
Interactions between Cytokines and the Pathogenesis of Prion Diseases: Insights and Implications
 ,
,  , ,
, , 
Abstract
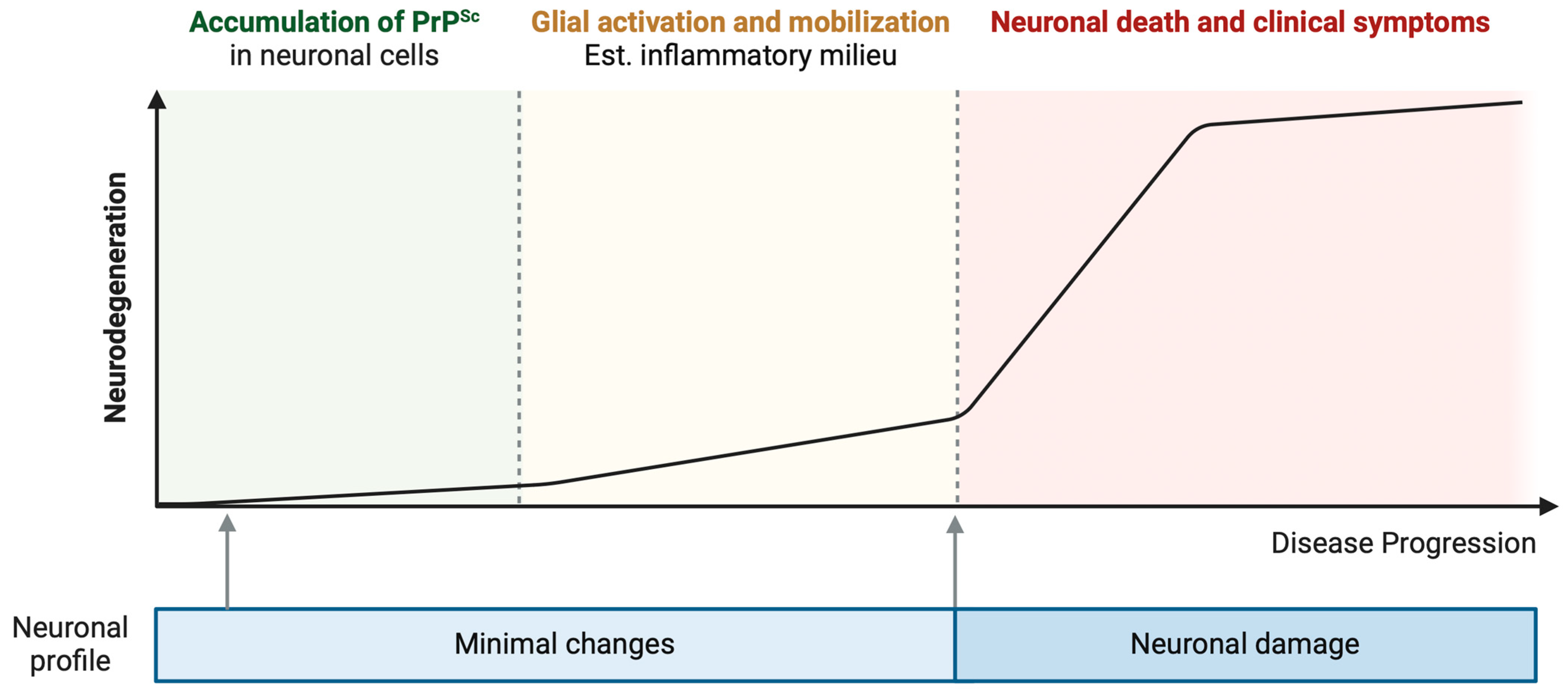
:1. Introduction
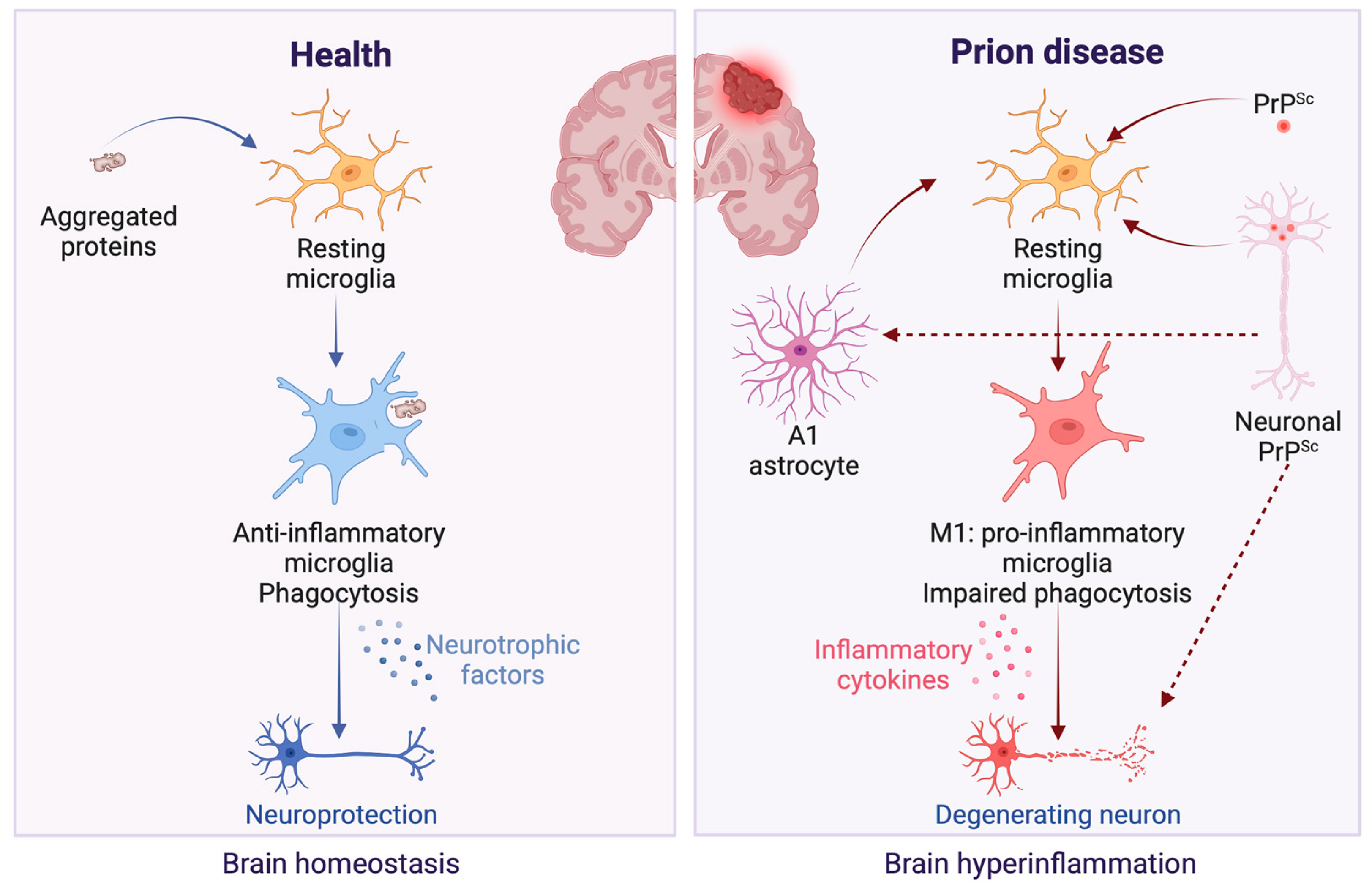
2. Immune Response Activation in Prion Infection
2.1. Glia Activation
2.2. Neuronal Response

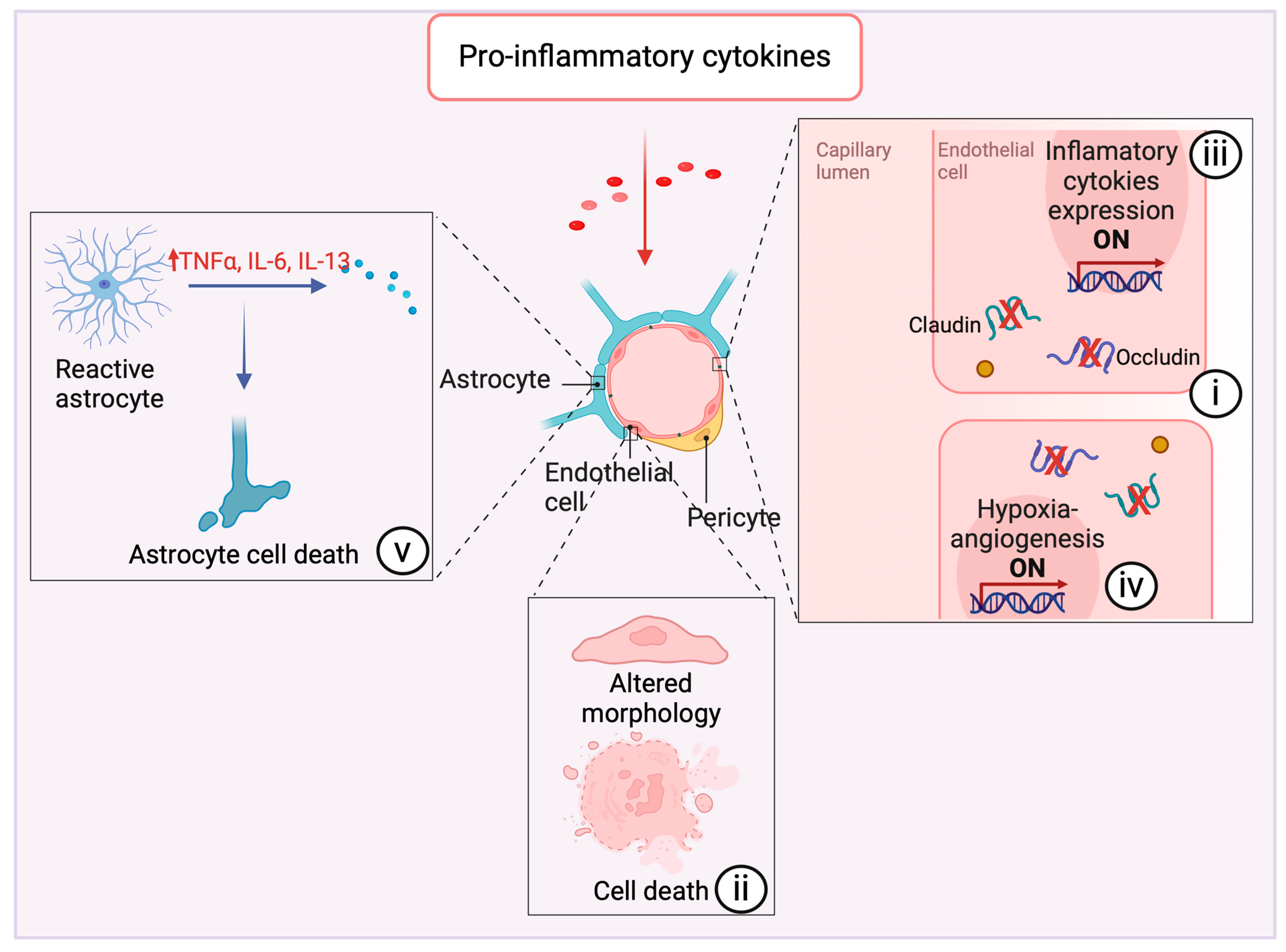
2.3. The Blood–Brain Barrier
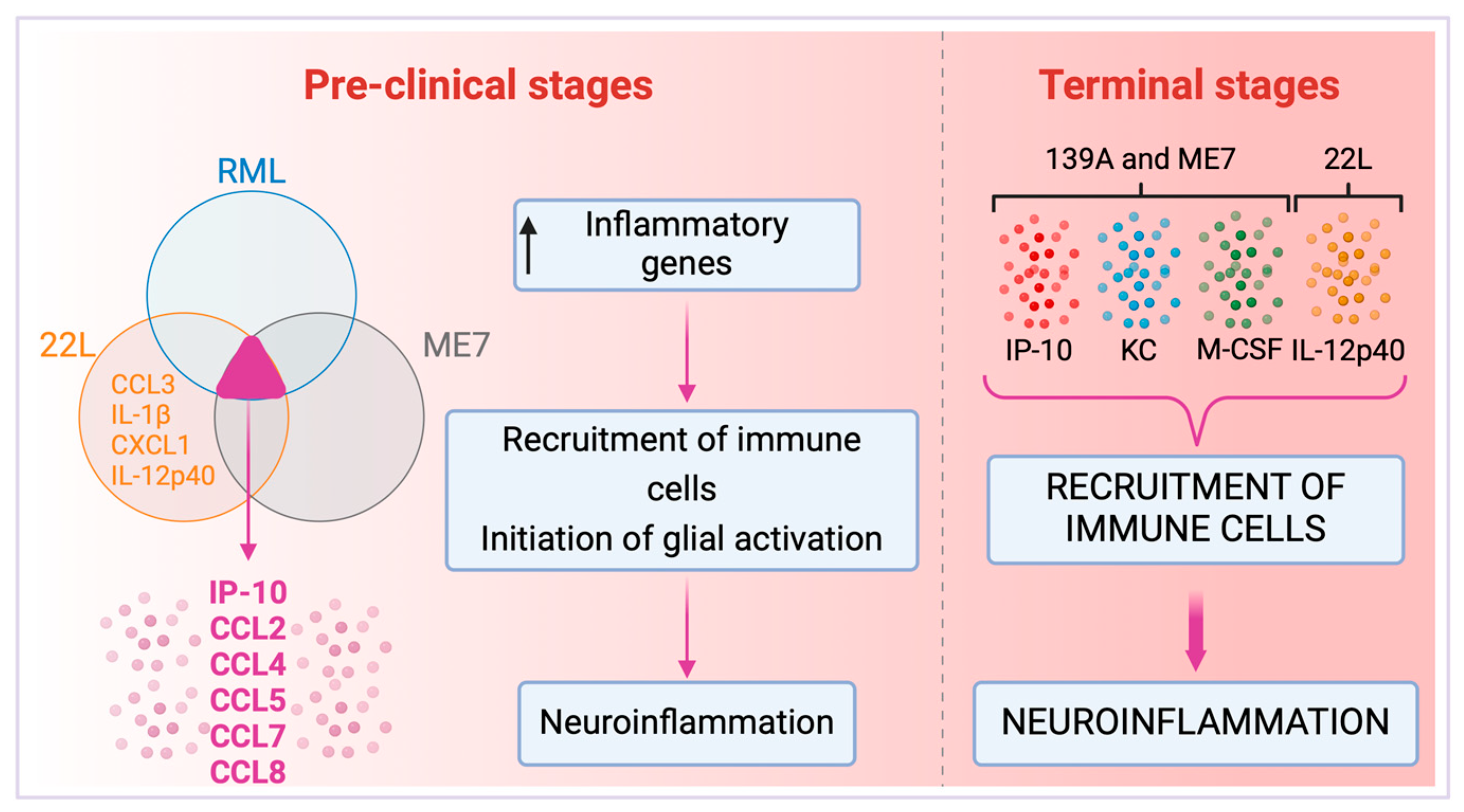
3. Cytokine Profiles
Cytokines and Prion Strains
4. Diagnostic and Therapeutic Potential
Immunomodulation
5. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef] [PubMed]
- Marin-Moreno, A.; Fernández-Borges, N.; Espinosa, J.C.; Andréoletti, O.; Torres, J.M. Transmission and Replication of Prions. Prog. Mol. Biol. Transl. Sci. 2017, 150, 181–201. [Google Scholar]
- Bradley, R. Bovine spongiform encephalopathy. Update. Acta Neurobiol. Exp. 2002, 62, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Chesebro, B. Prion protein and the transmissible spongiform encephalopathies. Trends Cell Biol. 1997, 7, 56–62. [Google Scholar] [CrossRef]
- Sikorska, B.; Liberski, P.P. Human prion diseases: From Kuru to variant Creutzfeldt-Jakob disease. Subcell. Biochem. 2012, 65, 457–496. [Google Scholar] [PubMed]
- Griffith, J.S. Nature of the scrapie agent: Self-replication and scrapie. Nature 1967, 215, 1043–1044. [Google Scholar] [CrossRef] [PubMed]
- Aucouturier, P.; Carp, R.I.; Carnaud, C.; Wisniewski, T. Prion diseases and the immune system. Clin. Immunol. 2000, 96, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Grégoire, S.; Bergot, A.S.; Féraudet, C.; Carnaud, C.; Aucouturier, P.; Rosset, M.B. The murine B cell repertoire is severely selected against endogenous cellular prion protein. J. Immunol. 2005, 175, 6443–6449. [Google Scholar] [CrossRef]
- Porter, D.D.; Porter, H.G.; Cox, N.A. Failure to demonstrate a humoral immune response to scrapie infection in mice. J. Immunol. 1973, 111, 1407–1410. [Google Scholar] [CrossRef]
- Barcikowska, M.; Liberski, P.P.; Boellaard, J.W.; Brown, P.; Gajdusek, D.C.; Budka, H. Microglia is a component of the prion protein amyloid plaque in the Gerstmann-Sträussler-Scheinker syndrome. Acta Neuropathol. 1993, 85, 623–627. [Google Scholar] [CrossRef]
- Lakkaraju, A.K.K.; Sorce, S.; Senatore, A.; Nuvolone, M.; Guo, J.; Schwarz, P.; Moos, R.; Pelczar, P.; Aguzzi, A. Glial activation in prion diseases is selectively triggered by neuronal PrP(Sc). Brain Pathol. 2022, 32, e13056. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Xie, W.-L.; Zhang, B.; Chen, L.-N.; Xu, Y.; Wang, K.; Ren, K.; Zhang, X.-M.; Chen, C.; Zhang, J.; et al. Brain microglia were activated in sporadic CJD but almost unchanged in fatal familial insomnia and G114V genetic CJD. Virol. J. 2013, 10, 216–219. [Google Scholar] [CrossRef] [PubMed]
- Sorce, S.; Nuvolone, M.; Russo, G.; Chincisan, A.; Heinzer, D.; Avar, M.; Pfammatter, M.; Schwarz, P.; Delic, M.; Müller, M.; et al. Genome-wide transcriptomics identifies an early preclinical signature of prion infection. PLoS Pathog. 2020, 16, e1008653. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.E.; Lawson, L.J.; Perry, V.H.; Fraser, H. Characterization of the microglial response in murine scrapie. Neuropathol. Appl. Neurobiol. 1994, 20, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Scheckel, C.; Imeri, M.; Schwarz, P.; Aguzzi, A. Ribosomal profiling during prion disease uncovers progressive translational derangement in glia but not in neurons. eLife 2020, 9, e62911. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. Microglia Are Critical in Host Defense against Prion Disease. J. Virol. 2018, 92, e00549-18. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.M.; Wijaya, C.A.W.; Mabbott, N.A. Discrimination of Prion Strain Targeting in the Central Nervous System via Reactive Astrocyte Heterogeneity in CD44 Expression. Front. Cell. Neurosci. 2019, 13, 411. [Google Scholar] [CrossRef] [PubMed]
