The GABAA Receptor α2 Subunit Activates a Neuronal TLR4 Signal in the Ventral Tegmental Area that Regulates Alcohol and Nicotine Abuse

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Antibodies
2.3. Cells, Plasmids, Transfection, and Reagents
2.4. Immunofluorescence
2.5. Immunoblotting
2.6. Co-Immunoprecipitation Assay
2.7. Small Interfering RNAs
2.8. Herpes Simplex Virus Type 1 (HSV-1)-Based Amplicon Vectors
2.9. Stereotaxic Procedures
2.10. Binge Drinking Paradigm
2.11. Kelley Nicotine Sensitization Paradigm
2.12. Statistics
3. Results
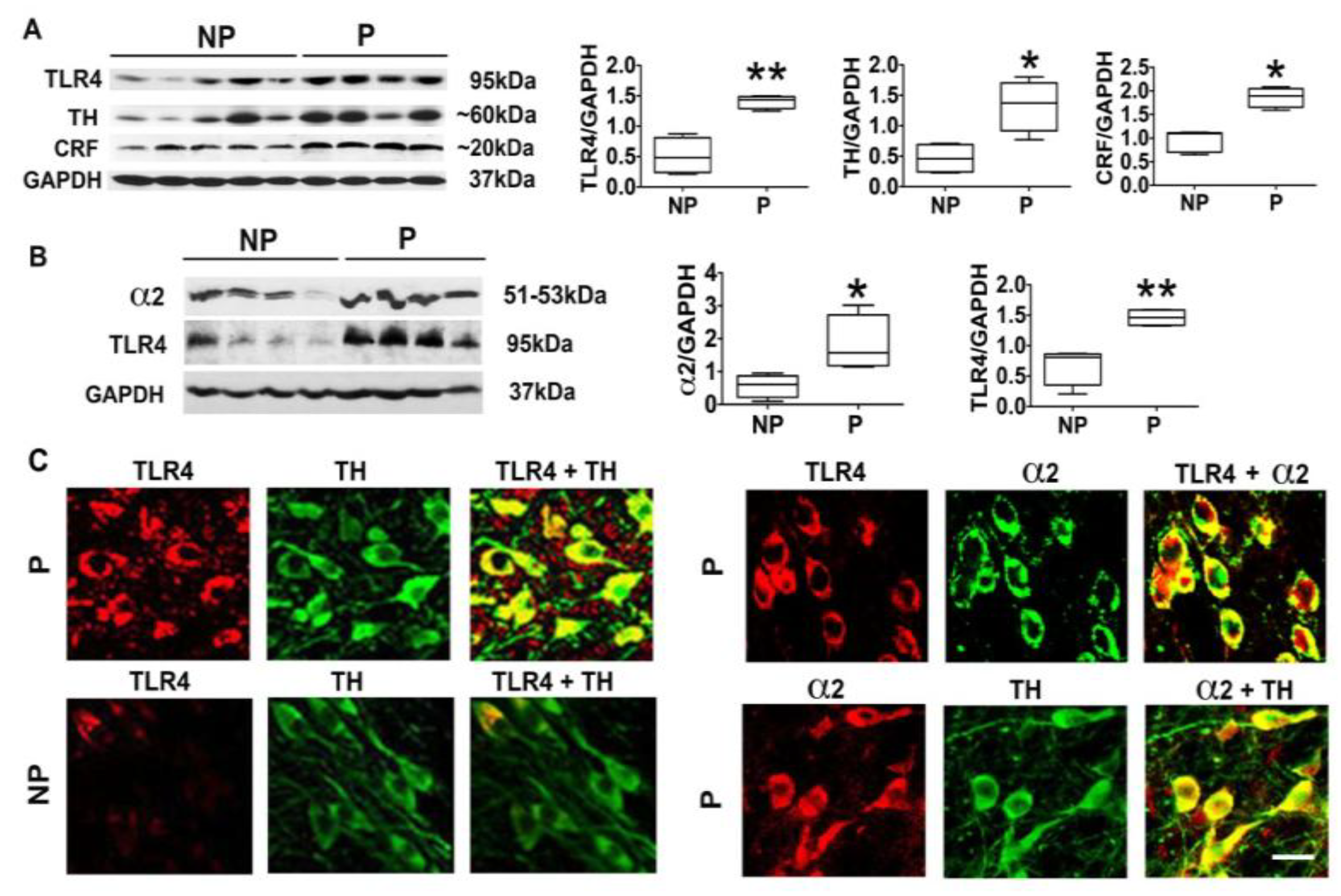
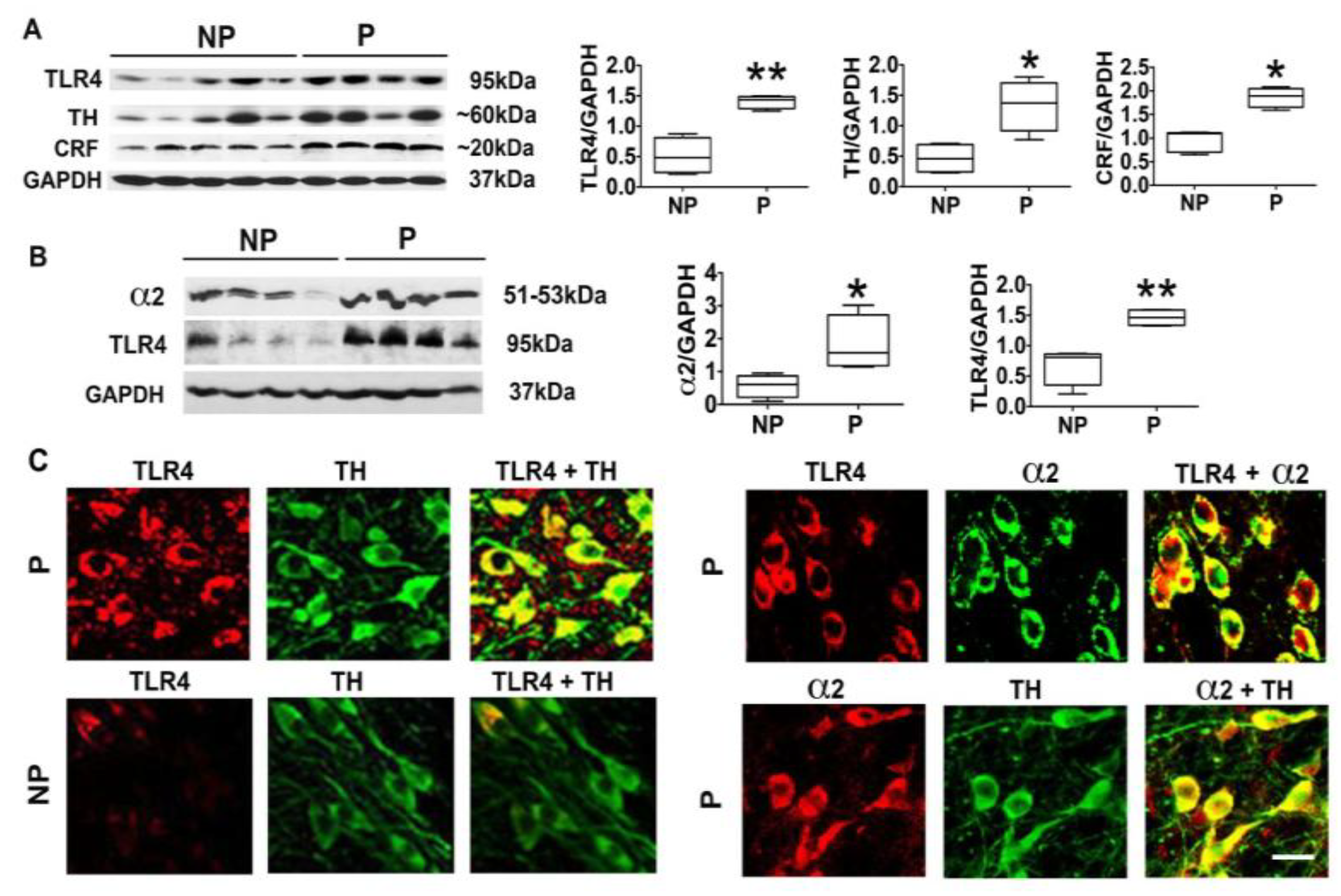
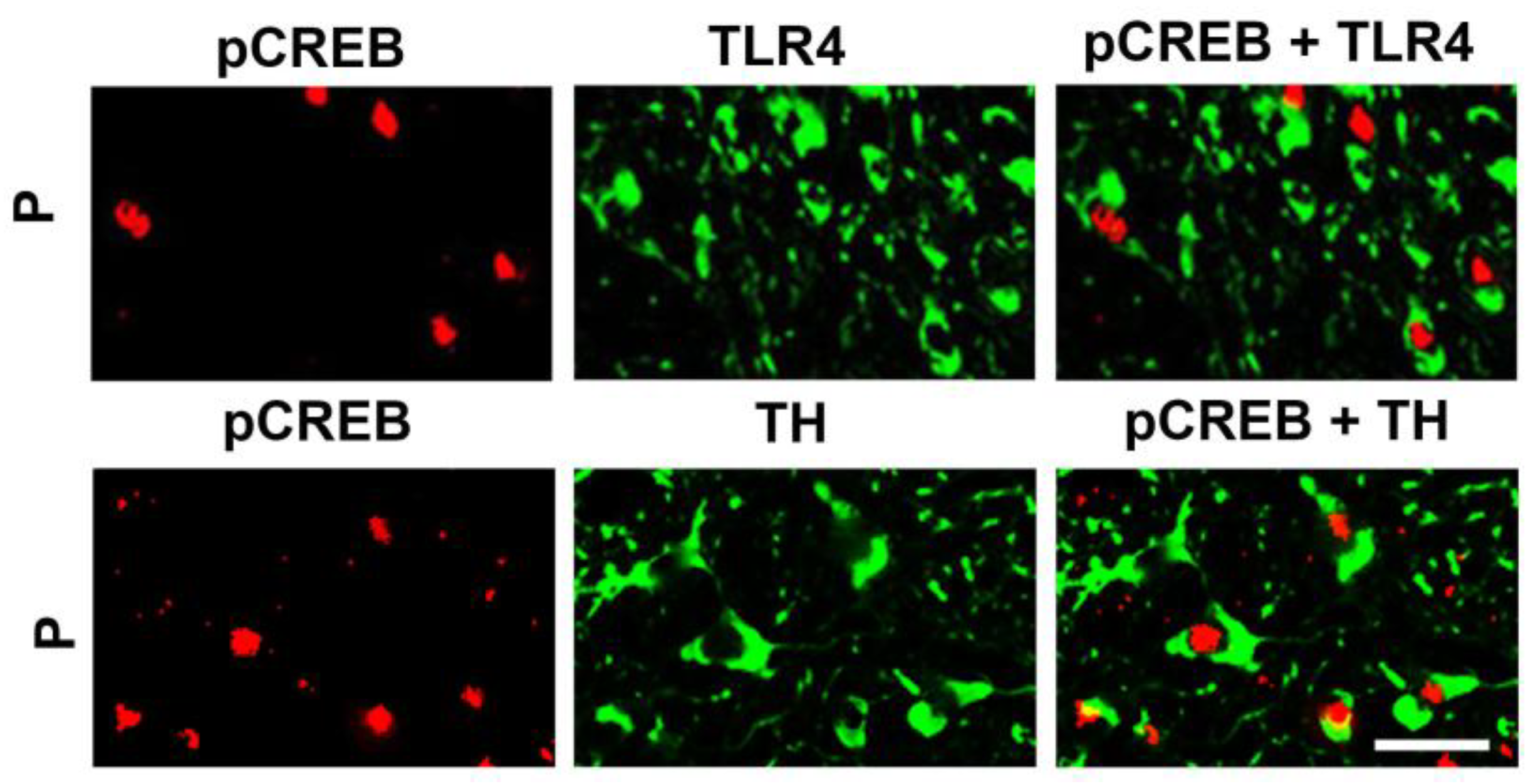
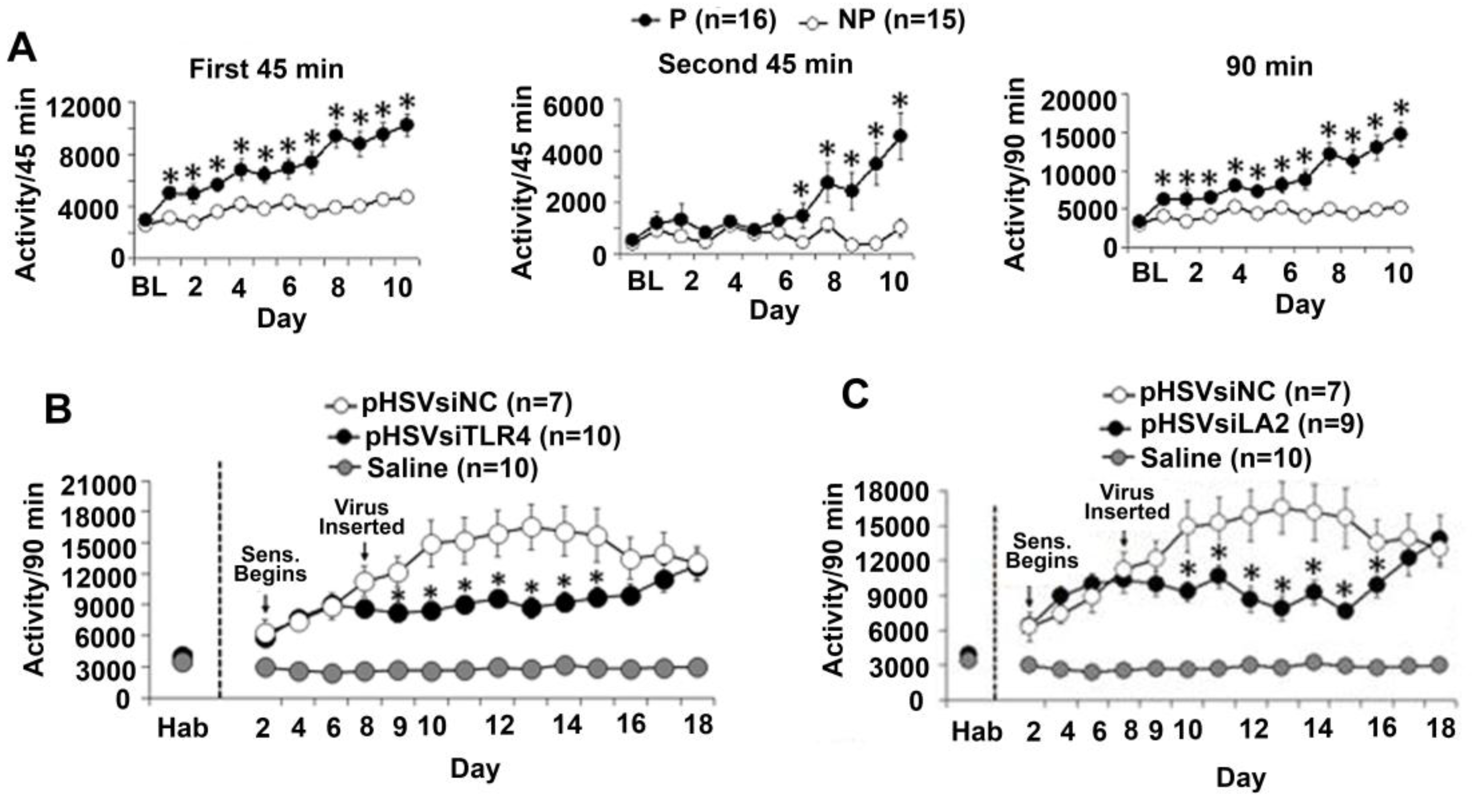
3.1. The P Rats Have Larger Numbers of VTA-Located TH+ Neurons that Co-Express TLR4 and α2 Than the NP Rats
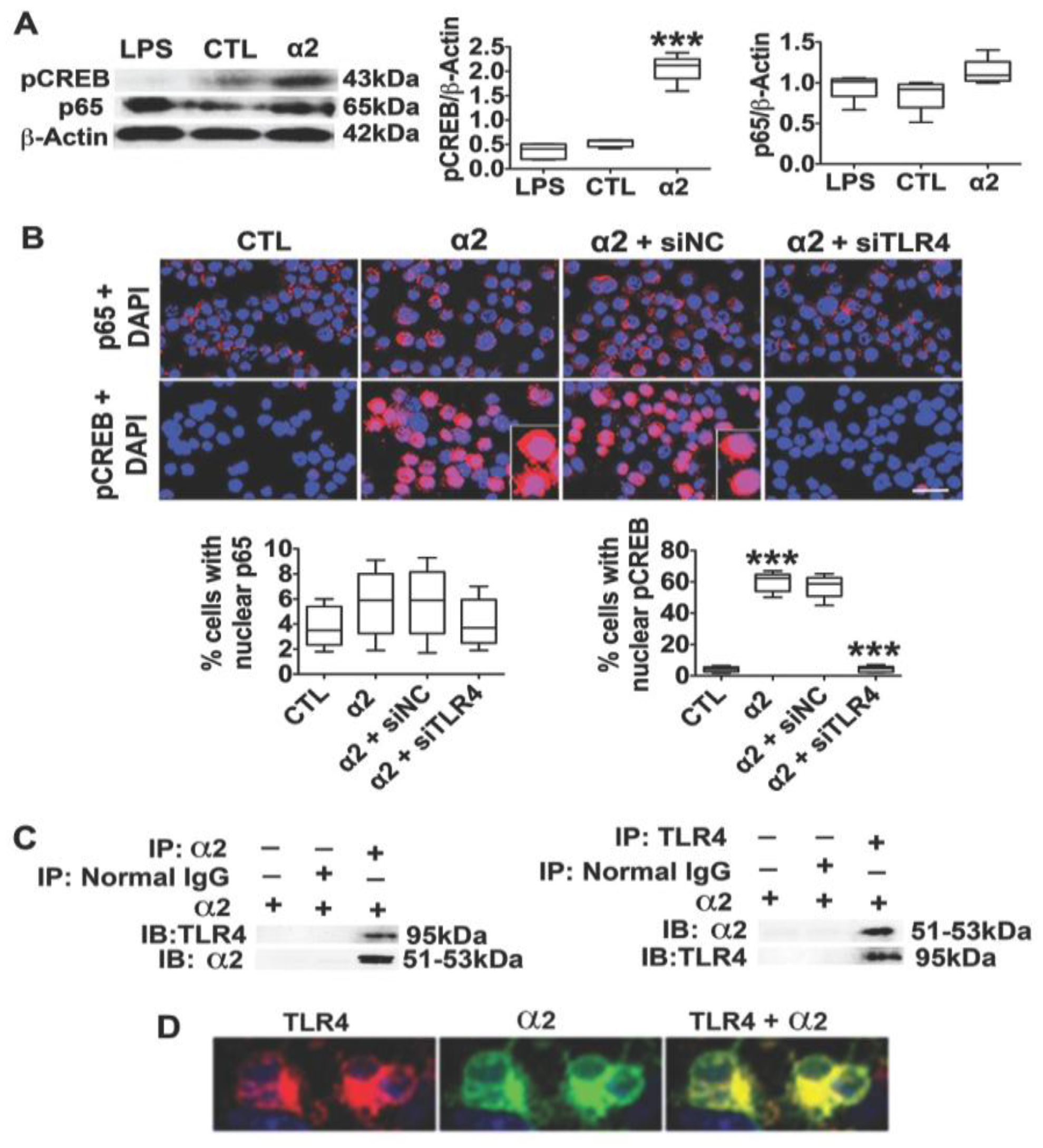
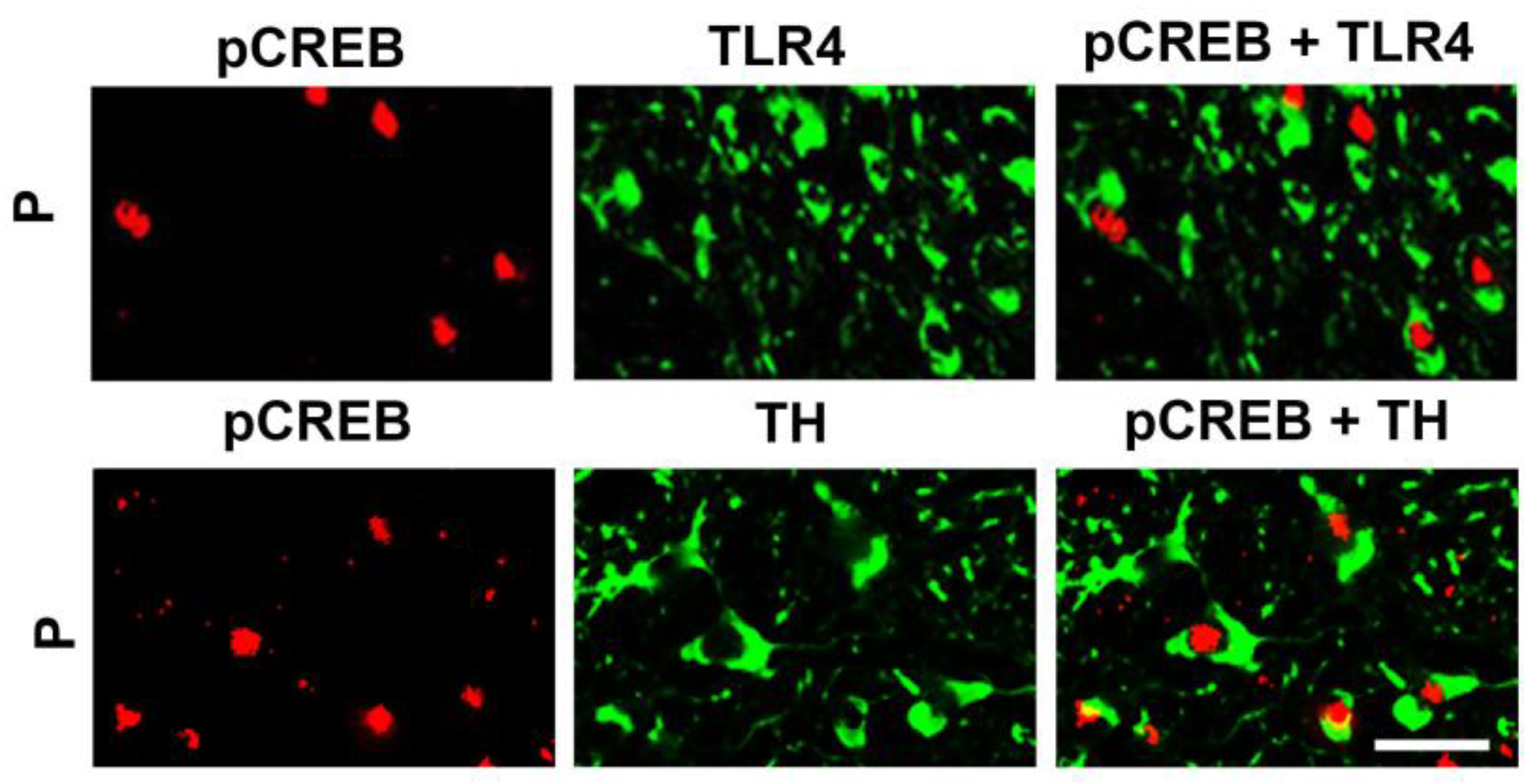
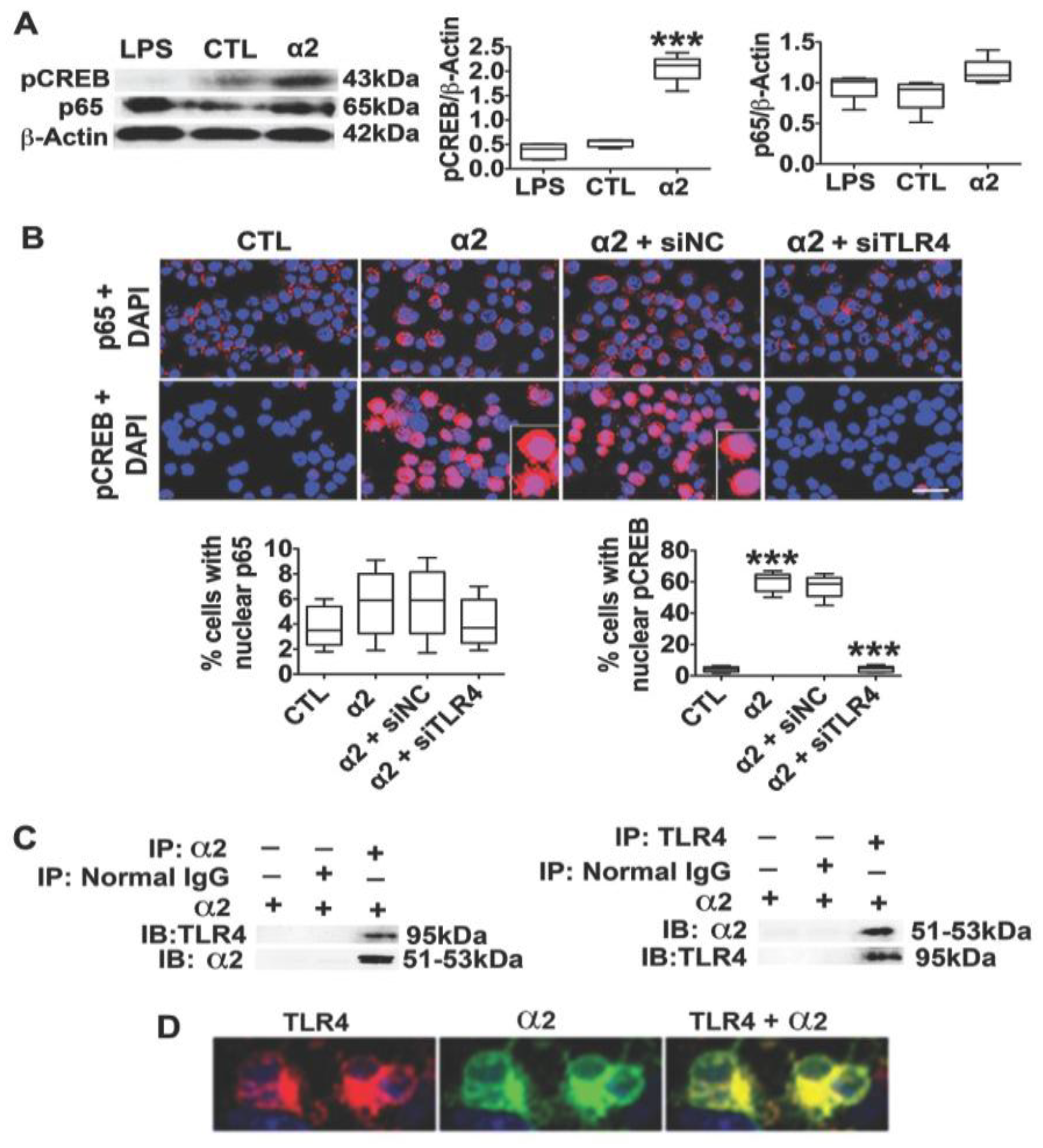
3.2. The Neuronal TLR4 Signal is Activated by α2 and it Culminates in Activated CREB, but not NF-kB
3.3. α2+ Binds TLR4, Likely at the Cell Surface
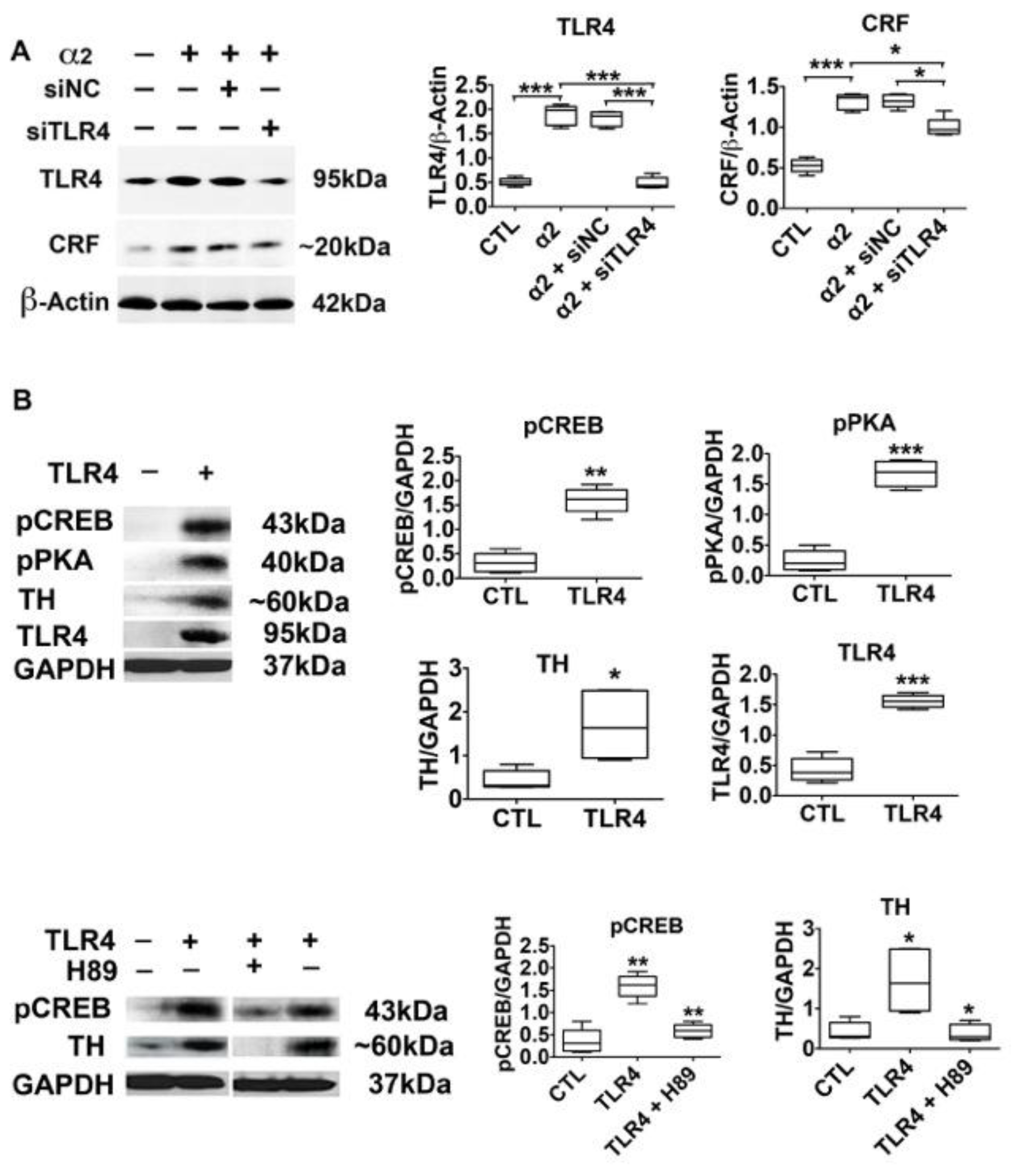
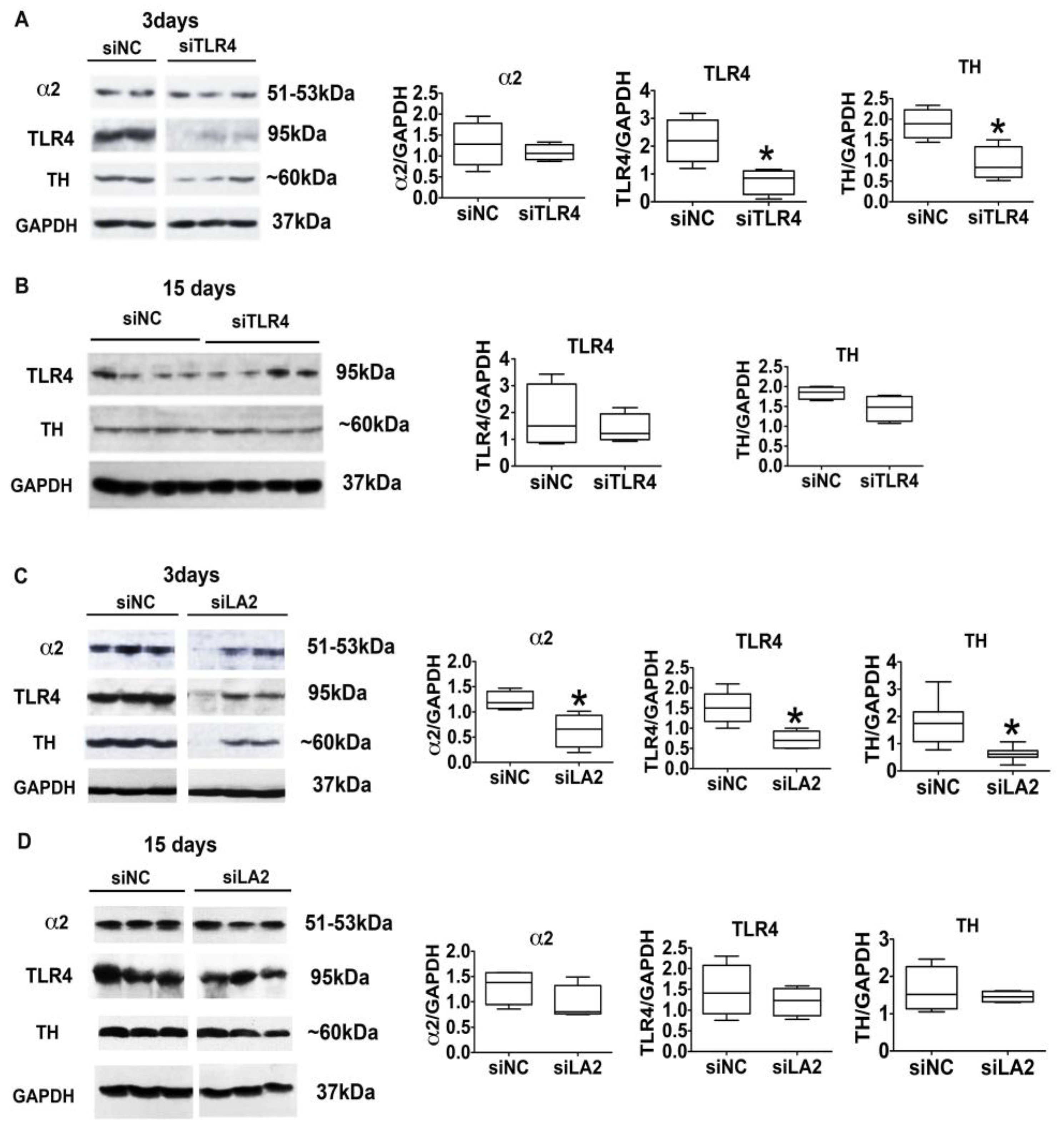
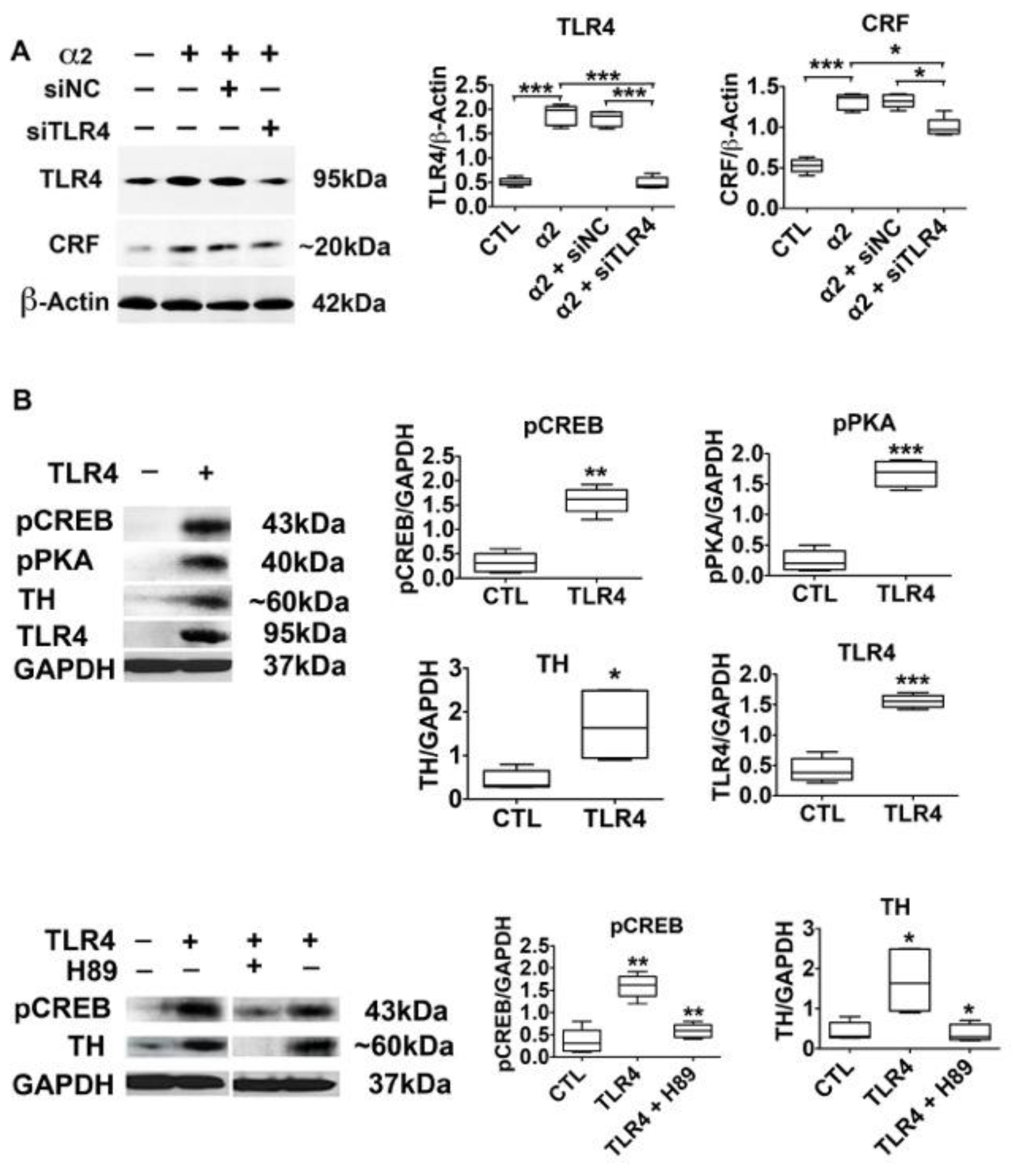
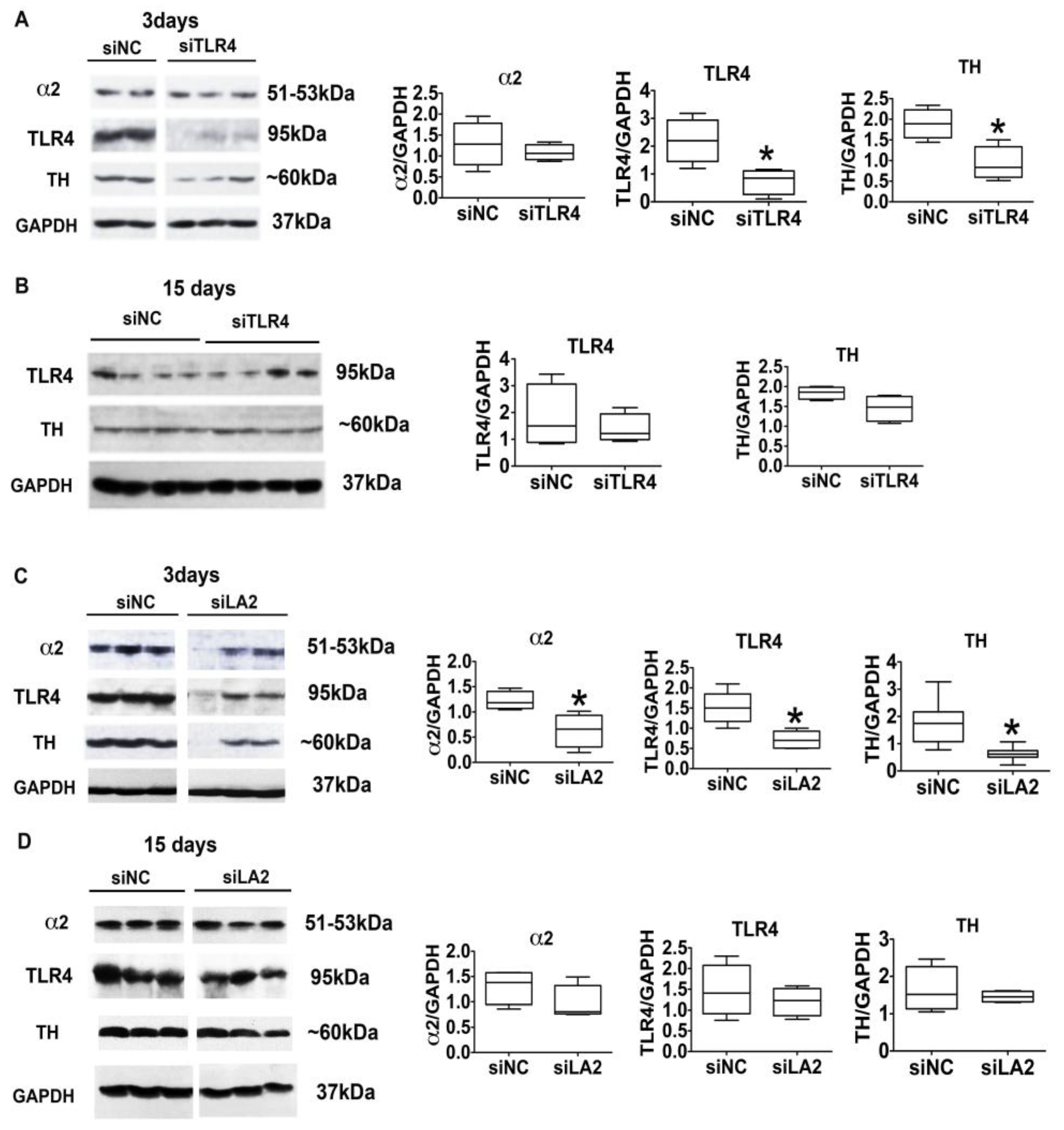
3.4. The α2-Activated TLR4 Signal Contributes to CRF and TH Expression
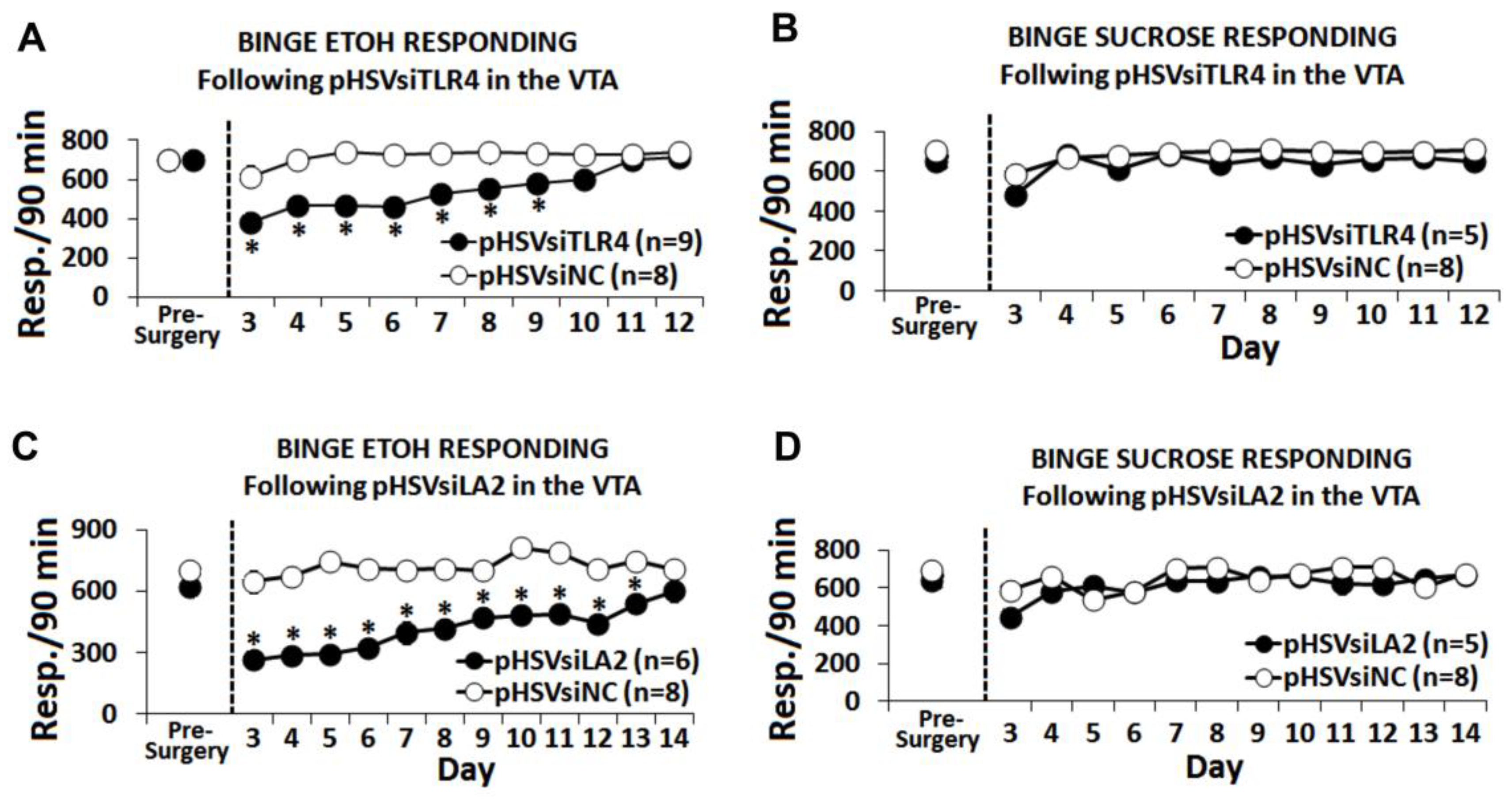
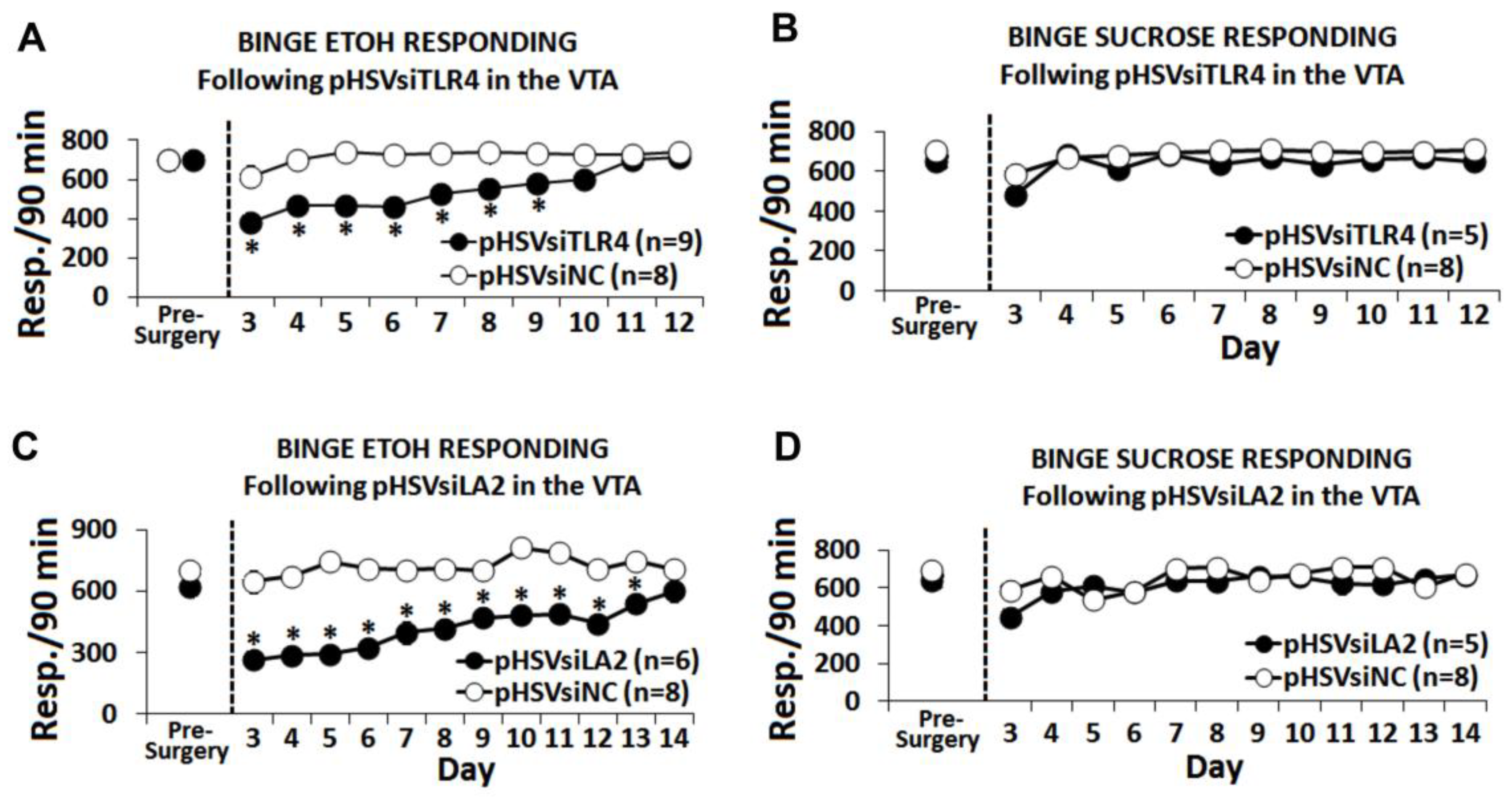
3.5. The α2/TLR4 Signal Controls Binge Drinking
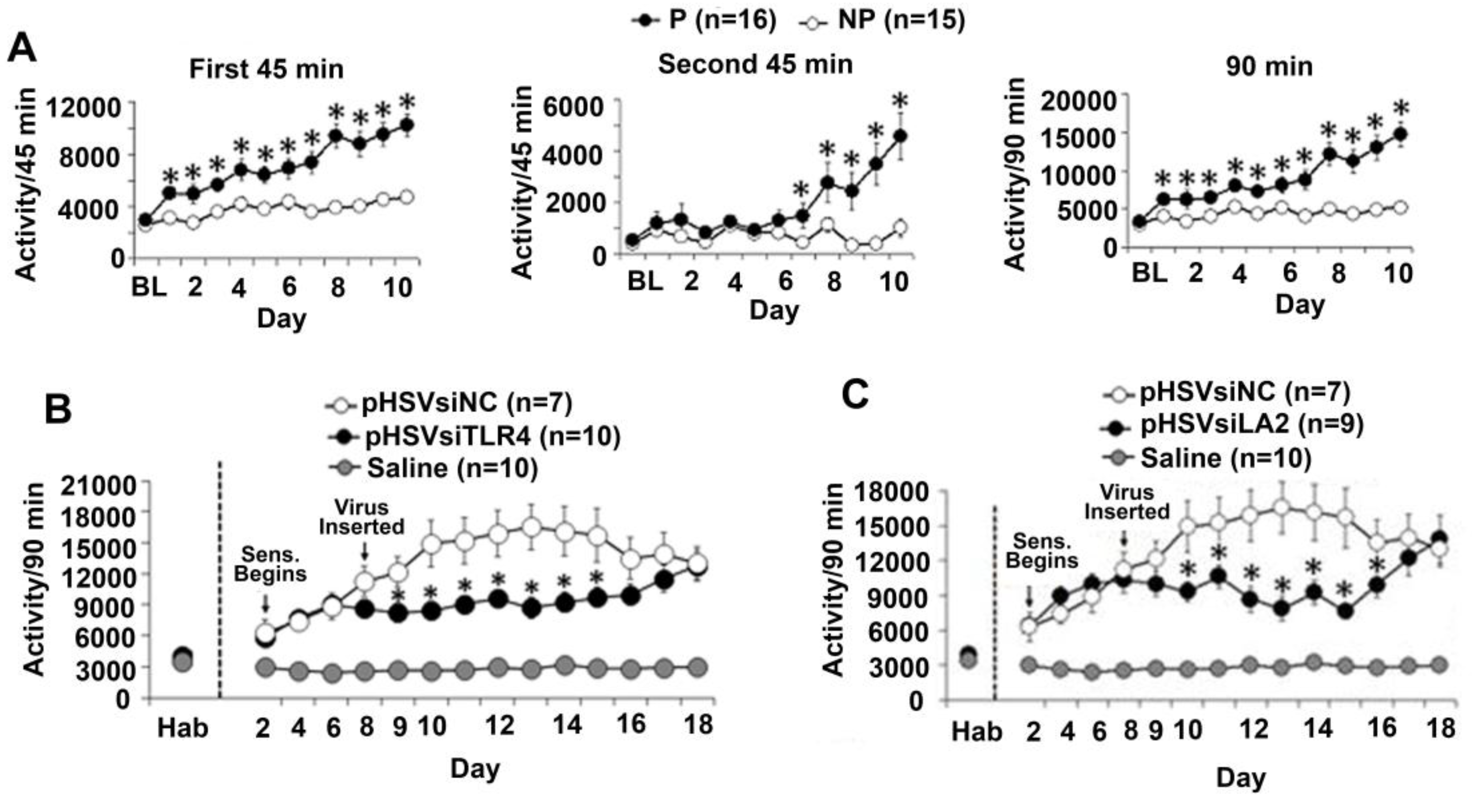
3.6. The α2/TLR4 Signal in the VTA Controls Nicotine Sensitization
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Town, M.; Naimi, T.S.; Mokdad, A.H.; Brewer, R.D. Health care access among U.S. adults who drink alcohol excessively: Missed opportunities for prevention. Prev. Chronic Dis. 2006, 3, A53. [Google Scholar] [PubMed]
- US Department of Health and Human Services. National Institute on Alcohol Abuse and Alcoholism NIAAA Council approves definition of binge drinking. In NIAAA Newsletter; US Department of Health and Human Services: Washington, DC, USA, 2004. [Google Scholar]
- Ducci, F.; Enoch, M.A.; Funt, S.; Virkkunen, M.; Albaugh, B.; Goldman, D. Increased anxiety and other similarities in temperament of alcoholics with and without antisocial personality disorder across three diverse populations. Alcohol 2007, 41, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Edenberg, H.J.; Dick, D.M.; Xuei, X.; Tian, H.; Almasy, L.; Bauer, L.O.; Crowe, R.R.; Goate, A.; Hesselbrock, V.; Jones, K.; et al. Variations in GABRA2, encoding the alpha 2 subunit of the GABA(A) receptor, are associated with alcohol dependence and with brain oscillations. Am. J. Hum. Genet. 2004, 74, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Chikritzhs, T.N.; Jonas, H.A.; Stockwell, T.R.; Heale, P.F.; Dietze, P.M. Mortality and life-years lost due to alcohol: A comparison of acute and chronic causes. Med. J. Aust. 2001, 174, 281–284. [Google Scholar] [PubMed]
- Naimi, T.S.; Brewer, R.D.; Mokdad, A.; Denny, C.; Serdula, M.K.; Marks, J.S. Binge drinking among US adults. JAMA 2003, 289, 70–75. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Ling, P.M. Impact of alcohol use and bar attendance on smoking and quit attempts among young adult bar patrons. Am. J. Public Health 2013, 103, e53–e61. [Google Scholar] [CrossRef] [PubMed]
