
The Role of the Thioredoxin System in Brain Diseases
by
 , , , and
, , , and
Geir Bjørklund
1,* ,
,
Lili Zou
2,*, 
Massimiliano Peana
3 ,
,
Christos T. Chasapis
4,
Tony Hangan
5,
Jun Lu
6 and
Michael Maes
7 1
Council for Nutritional and Environmental Medicine, Toften 24, 8610 Mo i Rana, Norway
2
Hubei Key Laboratory of Tumor Microenvironment and Immunotherapy, College of Basic Medical Sciences, China Three Gorges University, Yichang 443002, China
3
Department of Chemical, Physical, Mathematical and Natural Sciences, University of Sassari, Via Vienna 2, 07100 Sassari, Italy
4
Institute of Chemical Biology, National Hellenic Research Foundation, 11635 Athens, Greece
5
Faculty of Medicine, Ovidius University of Constanta, 900470 Constanta, Romania
6
School of Pharmaceutical Sciences, Southwest University, Chongqing 400715, China
7
Department of Psychiatry, Faculty of Medicine, Chulalongkorn University, Pathumwan, Bangkok 10330, Thailand
*
Authors to whom correspondence should be addressed.
Antioxidants 2022, 11(11), 2161; https://doi.org/10.3390/antiox11112161
Submission received: 28 September 2022
/
Revised: 23 October 2022
/
Accepted: 28 October 2022
/
Published: 31 October 2022
(This article belongs to the Special Issue 10th Anniversary of Antioxidants—Review Collection)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The thioredoxin system, consisting of thioredoxin (Trx), thioredoxin reductase (TrxR), and NADPH, plays a fundamental role in the control of antioxidant defenses, cell proliferation, redox states, and apoptosis. Aberrations in the Trx system may lead to increased oxidative stress toxicity and neurodegenerative processes. This study reviews the role of the Trx system in the pathophysiology and treatment of Alzheimer’s, Parkinson’s and Huntington’s diseases, brain stroke, and multiple sclerosis. Trx system plays an important role in the pathophysiology of those disorders via multiple interactions through oxidative stress, apoptotic, neuro-immune, and pro-survival pathways. Multiple aberrations in Trx and TrxR systems related to other redox systems and their multiple reciprocal relationships with the neurodegenerative, neuro-inflammatory, and neuro-oxidative pathways are here analyzed. Genetic and environmental factors (nutrition, metals, and toxins) may impact the function of the Trx system, thereby contributing to neuropsychiatric disease. Aberrations in the Trx and TrxR systems could be a promising drug target to prevent and treat neurodegenerative, neuro-inflammatory, neuro-oxidative stress processes, and related brain disorders.
1. Introduction
Neurological disorders, including acute stroke and chronic neurodegenerative diseases, present a significant cause of morbidity and mortality worldwide with the extreme medical and financial burden [1,2,3]. Oxidative stress toxicity contributes to the pathophysiology of most neurological diseases, and its magnitude is related to the ability of cellular antioxidants to neutralize the accumulating reactive oxygen species (ROS) [4,5,6,7]. To counteract the detrimental effects of ROS and consequent toxicity and restore the delicate redox hemostasis in the brain, cells are equipped with an endogenous antioxidant defense mechanism consisting of several antioxidant enzymes [8]. The thioredoxin (Trx) system is the major antioxidant system in the brain, and this thiol-dependent redox system comprises Trx, Trx reductase (TrxR), truncated Trx (Trx-80), and NADPH. This system is essential for maintaining the balance of the cellular redox status, and it is involved in regulating redox signaling [9]. Trx is a ubiquitous and multifunctional redox protein, executing its function through its antioxidative, protein-reducing, and signal-transducing activities, and its reduced state is maintained by TrxR [10,11]. Changes in the expression or activities of the Trx antioxidant system have been documented in the brains of patients with neuropsychiatric disorders and in animal models of those disorders [12,13,14,15]. The potential mechanisms of the Trx system components protecting against those diseases not only involve its antioxidant properties but also anti-apoptotic and anti-inflammatory as well as prosurvival pathways [12,13,14,15,16].