- Puoti, G.; Giaccone, G.; Mangieri, M.; Limido, L.; Fociani, P.; Zerbi, P.; Suardi, S.; Rossi, G.; Iussich, S.; Capobianco, R.; et al. Sporadic Creutzfeldt-Jakob disease: The extent of microglia activation is dependent on the biochemical type of PrPSc. J. Neuropathol. Exp. Neurol. 2005, 64, 902–909. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, H.; Koyama, S.; Yagita, K.; Shijo, M.; Matsuzono, K.; Hamasaki, H.; Kanemaru, T.; Okamoto, T.; Kai, K.; Aishima, S.; et al. Silence of resident microglia in GPI anchorless prion disease and activation of microglia in Gerstmann-Sträussler-Scheinker disease and sporadic Creutzfeldt-Jakob disease. J. Neuropathol. Exp. Neurol. 2022, 82, 38–48. [Google Scholar] [CrossRef]
- Michael, A.V.; Greenlee, J.J.; Harm, T.A.; Moore, S.J.; Zhang, M.; Lind, M.S.; Greenlee, M.H.W.; Smith, J.D. In Situ Temporospatial Characterization of the Glial Response to Prion Infection. Vet. Pathol. 2020, 57, 90–107. [Google Scholar] [CrossRef]
- Villacampa, N.; Almolda, B.; Vilella, A.; Campbell, I.L.; González, B.; Castellano, B. Astrocyte-targeted production of IL-10 induces changes in microglial reactivity and reduces motor neuron death after facial nerve axotomy. Glia 2015, 63, 1166–1184. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.C.; Lu, W.; Beckel, J.M.; Mitchell, C.H. Neuronal Release of Cytokine IL-3 Triggered by Mechanosensitive Autostimulation of the P2X7 Receptor Is Neuroprotective. Front. Cell. Neurosci. 2016, 10, 270. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.Y.; Hoffman, A.S.; Itoh, N.; Ao, Y.; Spence, R.; Sofroniew, M.V.; Voskuhl, R.R. Astrocyte CCL2 sustains immune cell infiltration in chronic experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2014, 274, 53–61. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Dong, H.; Huang, Y.; Lu, S.; Zhang, S.; Qian, Y.; Jin, W. Astrocyte-Derived CCL2 is Associated with M1 Activation and Recruitment of Cultured Microglial Cells. Cell. Physiol. Biochem. 2016, 38, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Ringheim, G.E.; Burgher, K.L.; Heroux, J.A. Interleukin-6 mRNA expression by cortical neurons in culture: Evidence for neuronal sources of interleukin-6 production in the brain. J. Neuroimmunol. 1995, 63, 113–123. [Google Scholar] [CrossRef]
- Kushwaha, R.; Li, Y.; Makarava, N.; Pandit, N.P.; Molesworth, K.; Birukov, K.G.; Baskakov, I.V. Reactive astrocytes associated with prion disease impair the blood brain barrier. bioRxiv 2023. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.T.; Mitchell, K.; Keller, J.M.; Iadarola, M.J. Peripheral inflammation increases Scya2 expression in sensory ganglia and cytokine and endothelial related gene expression in inflamed tissue. J. Neurochem. 2007, 103, 1628–1643. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.; Closhen, D.; Croxford, A.; White, R.; Kulig, P.; Pietrowski, E.; Bechmann, I.; Becher, B.; Luhmann, H.J.; Waisman, A.; et al. Cellular mechanisms of IL-17-induced blood-brain barrier disruption. FASEB J. 2010, 24, 1023–1034. [Google Scholar] [CrossRef]
- Labus, J.; Häckel, S.; Lucka, L.; Danker, K. Interleukin-1beta induces an inflammatory response and the breakdown of the endothelial cell layer in an improved human THBMEC-based in vitro blood-brain barrier model. J. Neurosci. Methods 2014, 228, 35–45. [Google Scholar] [CrossRef]
