.E. Spermatogonial stem cells and spermatogenesis in mice, monkeys and men. Stem. Cell Res. 2018, 29, 207–214. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Y.; Du, M.; Li, X.; Zhang, L.; Zhao, B.; Wang, N.; Dugarjaviin, M. Single-Cell Transcriptome Sequencing Reveals Molecular Expression Differences and Marker Genes in Testes during the Sexual Maturation of Mongolian Horses. Animals 2024, 14, 1258. https://doi.org/10.3390/ani14091258
Liu Y, Du M, Li X, Zhang L, Zhao B, Wang N, Dugarjaviin M. Single-Cell Transcriptome Sequencing Reveals Molecular Expression Differences and Marker Genes in Testes during the Sexual Maturation of Mongolian Horses. Animals. 2024; 14(9):1258. https://doi.org/10.3390/ani14091258
Chicago/Turabian StyleLiu, Yuanyi, Ming Du, Xinyu Li, Lei Zhang, Bilig Zhao, Na Wang, and Manglai Dugarjaviin. 2024. "Single-Cell Transcriptome Sequencing Reveals Molecular Expression Differences and Marker Genes in Testes during the Sexual Maturation of Mongolian Horses" Animals 14, no. 9: 1258. https://doi.org/10.3390/ani14091258