- Hurley, L.L.; Taylor, R.E.; Tizabi, Y. Positive and negative effects of alcohol and nicotine and their interactions: A mechanistic review. Neurotox. Res. 2012, 21, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Nurnberger, J.I., Jr.; Wiegand, R.; Bucholz, K.; O’Connor, S.; Meyer, E.T.; Reich, T.; Rice, J.; Schuckit, M.; King, L.; Petti, T.; et al. A family study of alcohol dependence: Coaggregation of multiple disorders in relatives of alcohol-dependent probands. Arch. Gen. Psychiatry 2004, 61, 1246–1256. [Google Scholar] [CrossRef] [PubMed]
- Volk, H.E.; Scherrer, J.F.; Bucholz, K.K.; Todorov, A.; Heath, A.C.; Jacob, T.; True, W.R. Evidence for specificity of transmission of alcohol and nicotine dependence in an offspring of twins design. Drug Alcohol Depend. 2007, 87, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.R.; Getachew, B.; Oster, S.M.; Dhaher, R.; Ding, Z.M.; Bell, R.L.; McBride, W.J.; Rodd, Z.A. Nicotine modulates alcohol-seeking and relapse by alcohol-preferring (P) rats in a time-dependent manner. Alcohol. Clin. Exp. Res. 2012, 36, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koob, G.F.; Buck, C.L.; Cohen, A.; Edwards, S.; Park, P.E.; Schlosburg, J.E.; Schmeichel, B.; Vendruscolo, L.F.; Wade, C.L.; Whitfield, T.W., Jr.; et al. Addiction as a stress surfeit disorder. Neuropharmacology 2014, 76 Pt B, 370–382. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Walter, T.J.; Coleman, L.G., Jr.; Vetreno, R.P. Toll-like receptor signaling and stages of addiction. Psychopharmacology 2017, 234, 1483–1498. [Google Scholar] [CrossRef] [PubMed]
- Dick, D.M.; Smith, G.; Olausson, P.; Mitchell, S.H.; Leeman, R.F.; O’Malley, S.S.; Sher, K. Understanding the construct of impulsivity and its relationship to alcohol use disorders. Addict. Biol. 2010, 15, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Caswell, A.J.; Celio, M.A.; Morgan, M.J.; Duka, T. Impulsivity as a Multifaceted Construct Related to Excessive Drinking Among UK Students. Alcohol Alcohol. 2016, 51, 77–83. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.L.; Rodd, Z.A.; Lumeng, L.; Murphy, J.M.; McBride, W.J. The alcohol-preferring P rat and animal models of excessive alcohol drinking. Addict. Biol. 2006, 11, 270–288. [Google Scholar] [CrossRef] [PubMed]
- Oberlin, B.G.; Grahame, N.J. High-alcohol preferring mice are more impulsive than low-alcohol preferring mice as measured in the delay discounting task. Alcohol. Clin. Exp. Res. 2009, 33, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, S.W.; Czachowski, C.L. Increased delay discounting tracks with a high ethanol-seeking phenotype and subsequent ethanol seeking but not consumption. Alcohol. Clin. Exp. Res. 2014, 38, 2607–2614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yang, A.R.; Kelly, T.; Puche, A.; Esoga, C.; June, H.L., Jr.; Elnabawi, A.; Merchenthaler, I.; Sieghart, W.; June, H.L., Sr.; et al. Binge alcohol drinking is associated with GABAA alpha2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proc. Natl. Acad. Sci. USA 2011, 108, 4465–4470. [Google Scholar] [CrossRef] [PubMed]
- June, H.L.; Liu, J.; Warnock, K.T.; Bell, K.A.; Balan, I.; Bollino, D.; Puche, A.; Aurelian, L. CRF-amplified neuronal TLR4/MCP-1 signaling regulates alcohol self-administration. Neuropsychopharmacology 2015, 40, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Aurelian, L.; Warnock, K.T.; Balan, I.; Puche, A.; June, H. TLR4 signaling in VTA dopaminergic neurons regulates impulsivity through tyrosine hydroxylase modulation. Transl. Psychiatry 2016, 6, e815. [Google Scholar] [CrossRef] [PubMed]