- van Kralingen, C.; Kho, D.T.; Costa, J.; Angel, C.E.; Graham, E.S. Exposure to inflammatory cytokines IL-1beta and TNFalpha induces compromise and death of astrocytes; implications for chronic neuroinflammation. PLoS ONE 2013, 8, e84269. [Google Scholar] [CrossRef] [PubMed]
- Tribouillard-Tanvier, D.; Race, B.; Striebel, J.F.; Carroll, J.A.; Phillips, K.; Chesebro, B. Early cytokine elevation, PrPres deposition, and gliosis in mouse scrapie: No effect on disease by deletion of cytokine genes IL-12p40 and IL-12p35. J. Virol. 2012, 86, 10377–10383. [Google Scholar] [CrossRef] [PubMed]
- Bradford, B.M.; McGuire, L.I.; Hume, D.A.; Pridans, C.; Mabbott, N.A. Microglia deficiency accelerates prion disease but does not enhance prion accumulation in the brain. Glia 2022, 70, 2169–2187. [Google Scholar] [CrossRef]
- Zhu, C.; Herrmann, U.S.; Falsig, J.; Abakumova, I.; Nuvolone, M.; Schwarz, P.; Frauenknecht, K.; Rushing, E.J.; Aguzzi, A. A neuroprotective role for microglia in prion diseases. J. Exp. Med. 2016, 213, 1047–1059. [Google Scholar] [CrossRef]
- Wang, Y.; Hartmann, K.; Thies, E.; Mohammadi, B.; Altmeppen, H.; Sepulveda-Falla, D.; Glatzel, M.; Krasemann, S. Loss of Homeostatic Microglia Signature in Prion Diseases. Cells 2022, 11, 2948. [Google Scholar] [CrossRef] [PubMed]
- Falsig, J.; Julius, C.; Margalith, I.; Schwarz, P.; Heppner, F.L.; Aguzzi, A. A versatile prion replication assay in organotypic brain slices. Nat. Neurosci. 2008, 11, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Thackray, A.M.; McKenzie, A.N.; Klein, M.A.; Lauder, A.; Bujdoso, R. Accelerated prion disease in the absence of interleukin-10. J. Virol. 2004, 78, 13697–13707. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Sepulveda-Falla, D.; Rose, I.V.L.; Madore, C.; Muth, C.; Matschke, J.; Butovsky, O.; Liddelow, S.; Glatzel, M.; Krasemann, S. Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol. Commun. 2019, 7, 83. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization from M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Ugalde, C.L.; Lewis, V.; Stehmann, C.; McLean, C.A.; Lawson, V.A.; Collins, S.J.; Hill, A.F. Markers of A1 astrocytes stratify to molecular sub-types in sporadic Creutzfeldt–Jakob disease brain. Brain Commun. 2020, 2, fcaa029. [Google Scholar] [CrossRef]
- Carroll, J.A.; Race, B.; Williams, K.; Striebel, J.; Chesebro, B. RNA-seq and network analysis reveal unique glial gene expression signatures during prion infection. Mol. Brain 2020, 13, 71. [Google Scholar] [CrossRef]
- Makarava, N.; Mychko, O.; Chang, J.C.-Y.; Molesworth, K.; Baskakov, I.V. The degree of astrocyte activation is predictive of the incubation time to prion disease. Acta Neuropathol. Commun. 2021, 9, 87. [Google Scholar] [CrossRef]
- Aguzzi, A.; Nuvolone, M.; Zhu, C. The immunobiology of prion diseases. Nat. Rev. Immunol. 2013, 13, 888–902. [Google Scholar] [CrossRef] [PubMed]
- Crespo, I.; Roomp, K.; Jurkowski, W.; Kitano, H.; del Sol, A. Gene regulatory network analysis supports inflammation as a key neurodegeneration process in prion disease. BMC Syst. Biol. 2012, 6, 132. [Google Scholar] [CrossRef]
- Banks, W. Blood-brain barrier transport of cytokines: A mechanism for neuropathology. Curr. Pharm. Des. 2005, 11, 973–984. [Google Scholar] [CrossRef] [PubMed]
- Bourgognon, J.M.; Cavanagh, J. The role of cytokines in modulating learning and memory and brain plasticity. Brain Neurosci. Adv. 2020, 4, 2398212820979802. [Google Scholar] [CrossRef]