The aims of the current study are to review the role of the Trx system in the pathophysiology and treatment of Alzheimer’s disease (AD), age-related decline in memory functions, Parkinson’s disease (PD), Huntington’s disease (HD), brain stroke, and multiple sclerosis (MS) [17,18,19]. We consequently reviewed the relevant pathways, including nitro-oxidative pathways, which play a crucial role in those disorders, the aberrations in the Trx system, and how the Trx system may modulate those pathways, thereby contributing to the pathophysiology of these different brain disorders. We reviewed the many synergistic and antagonistic interactions of the Trx system with the well-established pathways in those disorders and how environmental factors, including metals, dietary deficiencies, pesticides, and infections, may interfere with these interactions. In addition, we reviewed clinical and preclinical studies targeting the Trx system in those disorders to critically evaluate whether the Trx system could be a new drug target to treat these disorders [20].
A state-of-the-art review on the involvement of Trx-related signal pathways in the complex pathophysiology of neurological disorders will provide new insights into the mechanisms of the Trx family proteins in neuropsychiatric disorders, thereby revealing candidate biomarkers and new therapeutic drug targets, which may lead to clinical drug development for the treatment of various neuropsychiatric diseases.
2. The Methodology of the Review Analysis
The present review was performed according to a systematic search in the databases Science Direct, Web of Science, PubMed, Scopus, and CABI Direct for the years 2000 to 2021 (Figure 1). The keywords used were “brain disease”, “thioredoxin system”, “NADPH”, “thioredoxin reductase”, “thioredoxin” together with one of the following terms: “Alzheimer’s disease”, “Parkinson’s disease”, “Huntington’s disease”, “brain stroke”, or “multiple sclerosis”.
After a first screening, all potentially relevant articles were downloaded from the databases, and the applicable data were extracted and evaluated. Inclusion criteria for this study were: (1) Full-text availability; (2) English language for published full text; (3) Original studies and case reports about brain disease and the Trx system; (4) Research reporting the role of the Trx system, NADPH, TrxR, and Trx in brain disease. Relevant papers were selected and included in this review according to their titles, abstracts, and the mentioned search criteria. Redundant articles or data, conference abstracts, and letters were not evaluated [21].
3. The Thioredoxin System
Progressive neuronal dysfunctions may induce neurodegenerative diseases such as AD, PD, and HD. Oxidative damage may frequently occur in the brain because brain tissues consume a high amount of oxygen and contain oxidizable polyunsaturated fatty acids and redox-active metals [22,23,24]. Oxidative stress toxicity plays a critical role in the pathogenesis of several neurodegenerative disorders, especially in elderly individuals [12,25,26]. Moreover, clinical and preclinical studies suggest that altered redox homeostasis increases oxidative stress, as well as decreases levels of antioxidant defenses, may induce activation of stress-related pathways in the brain and peripheral tissues [26,27,28].