- Radcliffe, P.M.; Sterling, C.R.; Tank, A.W. Induction of tyrosine hydroxylase mRNA by nicotine in rat midbrain is inhibited by mifepristone. J. Neurochem. 2009, 109, 1272–1284. [Google Scholar] [CrossRef] [PubMed]
- Nisell, M.; Nomikos, G.G.; Svensson, T.H. Nicotine dependence, midbrain dopamine systems and psychiatric disorders. Pharmacol. Toxicol. 1995, 76, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Rahman, S.; Zhang, J.; Corrigall, W.A. Effects of acute and chronic nicotine on somatodendritic dopamine release of the rat ventral tegmental area: In vivo microdialysis study. Neurosci. Lett. 2003, 348, 61–64. [Google Scholar] [CrossRef]
- Balan, I.; Warnock, K.T.; Puche, A.; Gondre-Lewis, M.C.; Aurelian, L. Innately activated TLR4 signal in the nucleus accumbens is sustained by CRF amplification loop and regulates impulsivity. Brain. Behav. Immun. 2018, 69, 139–153. [Google Scholar] [CrossRef] [PubMed]
- Dedic, N.; Chen, A.; Deussing, J.M. The CRF family of neuropeptides and their receptors—Mediators of the central stress response. Curr. Mol. Pharmacol. 2018, 11, 4–31. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T.J.; Reed, C.; Pastor, R. Preclinical evidence implicating corticotropin-releasing factor signaling in ethanol consumption and neuroadaptation. Genes Brain Behav. 2015, 14, 98–135. [Google Scholar] [CrossRef] [PubMed]
- Le, A.D.; Wang, A.; Harding, S.; Juzytsch, W.; Shaham, Y. Nicotine increases alcohol self-administration and reinstates alcohol seeking in rats. Psychopharmacology 2003, 168, 216–221. [Google Scholar] [CrossRef] [PubMed]
- Le, A.D.; Li, Z.; Funk, D.; Shram, M.; Li, T.K.; Shaham, Y. Increased vulnerability to nicotine self-administration and relapse in alcohol-naive offspring of rats selectively bred for high alcohol intake. J. Neurosci. 2006, 26, 1872–1879. [Google Scholar] [CrossRef] [PubMed]
- De Biasi, M.; Dani, J.A. Reward, addiction, withdrawal to nicotine. Annu. Rev. Neurosci. 2011, 34, 105–130. [Google Scholar] [CrossRef] [PubMed]
- Di Chiara, G. Role of dopamine in the behavioural actions of nicotine related to addiction. Eur. J. Pharmacol. 2000, 393, 295–314. [Google Scholar] [CrossRef]
- Gonzales, R.A.; Job, M.O.; Doyon, W.M. The role of mesolimbic dopamine in the development and maintenance of ethanol reinforcement. Pharmacol. Ther. 2004, 103, 121–146. [Google Scholar] [CrossRef] [PubMed]
- Okun, E.; Griffioen, K.J.; Mattson, M.P. Toll-like receptor signaling in neural plasticity and disease. Trends Neurosci. 2011, 34, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Doyon, W.M.; Dong, Y.; Ostroumov, A.; Thomas, A.M.; Zhang, T.A.; Dani, J.A. Nicotine decreases ethanol-induced dopamine signaling and increases self-administration via stress hormones. Neuron 2013, 79, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Matsushita, N.; Kobayashi, K.; Kobayashi, K. Identification of GABAA receptor subunit variants in midbrain dopaminergic neurons. J. Neurochem. 2004, 89, 7–14. [Google Scholar] [CrossRef] [PubMed]
- McBride, W.J.; Li, T.K. Animal models of alcoholism: Neurobiology of high alcohol-drinking behavior in rodents. Crit. Rev. Neurobiol. 1998, 12, 339–369. [Google Scholar] [CrossRef] [PubMed]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates, 6th ed.; Academic Press: San Diego, CA, USA, 2009. [Google Scholar]