- Prieto, G.A.; Cotman, C.W. Cytokines and cytokine networks target neurons to modulate long-term potentiation. Cytokine Growth Factor Rev. 2017, 34, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Sawada, M.; Itoh, Y.; Suzumura, A.; Marunouchi, T. Expression of cytokine receptors in cultured neuronal and glial cells. Neurosci. Lett. 1993, 160, 131–134. [Google Scholar] [CrossRef]
- Gadient, R.; Otten, U. Identification of interleukin-6 (IL-6)-expressing neurons in the cerebellum and hippocampus of normal adult rats. Neurosci. Lett. 1994, 182, 243–246. [Google Scholar] [CrossRef]
- Schöbitz, B.; de Kloet, E.R.; Sutanto, W.; Holsboer, F. Cellular localization of interleukin 6 mRNA and interleukin 6 receptor mRNA in rat brain. Eur. J. Neurosci. 1993, 5, 1426–1435. [Google Scholar] [CrossRef]
- Breder, C.D.; Dinarello, C.A.; Saper, C.B. Interleukin-1 immunoreactive innervation of the human hypothalamus. Science 1988, 240, 321–324. [Google Scholar] [CrossRef]
- Boulanger, L.M. Immune proteins in brain development and synaptic plasticity. Neuron 2009, 64, 93–109. [Google Scholar] [CrossRef] [PubMed]
- Galic, M.A.; Riazi, K.; Pittman, Q.J. Cytokines and brain excitability. Front. Neuroendocr. 2012, 33, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Park, K.M.; Bowers, W.J. Tumor necrosis factor-alpha mediated signaling in neuronal homeostasis and dysfunction. Cell. Signal. 2010, 22, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Vereyken, E.J.F.; Bajova, H.; Chow, S.; De Graan, P.N.E.; Gruol, D.L. Chronic interleukin-6 alters the level of synaptic proteins in hippocampus in culture and in vivo. Eur. J. Neurosci. 2007, 25, 3605–3616. [Google Scholar] [CrossRef] [PubMed]
- Breder, C.D.; Hazuka, C.; Ghayur, T.; Klug, C.; Huginin, M.; Yasuda, K.; Teng, M.; Saper, C.B. Regional induction of tumor necrosis factor alpha expression in the mouse brain after systemic lipopolysaccharide administration. Proc. Natl. Acad. Sci. USA 1994, 91, 11393–11397. [Google Scholar] [CrossRef] [PubMed]
- Schlachetzki, J.C.M.; Süβ, P.; Lana, A.J. Chronic peripheral inflammation: A possible contributor to neurodegenerative diseases. Neural Regen. Res. 2021, 16, 1711–1714. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wang, G.; Zhang, F. Role of Peripheral Immune Cells-Mediated Inflammation on the Process of Neurodegenerative Diseases. Front. Immunol. 2020, 11, 582825. [Google Scholar] [CrossRef]
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef]
- Mott, R.T.; Ait-Ghezala, G.; Town, T.; Mori, T.; Vendrame, M.; Zeng, J.; Ehrhart, J.; Mullan, M.; Tan, J. Neuronal expression of CD22: Novel mechanism for inhibiting microglial proinflammatory cytokine production. Glia 2004, 46, 369–379. [Google Scholar] [CrossRef]
- Tan, J.; Town, T.; Mori, T.; Wu, Y.; Saxe, M.; Crawford, F.; Mullan, M. CD45 opposes beta-amyloid peptide-induced microglial activation via inhibition of p44/42 mitogen-activated protein kinase. J. Neurosci. 2000, 20, 7587–7594. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, L.; Engelhardt, B. Immune cell trafficking across the blood-brain barrier in the absence and presence of neuroinflammation. Vasc. Biol. 2020, 2, H1–H18. [Google Scholar] [CrossRef] [PubMed]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Dijkhuizen, R.M.; Reiss, Y.; Plate, K.H.; Agalliu, D.; Constantin, G. Functional morphology of the blood–brain barrier in health and disease. Acta Neuropathol. 2018, 135, 311–336. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Keep, R.F.; Wang, M.M.; Jankovic, I.; Andjelkovic, A.V. Caveolae-mediated internalization of occludin and claudin-5 during CCL2-induced tight junction remodeling in brain endothelial cells. J. Biol. Chem. 2009, 284, 19053–19066. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; Williams, A.; Farquhar, C.F.; Pasparakis, M.; Kollias, G.; Bruce, M.E. Tumor necrosis factor alpha-deficient, but not interleukin-6-deficient, mice resist peripheral infection with scrapie. J. Virol. 2000, 74, 3338–3344. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.; Ahmad, N.; Srivastava, R.; Hemmer, B. TNF-alpha induced NFkappaB signaling and p65 (RelA) overexpression repress Cldn5 promoter in mouse brain endothelial cells. Cytokine 2012, 57, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gutierrez, R.; Morales, R. The prion-like phenomenon in Alzheimer’s disease: Evidence of pathology transmission in humans. PLoS Pathog. 2020, 16, e1009004. [Google Scholar] [CrossRef] [PubMed]
- Vaquer-Alicea, J.; Diamond, M.I. Propagation of Protein Aggregation in Neurodegenerative Diseases. Annu. Rev. Biochem. 2019, 88, 785–810. [Google Scholar] [CrossRef]
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; van Horssen, J.; de Vries, H.E.; Rozemuller, A.J. Neuroinflammation and blood-brain barrier changes in capillary amyloid angiopathy. Neurodegener. Dis. 2012, 10, 329–331. [Google Scholar] [CrossRef]
- Yarlagadda, A.; Alfson, E.; Clayton, A.H. The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry 2009, 6, 18–22. [Google Scholar] [PubMed]
- Chen, A.Q.; Fang, Z.; Chen, X.L.; Yang, S.; Zhou, Y.F.; Mao, L.; Xia, Y.P.; Jin, H.J.; Li, Y.N.; You, M.F.; et al. Microglia-derived TNF-α mediates endothelial necroptosis aggravating blood brain–barrier disruption after ischemic stroke. Cell Death Dis. 2019, 10, 487. [Google Scholar] [CrossRef]
- Ni, Y.; Teng, T.; Li, R.; Simonyi, A.; Sun, G.Y.; Lee, J.C. TNFalpha alters occludin and cerebral endothelial permeability: Role of p38MAPK. PLoS ONE 2017, 12, e0170346. [Google Scholar] [CrossRef] [PubMed]
- Argaw, A.T.; Zhang, Y.; Snyder, B.J.; Zhao, M.-L.; Kopp, N.; Lee, S.C.; Raine, C.S.; Brosnan, C.F.; John, G.R. IL-1beta regulates blood-brain barrier permeability via reactivation of the hypoxia-angiogenesis program. J. Immunol. 2006, 177, 5574–5584. [Google Scholar] [CrossRef]
- Bartz, J.C. Prion Strain Diversity. Cold Spring Harb. Perspect. Med. 2016, 6, a024349. [Google Scholar] [CrossRef]
- Manka, S.W.; Wenborn, A.; Betts, J.; Joiner, S.; Saibil, H.R.; Collinge, J.; Wadsworth, J.D.F. A structural basis for prion strain diversity. Nat. Chem. Biol. 2023, 19, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Makarava, N.; Chang, J.C.-Y.; Kushwaha, R.; Baskakov, I.V. Region-Specific Response of Astrocytes to Prion Infection. Front. Neurosci. 2019, 13, 1048. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.; Riccardi, G.; Iacobone, F.; Chiarotti, F.; Pirisinu, L.; Vanni, I.; Marcon, S.; D’agostino, C.; Giovannelli, M.; Parchi, P.; et al. Strain-Dependent Morphology of Reactive Astrocytes in Human- and Animal-Vole-Adapted Prions. Biomolecules 2023, 13, 757. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.A.; Striebel, J.F.; Rangel, A.; Woods, T.; Phillips, K.; Peterson, K.E.; Race, B.; Chesebro, B. Prion Strain Differences in Accumulation of PrPSc on Neurons and Glia Are Associated with Similar Expression Profiles of Neuroinflammatory Genes: Comparison of Three Prion Strains. PLoS Pathog. 2016, 12, e1005551. [Google Scholar] [CrossRef]
- Durrant, D.M.; Williams, J.L.; Daniels, B.P.; Klein, R.S. Chemokines Referee Inflammation within the Central Nervous System during Infection and Disease. Adv. Med. 2014, 2014, 806741. [Google Scholar] [CrossRef]