The thioredoxin (Trx) system consists of Trx, thioredoxin reductase (TrxR), and NADPH, which are present in various organisms from Archea to humans [29,30]. The Trx system has various functions protecting against oxidative stress and, consequently, serves as a critical antioxidant system in the central nervous system (CNS), DNA synthesis, and apoptosis, thereby maintaining the redox balance in the brain [11,31]. Mammalian cells possess two Trx systems, the cytosolic Trx1 and the mitochondrial Trx2 system. Trx2 has only the two cysteines in its active site, whereas Trx1 has three additional extra cysteines, which play a role in the redox regulation of activity and NO signaling [32]. Trxs, with a dithiol/disulfide active site (-CGPC-), are the main cellular protein disulfide reductases, which act as electron donors for enzymes, including ribonucleotide reductases (RNRs), thioredoxin peroxidases (peroxiredoxins, Prxs), and methionine sulfoxide reductases (MSRs). They play critical roles in pleiotropic cellular effects, such as controlling cell proliferation, redox states, and apoptosis [29,33]. Also, Trx is a serum antioxidant that has been evaluated in the etiology of schizophrenia [34]. Human cytosolic Trx, which is the 12-kDa protein disulfide reductase with the Cys-Gly-Pro-Cys active site and a key component of cellular redox biochemistry and regulation, acts as cocytokine upon leaderless secretion. A 10-kDa C-terminally Trx80 comprising the 80 or 84 N-terminal amino acids is also secreted and present in plasma, where it originally was identified as eosinophilic cytotoxicity enhancing factor [35]. Moreover, TrxRs in higher eukaryotes, including cytosolic TrxR1 and mitochondrial TrxR2, are selenium-dependent dimeric flavoproteins (112–130 kDa). TrxR1 shows a broad substrate specificity that reduces non-disulfide substrates such as hydroperoxides, vitamin C, or selenite. In contrast, TrxRs in bacteria, fungi, and plants are specific dimeric 70 kDa flavoproteins with a redox-active disulfide/dithiol site [29,36]. Three TrxRs are found in mammalian cells, cytosolic TrxR1, mitochondrial TrxR2, and a testis-specific thioredoxin glutathione reductase (TGR) [37]. All mammalian TrxR isozymes are homologous to glutathione reductase (GR) and contain a conserved C-terminal elongation with a cysteine-selenocysteine sequence forming a redox-active selenenyl sulfide/selenol thiol active site.
One of the important endogenous molecules to interact with Trx is thioredoxin interacting protein (TXNIP), which is a negative regulator of Trx function [38]. Cys63 and Cys247 in TXNIP can form the mixed disulfide bond with Trx active site thiols and suppress the activity of Trx and result in oxidative stress [39]. TXNIP located in the nucleus under normal conditions. In response to oxidative stress TXNIP can shuttle into cytosol or mitochondria, which binds and oxidizes Trx1/Trx2, reducing the binding of Trx1/Trx2 with ASK1 and resulting in a ASK1-mediated signaling pathway [40,41]. TXNIP has been found to bind to inflammasome in response to ROS production [42,43]. Trx binds to TXNIP in the steady state. On stimulation of inflammasome activators, ROS will be produced, resulting in the dissociation of TXNIP from Trx and the binding to NLRP3. Then the NLRP3 inflammasome is activated, and the active, mature interleukin 1β (IL-1β) can be produced and secreted after the cleavage from pro-IL-1β precursor by caspase-1 [42]. TXNIP is also shown to be induced by endoplasmic reticulum (ER) stress, to activate IL-1β production via inflammasome, and to mediate ER stress-induced β cell death [44]. In addition, recent study showed that treatment with proteasome inhibitors reversed GAS-induced Txnip degradation. These results indicate that TXNIP may be a potential target against brain disease.
Selenium-based TrxR/Trx and sulfur-based GSH systems are both interactively involved in providing antioxidant activity in cells and tissues, but differences in their relative contributions are evident. Thus, the GSH concentration in neurons is in the range of 0.2 mM [45,46], and this contrasts with the concentrations in hepatocytes, namely around 10 mM [47] or a 50-fold difference. Brain levels of GSH and its precursor cysteine are nearly 10-fold lower than in plasma [48], while selenium content in the brain is only about half of the selenium in plasma [49,50], despite a higher level of aerobic metabolism. Glutathione (GSH) increases the redox impact of neurotrophic factors, which exert epigenetic effects on gene expression via changes in DNA and histone methylation [51]. Accordingly, a normal function of the Trx system is crucial for maintaining brain redox status and for allowing neurotrophic factor signaling to affect gene expression. The latter is particularly crucial during neurodevelopment [52].