- Nelson, J.W.; Zhu, J.; Smith, C.C.; Kulka, M.; Aurelian, L. ATP and SH3 binding sites in the protein kinase of the large subunit of herpes simplex virus type 2 of ribonucleotide reductase (ICP10). J. Biol. Chem. 1996, 271, 17021–17027. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Yu, Y.X.; Kulka, M.; Aurelian, L. A novel human gene similar to the protein kinase (PK) coding domain of the large subunit of herpes simplex virus type 2 ribonucleotide reductase (ICP10) codes for a serine-threonine PK and is expressed in melanoma cells. J. Biol. Chem. 2000, 275, 25690–25699. [Google Scholar] [CrossRef] [PubMed]
- Poulopoulos, A.; Aramuni, G.; Meyer, G.; Soykan, T.; Hoon, M.; Papadopoulos, T.; Zhang, M.; Paarmann, I.; Fuchs, C.; Harvey, K.; et al. Neuroligin 2 drives postsynaptic assembly at perisomatic inhibitory synapses through gephyrin and collybistin. Neuron 2009, 63, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Saydam, O.; Glauser, D.L.; Heid, I.; Turkeri, G.; Hilbe, M.; Jacobs, A.H.; Ackermann, M.; Fraefel, C. Herpes simplex virus 1 amplicon vector-mediated siRNA targeting epidermal growth factor receptor inhibits growth of human glioma cells in vivo. Mol. Ther. 2005, 12, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.C.; Foster, K.L.; McKay, P.F.; Carroll, M.R.; Seyoum, R.; Woods, J.E., II; Grey, C.; Jones, C.M.; McCane, S.; Cummings, R.; et al. The GABA(A) receptor alpha1 subtype in the ventral pallidum regulates alcohol-seeking behaviors. J. Neurosci. 2002, 22, 3765–3775. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.L.; Rodd, Z.A.; Engleman, E.A.; Toalston, J.E.; McBride, W.J. Scheduled access alcohol drinking by alcohol-preferring (P) and high-alcohol-drinking (HAD) rats: Modeling adolescent and adult binge-like drinking. Alcohol 2014, 48, 225–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- June, H.L.; Eiler, W.J.A. Dopaminergic and GABAergic regulation of alcohol-motivated behaviors: novel neuroanatomical substrates. In Handbook of Contemporary Neuropharmacology; John Wiley & Sons, Inc.: Francisco, CA, USA, 2007. [Google Scholar]
- Stephens, D.N.; Duka, T. Cognitive and emotional consequences of binge drinking: Role of amygdala and prefrontal cortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2008, 363, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.E.; Holahan, M.R.; Landry, C.F.; Kelley, A.E. Morphine-associated environmental cues elicit conditioned gene expression. Synapse 2000, 37, 146–158. [Google Scholar] [CrossRef]
- DiFranza, J.R.; Wellman, R.J. Sensitization to nicotine: How the animal literature might inform future human research. Nicot. Tob. Res. 2007, 9, 9–20. [Google Scholar] [CrossRef] [PubMed]
- McBride, W.J.; Kimpel, M.W.; McClintick, J.N.; Ding, Z.M.; Hauser, S.R.; Edenberg, H.J.; Bell, R.L.; Rodd, Z.A. Changes in gene expression within the ventral tegmental area following repeated excessive binge-like alcohol drinking by alcohol-preferring (P) rats. Alcohol 2013, 47, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Gondre-Lewis, M.C.; Warnock, K.T.; Wang, H.; June, H.L., Jr.; Bell, K.A.; Rabe, H.; Tiruveedhula, V.V.; Cook, J.; Luddens, H.; Aurelian, L.; et al. Early life stress is a risk factor for excessive alcohol drinking and impulsivity in adults and is mediated via a CRF/GABA(A) mechanism. Stress 2016, 19, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Theberge, F.R.; Li, X.; Kambhampati, S.; Pickens, C.L.; St Laurent, R.; Bossert, J.M.; Baumann, M.H.; Hutchinson, M.R.; Rice, K.C.; Watkins, L.R.; et al. Effect of chronic delivery of the Toll-like receptor 4 antagonist (+)-naltrexone on incubation of heroin craving. Biol. Psychiatry 2013, 73, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Bonci, A.; Bernardi, G.; Grillner, P.; Mercuri, N.B. The dopamine-containing neuron: Maestro or simple musician in the orchestra of addiction? Trends Pharmacol. Sci. 2003, 24, 172–177. [Google Scholar] [CrossRef]