- Chen, J.; Chen, C.; Hu, C.; Liu, L.; Xia, Y.; Wang, L.; Yang, W.; Wu, H.-Y.; Zhou, W.; Xiao, K.; et al. IP10, KC and M-CSF Are Remarkably Increased in the Brains from the Various Strains of Experimental Mice Infected with Different Scrapie Agents. Virol. Sin. 2020, 35, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leukoc. Biol. 2016, 100, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Sharief, M.; Green, A.; Dick, J.R.; Gawler, J.; Thompson, E. Heightened intrathecal release of proinflammatory cytokines in Creutzfeldt–Jakob disease. Neurology 1999, 52, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Mabbott, N.A.; McGovern, G.; Jeffrey, M.; Bruce, M.E. Temporary blockade of the tumor necrosis factor receptor signaling pathway impedes the spread of scrapie to the brain. J. Virol. 2002, 76, 5131–5139. [Google Scholar] [CrossRef] [PubMed]
- Hennessy, E.; Gormley, S.; Lopez-Rodriguez, A.B.; Murray, C.; Murray, C.; Cunningham, C. Systemic TNF-α produces acute cognitive dysfunction and exaggerated sickness behavior when superimposed upon progressive neurodegeneration. Brain Behav. Immun. 2017, 59, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Hafner-Bratkovič, I.; Benčina, M.; Fitzgerald, K.A.; Golenbock, D.; Jerala, R. NLRP3 inflammasome activation in macrophage cell lines by prion protein fibrils as the source of IL-1β and neuronal toxicity. Cell. Mol. Life Sci. 2012, 69, 4215–4228. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.; Schwarz, A.; Neidhold, S.; Burwinkel, M.; Riemer, C.; Simon, D.; Kopf, M.; Otto, M.; Baier, M. Role of interleukin-1 in prion disease-associated astrocyte activation. Am. J. Pathol. 2004, 165, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.T.; Betmouni, S.; Perry, V.H. Absence of detectable IL-1beta production in murine prion disease: A model of chronic neurodegeneration. J. Neuropathol. Exp. Neurol. 2001, 60, 173–182. [Google Scholar] [CrossRef]
- Burwinkel, M.; Schwarz, A.; Riemer, C.; Schultz, J.; van Landeghem, F.; Baier, M. Rapid disease development in scrapie-infected mice deficient for CD40 ligand. EMBO Rep. 2004, 5, 527–531. [Google Scholar] [CrossRef]
- Abdel-Haq, N.; Hao, H.-N.; Lyman, W.D. Cytokine regulation of CD40 expression in fetal human astrocyte cultures. J. Neuroimmunol. 1999, 101, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Daoussis, D.; Andonopoulos, A.P.; Liossis, S.-N.C. Targeting CD40L: A promising therapeutic approach. Clin. Diagn. Lab. Immunol. 2004, 11, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.; Cheng, X.; Truong, B.; Sun, L.; Yang, X.; Wang, H. Molecular basis and therapeutic implications of CD40/CD40L immune checkpoint. Pharmacol. Ther. 2021, 219, 107709. [Google Scholar] [CrossRef] [PubMed]
- Tamgüney, G.; Giles, K.; Glidden, D.V.; Lessard, P.; Wille, H.; Tremblay, P.; Groth, D.F.; Yehiely, F.; Korth, C.; Moore, R.C.; et al. Genes contributing to prion pathogenesis. J. Gen. Virol. 2008, 89 Pt 7, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Basu, A.; Krady, J.K.; O’Malley, M.; Styren, S.D.; DeKosky, S.T.; Levison, S.W. The type 1 interleukin-1 receptor is essential for the efficient activation of microglia and the induction of multiple proinflammatory mediators in response to brain injury. J. Neurosci. 2002, 22, 6071–6082. [Google Scholar] [CrossRef] [PubMed]
- Bertani, I.; Iori, V.; Trusel, M.; Maroso, M.; Foray, C.; Mantovani, S.; Tonini, R.; Vezzani, A.; Chiesa, R. Inhibition of IL-1beta Signaling Normalizes NMDA-Dependent Neurotransmission and Reduces Seizure Susceptibility in a Mouse Model of Creutzfeldt-Jakob Disease. J. Neurosci. 2017, 37, 10278–10289. [Google Scholar] [CrossRef] [PubMed]
- Luís, J.P.; Simões, C.J.V.; Brito, R.M.M. The Therapeutic Prospects of Targeting IL-1R1 for the Modulation of Neuroinflammation in Central Nervous System Disorders. Int. J. Mol. Sci. 2022, 23, 1731. [Google Scholar] [CrossRef] [PubMed]