Levels of GPx1 and GR in brain mitochondria are low [53], whereas the 2-Cys Prxs, viz. Prx1, Prx2, Prx3, Prx4, and Prx5, are highly expressed in the brain and some in brain mitochondria [54,55,56,57,58,59,60,61,62]. This suggests that they may protect mitochondrial DNA in the brain against damage caused by oxidative and nitrative stress and the formation of mutagenic aldehydes as secondary products of lipid peroxidation, such as acrolein [63,64,65] and crotonaldehyde [63,64,65]. 1-Cys Prx (Prx6) is also expressed in the brain [60,66], mainly in glial cells [66,67] and not to any significant extent in neurons [67].
The relative contributions of different scavenging enzymes to H2O2 removal from isolated mitochondria from brain cells have been measured through in vitro experiments, using polarographic methods for real-time detection of steady-state H2O2 levels [68]. This research showed that isolated rat brain mitochondria display significant rates of exogenous H2O2 removal (9–12 nmol/min/mg of protein) in the presence of substrates, indicating a respiration-dependent process [68]. GSH-dependent systems showed only minimal contributions: a 25% decrease in GR inhibition and no effect by GPx inhibition [68]. In contrast, inhibitors of TrxRs, including auranofin, attenuate H2O2 removal rates in mitochondria by 80% [68]. Furthermore, a 50% decrease in H2O2 removal was observed following the oxidation of Prx [68]. Decreases in H2O2 removal are observed in intact dopaminergic neurons with TrxRs inhibition, implicating this mechanism in whole-cell systems [68]. In another study, the specific inhibitors auranofin and 1-chloro-2,4-dinitrobenzene (DNCB) were used to determine the relative contributions of Trx2-dependent and GSH-dependent systems to H2O2 removal from rat brain mitochondria [69]. The contributions of Trx2-dependent and GSH-dependent systems to ROS detoxification in rat brain mitochondria were 60 ± 20% and 20 ± 15%, respectively [69]. As revealed by aminotriazole inhibition, catalase only contributed to a non-significant extent [69].
4. Synergistic and Antagonistic Interactions between Aberrations in Different Signal Transduction Systems
Before discussing the oxidative pathophysiology of the various brain disorders and the involvement of the Trx system in these disorders, we would stress that—as we will show–alterations in the Trx system show many interactions with different signal transduction networks and that combined with these networks the Trx system may induce multiple protective as well as pathophysiological responses. In analogy, synergistic and antagonistic interactions in serotoninergic, dopaminergic, adrenergic, and GABAergic systems may induce more detrimental consequences when all neurotransmitters are simultaneously disturbed. It follows that pharmacological interventions targeting only one of the systems cannot be expected to be effective. In principle, this is similar to other areas of biology, where multiple controls, circular feedback systems, non-linear synergistic cooperation and interdependency of subsystems, and apparent redundancies can be found. Multiple interacting signal pathways, which are present in all nucleated cells of the human body, play a crucial role in controlling cell differentiation and morphogenesis during embryonic development; and multiple genes participate in the control of apparently simple morphological features, including body length [70,71]. There are very fuzzy interactions that are remarkably efficient in the regulatory control of human features and, in fact, quite different from the control systems that engineers design to control chemical processes in chemical factories. However, the reductionist “control engineer” way of thinking is still widespread in medicine—as if the human body were nothing but an expensive new car full of the most recent high-tech gadgets. Therefore, it is important to consider that the associations of the Trx system with the neuropsychiatric disorders discussed here should be interpreted with regard to its many synergistic and antagonistic interactions, feedback, and regulatory effects on oxidative stress toxicity, antioxidant defenses, and apoptotic, immune-inflammatory and prosurvival pathways.