- Tsuchiya, M.; Piras, V.; Choi, S.; Akira, S.; Tomita, M.; Giuliani, A.; Selvarajoo, K. Emergent genome-wide control in wildtype and genetically mutated lipopolysaccarides-stimulated macrophages. PLoS ONE 2009, 4, e4905. [Google Scholar] [CrossRef] [PubMed]
- Morita, N.; Yamazaki, T.; Murakami, Y.; Fukui, R.; Yamai, I.; Ichimonji, I.; Nakashima, A.; Nagaoka, F.; Takagi, H.; Miyake, K.; et al. C4b-binding protein negatively regulates TLR4/MD-2 response but not TLR3 response. FEBS Lett. 2017, 591, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekaran, I.R.; Norton, R.S.; Schmitz-Peiffer, C. Characterisation of peptide interactions that regulate PKCepsilon activation. FEBS Lett. 2018, 592, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Faraz, M.; Herdenberg, C.; Holmlund, C.; Henriksson, R.; Hedman, H. A protein interaction network centered on leucine-rich repeats and immunoglobulin-like domains 1 (LRIG1) regulates growth factor receptors. J. Biol. Chem. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, K.; Suda, T. Regulatory mechanisms underlying corticotropin-releasing factor gene expression in the hypothalamus. Endocr. J. 2009, 56, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, B.Y.; Husnain, O.; Stafford, R.; Howard, M.; Gujar, A.S.; Moradiya, V.; Patel, K.K.; Sihotra, S. Hyperphosphorylation of CREB in human dopaminergic neurons: A kinetic study of cellular distribution of total CREB and phospho-CREB following oxidative stress. Neuroreport 2013, 24, 757–762. [Google Scholar] [CrossRef] [PubMed]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The role of the transcription factor CREB in immune function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Banerjee, K.; Baker, H.; Cave, J.W. Nucleotide sequence conservation of novel and established cis-regulatory sites within the tyrosine hydroxylase gene promoter. Front. Biol. (Beijing) 2015, 10, 74–90. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, J.J.; Crews, L.; Roffler-Tarlov, S.; Chikaraishi, D.M. 4.5 kb of the rat tyrosine hydroxylase 5′ flanking sequence directs tissue specific expression during development and contains consensus sites for multiple transcription factors. Mol. Brain Res. 1999, 74, 1–14. [Google Scholar] [CrossRef]
- Gueorguiev, V.D.; Cheng, S.Y.; Sabban, E.L. Prolonged activation of cAMP-response element-binding protein and ATF-2 needed for nicotine-triggered elevation of tyrosine hydroxylase gene transcription in PC12 cells. J. Biol. Chem. 2006, 281, 10188–10195. [Google Scholar] [CrossRef] [PubMed]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Sands, W.A.; Palmer, T.M. Regulating gene transcription in response to cyclic AMP elevation. Cell Sign. 2008, 20, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Moore, M.J.; Kanter, J.R.; Jones, K.C.; Taylor, S.S. Phosphorylation of the catalytic subunit of protein kinase A. Autophosphorylation versus phosphorylation by phosphoinositide-dependent kinase-1. J. Biol. Chem. 2002, 277, 47878–47884. [Google Scholar] [CrossRef] [PubMed]
- Corrigall, W.A. Nicotine self-administration in animals as a dependence model. Nicot. Tob. Res. 1999, 1, 11–20. [Google Scholar] [CrossRef]