- Bartels, A.L.; Leenders, K.L. Cyclooxygenase and neuroinflammation in Parkinson’s disease neurodegeneration. Curr. Neuropharmacol. 2010, 8, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Pasinetti, G.M.; Aisen, P.S. Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer’s disease brain. Neuroscience 1998, 87, 319–324. [Google Scholar] [CrossRef]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef]
- Williams, A.; Van Dam, A.-M.; Ritchie, D.; Eikelenboom, P.; Fraser, H. Immunocytochemical appearance of cytokines, prostaglandin E2 and lipocortin-1 in the CNS during the incubation period of murine scrapie correlates with progressive PrP accumulations. Brain Res. 1997, 754, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Carlson, N.G.; Rojas, M.A.; Redd, J.W.; Tang, P.; Wood, B.; Hill, K.E.; Rose, J.W. Cyclooxygenase-2 expression in oligodendrocytes increases sensitivity to excitotoxic death. J. Neuroinflamm. 2010, 7, 25. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.T.; Perry, V.H.; Minghetti, L. Cyclooxygenase-2 is highly expressed in microglial-like cells in a murine model of prion disease. Glia 2000, 29, 392–396. [Google Scholar] [CrossRef]
- Ferrer, M.D.; Busquests-Cortes, C.; Capó, X.; Tejada, S.; Tur, J.A.; Pons, A.; Sureda, A. Cyclooxygenase-2 Inhibitors as a Therapeutic Target in Inflammatory Diseases. Curr. Med. Chem. 2019, 26, 3225–3241. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Shi, Q.; Xiao, K.; Wang, J.; Chen, C.; Gao, L.-P.; Gao, C.; Dong, X.-P. Stimulations of the Culture Medium of Activated Microglia and TNF-Alpha on a Scrapie-Infected Cell Line Decrease the Cell Viability and Induce Marked Necroptosis That Also Occurs in the Brains from the Patients of Human Prion Diseases. ACS Chem. Neurosci. 2019, 10, 1273–1283. [Google Scholar] [CrossRef] [PubMed]
- Ezpeleta, J.; Boudet-Devaud, F.; Pietri, M.; Baudry, A.; Baudouin, V.; Alleaume-Butaux, A.; Dagoneau, N.; Kellermann, O.; Launay, J.-M.; Schneider, B. Protective role of cellular prion protein against TNFalpha-mediated inflammation through TACE alpha-secretase. Sci. Rep. 2017, 7, 7671. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.R.; Mu, T.-C.; Gao, Z.-X.; Wang, J.; Sy, M.-S.; Li, C.-Y. Prion protein is required for tumor necrosis factor alpha (TNFalpha)-triggered nuclear factor kappaB (NF-kappaB) signaling and cytokine production. J. Biol. Chem. 2017, 292, 18747–18759. [Google Scholar] [CrossRef]
- Evangelatos, G.; Bamias, G.; Kitas, G.D.; Kollias, G.; Sfikakis, P.P. The second decade of anti-TNF-a therapy in clinical practice: New lessons and future directions in the COVID-19 era. Rheumatol. Int. 2022, 42, 1493–1511. [Google Scholar] [CrossRef]
- Paik, P.K.; Luo, J.; Ai, N.; Kim, R.; Ahn, L.; Biswas, A.; Coker, C.; Ma, W.; Wong, P.; Buonocore, D.J.; et al. Phase I trial of the TNF-α inhibitor certolizumab plus chemotherapy in stage IV lung adenocarcinomas. Nat. Commun. 2022, 13, 6095. [Google Scholar] [CrossRef]
- Chen, Y.; Qin, C.; Huang, J.; Tang, X.; Liu, C.; Huang, K.; Xu, J.; Guo, G.; Tong, A.; Zhou, L. The role of astrocytes in oxidative stress of central nervous system: A mixed blessing. Cell Prolif. 2020, 53, e12781. [Google Scholar] [CrossRef]
- González-Reyes, R.E.; Nava-Mesa, M.O.; Vargas-Sánchez, K.; Ariza-Salamanca, D.; Mora-Muñoz, L. Involvement of Astrocytes in Alzheimer’s Disease from a Neuroinflammatory and Oxidative Stress Perspective. Front. Mol. Neurosci. 2017, 10, 427. [Google Scholar] [CrossRef] [PubMed]
- Solleiro-Villavicencio, H.; Rivas-Arancibia, S. Effect of Chronic Oxidative Stress on Neuroinflammatory Response Mediated by CD4+T Cells in Neurodegenerative Diseases. Front. Cell. Neurosci. 2018, 12, 114. [Google Scholar] [CrossRef] [PubMed]
- Vasconcelos, A.R.; Santos, N.B.D.; Scavone, C.; Munhoz, C.D. Nrf2/ARE Pathway Modulation by Dietary Energy Regulation in Neurological Disorders. Front. Pharmacol. 2019, 10, 33. [Google Scholar] [CrossRef] [PubMed]
- Calkins, M.J.; Johnson, D.A.; Townsend, J.A.; Vargas, M.R.; Dowell, J.A.; Williamson, T.P.; Kraft, A.D.; Lee, J.-M.; Li, J.; Johnson, J.A. The Nrf2/ARE pathway as a potential therapeutic target in neurodegenerative disease. Antioxid. Redox Signal. 2009, 11, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Gan, L.; Johnson, J.A. Oxidative damage and the Nrf2-ARE pathway in neurodegenerative diseases. Biochim. Biophys. Acta 2014, 1842, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Karagianni, K.; Pettas, S.; Kanata, E.; Lioulia, E.; Thune, K.; Schmitz, M.; Tsamesidis, I.; Lymperaki, E.; Xanthopoulos, K.; Sklaviadis, T.; et al. Carnosic Acid and Carnosol Display Antioxidant and Anti-Prion Properties in In Vitro and Cell-Free Models of Prion Diseases. Antioxidants 2022, 11, 726. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Barger, S.W.; Griffin, W.S.T. Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J. Neurosci. 2003, 23, 1605–1611. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, E.; Banks, W.; Kastin, A. Blood-borne interleukin-1 receptor antagonist crosses the blood-brain barrier. J. Neuroimmunol. 1994, 55, 153–160. [Google Scholar] [CrossRef]
- Freeman, B.D.; Buchman, T.G. Interleukin-1 receptor antagonist as therapy for inflammatory disorders. Expert Opin. Biol. Ther. 2001, 1, 301–308. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | Source | Pro-Inflammatory or Anti-Inflammatory? | Reference |
|---|---|---|---|
| IL-10 | Activated astrocytes and microglia; neurons | Anti-inflammatory | [21] |
| IL-1α | Activated microglia | Pro-inflammatory | [22] |
| TNF-α | Activated microglia | Pro-inflammatory | [22] |
| Neurons | Anti-inflammatory | [23] | |
| C1q | Activated microglia | Pro-inflammatory | [22] |
| CCL2 | Reactive A1 astrocytes | Pro-inflammatory | [24,25] |
| IL-3 | Neurons | Anti-inflammatory | [23] |
| IL-4 | Neurons | Anti-inflammatory | [23] |
| IL-6 | Neurons; astrocytes | Pro-inflammatory | [26,27] |
| MCP-1 | Neurons | Pro-inflammatory | [28] |
| IL-17A | Th17 lymphocytes | Pro-inflammatory | [29] |
| IL-22 | Th17 lymphocytes | Pro-inflammatory | [29] |
| IL-8 | endothelial cell layer | Pro-inflammatory | [30] |
| IL-1 | Activated astrocytes | Pro-inflammatory | [30] |
| IL-13 | Activated astrocytes | Anti-inflammatory | [31] |
| IL-12p40 | Brain homogenates (22L-strain-infected mice) | Pro-inflammatory | [32] * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Assis-de-Lemos, G.; Moura-do-Nascimento, R.; Amaral-do-Nascimento, M.; Miceli, A.C.; Vieira, T.C.R.G. Interactions between Cytokines and the Pathogenesis of Prion Diseases: Insights and Implications. Brain Sci. 2024, 14, 413. https://doi.org/10.3390/brainsci14050413
Assis-de-Lemos G, Moura-do-Nascimento R, Amaral-do-Nascimento M, Miceli AC, Vieira TCRG. Interactions between Cytokines and the Pathogenesis of Prion Diseases: Insights and Implications. Brain Sciences. 2024; 14(5):413. https://doi.org/10.3390/brainsci14050413
Chicago/Turabian StyleAssis-de-Lemos, Gabriela, Rayanne Moura-do-Nascimento, Manuela Amaral-do-Nascimento, Ana C. Miceli, and Tuane C. R. G. Vieira. 2024. "Interactions between Cytokines and the Pathogenesis of Prion Diseases: Insights and Implications" Brain Sciences 14, no. 5: 413. https://doi.org/10.3390/brainsci14050413