- Uhl, G.R.; Drgon, T.; Johnson, C.; Fatusin, O.O.; Liu, Q.R.; Contoreggi, C.; Li, C.Y.; Buck, K.; Crabbe, J. “Higher order” addiction molecular genetics: Convergent data from genome-wide association in humans and mice. Biochem. Pharmacol. 2008, 75, 98–111. [Google Scholar] [CrossRef] [PubMed]
- Ducci, F.; Goldman, D. Genetic approaches to addiction: Genes and alcohol. Addiction 2008, 103, 1414–1428. [Google Scholar] [CrossRef] [PubMed]
- Blednov, Y.A.; Benavidez, J.M.; Geil, C.; Perra, S.; Morikawa, H.; Harris, R.A. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav. Immun. 2011, 25 (Suppl. 1), S92–S105. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Lousberg, E.L.; Moldenhauer, L.M.; Hayball, J.D.; Coller, J.K.; Rice, K.C.; Watkins, L.R.; Somogyi, A.A.; Hutchinson, M.R. Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br. J. Pharmacol. 2012, 165, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Bajo, M.; Montgomery, S.E.; Cates, L.N.; Nadav, T.; Delucchi, A.M.; Cheng, K.; Yin, H.; Crawford, E.F.; Roberts, A.J.; Roberto, M. Evaluation of TLR4 Inhibitor, T5342126, in Modulation of Ethanol-Drinking Behavior in Alcohol-Dependent Mice. Alcohol Alcohol. 2016, 51, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Montesinos, J.; Alfonso-Loeches, S.; Guerri, C. Impact of the Innate Immune Response in the Actions of Ethanol on the Central Nervous System. Alcohol. Clin. Exp. Res. 2016, 40, 2260–2270. [Google Scholar] [CrossRef] [PubMed]
- Blednov, Y.A.; Black, M.; Chernis, J.; Da Costa, A.; Mayfield, J.; Harris, R.A. Ethanol Consumption in Mice Lacking CD14, TLR2, TLR4, or MyD88. Alcohol. Clin. Exp. Res. 2017, 41, 516–530. [Google Scholar] [CrossRef] [PubMed]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef] [PubMed]
- Teng, X.; Dayhoff-Brannigan, M.; Cheng, W.C.; Gilbert, C.E.; Sing, C.N.; Diny, N.L.; Wheelan, S.J.; Dunham, M.J.; Boeke, J.D.; Pineda, F.J.; et al. Genome-wide consequences of deleting any single gene. Mol. Cell 2013, 52, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Akira, S. Toll-like receptors. Curr. Protoc. Immunol. 2015, 109, 1–10. [Google Scholar]
- Harris, R.A.; Bajo, M.; Bell, R.L.; Blednov, Y.A.; Varodayan, F.P.; Truitt, J.M.; de Guglielmo, G.; Lasek, A.W.; Logrip, M.L.; Vendruscolo, L.F.; et al. Genetic and Pharmacologic Manipulation of TLR4 Has Minimal Impact on Ethanol Consumption in Rodents. J. Neurosci. 2017, 37, 1139–1155. [Google Scholar] [CrossRef] [PubMed]
- Gaydos, J.; McNally, A.; Guo, R.; Vandivier, R.W.; Simonian, P.L.; Burnham, E.L. Alcohol abuse and smoking alter inflammatory mediator production by pulmonary and systemic immune cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L507–L518. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhang, Y.; Cardell, L.O. Nicotine exaggerates LPS-induced airway hyperreactivity via JNK-mediated up-regulation of Toll-like receptor 4. Am. J. Respir. Cell Mol. Biol. 2014, 51, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Lacey, M.G. Neurotransmitter receptors and ionic conductances regulating the activity of neurones in substantia nigra pars compacta and ventral tegmental area. Prog. Brain Res. 1993, 99, 251–276. [Google Scholar] [PubMed]
- Aurelian, L. Herpes simplex viruses: General features. In Encyclopedia of Virology, 3rd ed.; Mahy, B.W.J., van Regenmortel, M.H., Eds.; Elsevier: New York, NY, USA, 2014; pp. 383–397. [Google Scholar]
- Berges, B.K.; Wolfe, J.H.; Fraser, N.W. Transduction of brain by herpes simplex virus vectors. Mol. Ther. 2007, 15, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Braun, E.; Tsalenchuck, Y.; Panet, A.; Steiner, I. Restrictions that control herpes simplex virus type 1 infection in mouse brain ex vivo. J. Gen. Virol. 2011, 92 Pt 10, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.; Bowers, W.J. Targeting the central nervous system with herpes simplex virus/Sleeping Beauty hybrid amplicon vectors. Curr. Gene Ther. 2011, 11, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Bankiewicz, K.S.; Federoff, H.J. Gene therapy for the treatment of Parkinson’s disease: The nature of the biologics expands the future indications. Pharmaceuticals (Basel) 2012, 5, 553–590. [Google Scholar] [CrossRef] [PubMed]
- Manservigi, R.; Argnani, R.; Marconi, P. HSV Recombinant Vectors for Gene Therapy. Open Virol. J. 2010, 4, 123–156. [Google Scholar] [CrossRef] [PubMed]
- Perkins, D.; Gyure, K.A.; Pereira, E.F.; Aurelian, L. Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J. Neurovirol. 2003, 9, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Saeki, Y. Stable CNS gene delivery with Sleeping Beauty armed with a high-capacity HSV virion. Mol. Ther. 2006, 13, 457–458. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Chiocca, E.A.; Saeki, Y. Stable transgene expression from HSV amplicon vectors in the brain: Potential involvement of immunoregulatory signals. Mol. Ther. 2008, 16, 1727–1736. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.W.; Smith, R.M.; Pari, G.; Wobeser, W.; Rossiter, J.P.; Jackson, A.C. Herpes simplex encephalitis. Can. J. Neurol. Sci. 2005, 32, 246–247. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Cighetti, R.; Peri, F. Molecular simplification of lipid a structure: TLR4-modulating cationic and anionic amphiphiles. Mol. Immunol. 2015, 63, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Jackson, K.J.; Walters, C.L.; Damaj, M.I. Beta 2 subunit-containing nicotinic receptors mediate acute nicotine-induced activation of calcium/calmodulin-dependent protein kinase II-dependent pathways in vivo. J. Pharmacol. Exp. Ther. 2009, 330, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Lowery-Gionta, E.G.; Navarro, M.; Li, C.; Pleil, K.E.; Rinker, J.A.; Cox, B.R.; Sprow, G.M.; Kash, T.L.; Thiele, T.E. Corticotropin releasing factor signaling in the central amygdala is recruited during binge-like ethanol consumption in C57BL/6J mice. J. Neurosci. 2012, 32, 3405–3413. [Google Scholar] [CrossRef] [PubMed]
- Burke, A.R.; Miczek, K.A. Stress in adolescence and drugs of abuse in rodent models: Role of dopamine, CRF, and HPA axis. Psychopharmacology 2014, 231, 1557–1580. [Google Scholar] [CrossRef] [PubMed]
- Balan, I.; Beattie, M.; O’Buckley, T.; Aurelian, L.; Morrow, A.L. Endogenous Neurosteroid (3α,5α)3-Hydroxypregnan-20-one Inhibits Toll-like-4 Receptor Signaling in Immune Cells and Rat Brain. Neuropsychopharmacology. Submitted.
- Middleton, L.S.; Apparsundaram, S.; King-Pospisil, K.A.; Dwoskin, L.P. Nicotine increases dopamine transporter function in rat striatum through a trafficking-independent mechanism. Eur. J. Pharmacol. 2007, 554, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Kirch, D.G.; Alho, A.M.; Wyatt, R.J. Hypothesis: A nicotine-dopamine interaction linking smoking with Parkinson’s disease and tardive dyskinesia. Cell Mol. Neurobiol. 1988, 8, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Fuenzalida, J.; Galaz, P.; Araya, K.A.; Slater, P.G.; Blanco, E.H.; Campusano, J.M.; Ciruela, F.; Gysling, K. Dopamine D1 and corticotrophin-releasing hormone type-2alpha receptors assemble into functionally interacting complexes in living cells. Br. J. Pharmacol. 2014, 171, 5650–5664. [Google Scholar] [CrossRef] [PubMed]
- Fuxe, K.; Tarakanov, A.; Romero Fernandez, W.; Ferraro, L.; Tanganelli, S.; Filip, M.; Agnati, L.F.; Garriga, P.; Diaz-Cabiale, Z.; Borroto-Escuela, D.O. Diversity and Bias through Receptor-Receptor Interactions in GPCR Heteroreceptor Complexes. Focus on Examples from Dopamine D2 Receptor Heteromerization. Front. Endocrinol. (Lausanne) 2014, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Ozinsky, A.; Underhill, D.M.; Fontenot, J.D.; Hajjar, A.M.; Smith, K.D.; Wilson, C.B.; Schroeder, L.; Aderem, A. The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 13766–13771. [Google Scholar] [CrossRef] [PubMed]
- Kimman, T.G.; Banus, S.; Reijmerink, N.; Reimerink, J.; Stelma, F.F.; Koppelman, G.H.; Thijs, C.; Postma, D.S.; Kerkhof, M. Association of interacting genes in the toll-like receptor signaling pathway and the antibody response to pertussis vaccination. PLoS ONE 2008, 3, e3665. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lizarbe, S.; Montesinos, J.; Guerri, C. Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J. Neurochem. 2013, 126, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Rocha, D.M.; Caldas, A.P.; Oliveira, L.L.; Bressan, J.; Hermsdorff, H.H. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis 2016, 244, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Su, L.; Morin, M.D.; Jones, B.T.; Whitby, L.R.; Surakattula, M.M.; Huang, H.; Shi, H.; Choi, J.H.; Wang, K.W.; et al. TLR4/MD-2 activation by a synthetic agonist with no similarity to LPS. Proc. Natl. Acad. Sci. USA 2016, 113, E884–E893. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.Y.; Crews, F.T. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS ONE 2014, 9, e87915. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, M.F.; Boelens, W.C.; Joosten, L.A.; Abdollahi-Roodsaz, S.; Geurts, J.; Wunderink, L.U.; Schreurs, B.W.; van den Berg, W.B.; Radstake, T.R. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J. Immunol. 2006, 176, 7021–7027. [Google Scholar] [CrossRef] [PubMed]
- Klasener, K.; Yang, J.; Reth, M. Study B Cell Antigen Receptor Nano-Scale Organization by In Situ Fab Proximity Ligation Assay. Methods Mol. Biol. 2018, 1707, 171–181. [Google Scholar] [PubMed]
- Morud, J.; Adermark, L.; Perez-Alcazar, M.; Ericson, M.; Soderpalm, B. Nicotine produces chronic behavioral sensitization with changes in accumbal neurotransmission and increased sensitivity to re-exposure. Addict. Biol. 2016, 21, 397–406. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balan, I.; Warnock, K.T.; Puche, A.; Gondre-Lewis, M.C.; June, H.; Aurelian, L. The GABAA Receptor α2 Subunit Activates a Neuronal TLR4 Signal in the Ventral Tegmental Area that Regulates Alcohol and Nicotine Abuse. Brain Sci. 2018, 8, 72. https://doi.org/10.3390/brainsci8040072
Balan I, Warnock KT, Puche A, Gondre-Lewis MC, June H, Aurelian L. The GABAA Receptor α2 Subunit Activates a Neuronal TLR4 Signal in the Ventral Tegmental Area that Regulates Alcohol and Nicotine Abuse. Brain Sciences. 2018; 8(4):72. https://doi.org/10.3390/brainsci8040072
Chicago/Turabian StyleBalan, Irina, Kaitlin T. Warnock, Adam Puche, Marjorie C. Gondre-Lewis, Harry June, and Laure Aurelian. 2018. "The GABAA Receptor α2 Subunit Activates a Neuronal TLR4 Signal in the Ventral Tegmental Area that Regulates Alcohol and Nicotine Abuse" Brain Sciences 8, no. 4: 72. https://doi.org/10.3390/brainsci8040072