5. Alzheimer’s Disease
5.1. Pathophysiology of Alzheimer’s Disease
Alzheimer’s disease (AD) is the most critical neurodegenerative disorder of the central nervous system, accounting for more than 60–80% of all dementia cases [17,72] and is a severe healthcare challenge in developed countries in elderly individuals [73,74]. The prevalence estimates indicate that AD affects approximately 15 million persons in the United States [75] and will affect more than 115 million individuals worldwide [76] by the year 2050. According to the Alzheimer’s Association, 13% of older adults over 65 suffer from AD in developed countries [77]. AD progressively affects individuals aged 65 years and older and is the sixth leading cause of early death [78,79]. AD is defined by impairments in memory and executive functions and is accompanied by senile plaques consisting of an accumulation of extracellular amyloid β (Aβ) as well as intracellular neurofibrillary tangles (NFTs) containing a high accumulation of phosphorylated TAU protein, and by loss of neurons, mostly in the hippocampus and cortex [74,79,80,81,82]. Age is the most critical risk factor influencing the heterogeneity and topography of neurodegeneration of Alzheimer’s disease. With a growing elderly population, millions more will be affected by this disease [83,84]. Oxidative stress has been involved in age-related neurodegenerative disorders characterized by progressive neuron dysfunction and cell death. Also, synaptic dysfunctions are involved in AD’s behavioral symptoms and pathophysiology [85] and are strongly associated with oxidative stress toxicity [86]. Additionally, oxidative stress is related to mitochondrial and endoplasmic reticulum dysfunctions, inducing protein misfolding and neuronal apoptosis [74,87].
Various factors may increase the risk of AD, including cardiovascular disease, high blood pressure, diabetes, obesity, high cholesterol, chronic kidney disease, and psychiatric disorders [79,88]. Moreover, familial forms of the disease are linked to genetic mutations in the amyloid precursor protein (APP) gene, presenilin (PS) 1 and 2 genes [89,90], as well as genetic variants in apolipoprotein E allele type 4 (APOE4), which elevates disease risk in most populations [79,91,92,93]. On the other hand, some environmental factors may have a critical role in the pathophysiology and pathogenesis of AD, such as metals, dietary deficiencies, pesticides, and infections [94]. APP is cleaved by γ-secretase and α-secretase to make a soluble, non-amylogenic peptide [95]. However, in AD, APP can be cleaved by γ-secretase and β-secretase to produce the insoluble peptides Aβ40 and Aβ42 as the main shape of amyloid in the cerebral cortex and hippocampus, and while the disease progresses, they spread all over the brain [96]. Also, in AD, neuronal tissue lesions are often associated with the hyperphosphorylation of TAU proteins, producing the NFTs, which aggregate in the neuron’s somatic-dendritic portion [97].
5.2. Trx and TrxR in Alzheimer’s Disease
TrxR1 and TrxR2 play critical roles in brain functions. Both TrxRs decrease oxidative stress, neutralize hydrogen peroxide (H2O2), and regulate redox-sensitive transcription factors that inhibit cellular transcription mechanisms [98]. This protein family may be a protective factor in AD [99]. In brain tissues of AD patients, low levels of Trx1 are observed [97,100] in association with the accumulation of Aβ peptide, and, therefore, it was proposed that Trx1 is a critical protective factor in AD [99]. Decreased expression of Trx1 is observed in both the frontal cortex and the hippocampus CA1 regions of patients suffering from AD [101]. Moreover, treatment with the β-amyloid peptide in human SH-SY5Y neuroblastoma cells may induce early and time-dependent oxidation in Trx1 and Grx1 while Trx1 overexpression effectively preserved cells from Aβ peptide-related toxicity. Trx expression is induced by oxidative stress, thereby regulating redox status and the functions of signaling proteins [102]. Furthermore, TrxR plays a protective role against Aβ toxicity in neuronal cultures and primary hippocampal cultures. Indeed, Trx or TrxR may induce a significant concentration-dependent improvement in cell survival against Aβ-mediated toxicity [99]. Also, administration of reduced Trx to apoptotic cultures of cerebellar granule cells induced a marked reduction in cell death, indicating that a well-functioning Trx/TrxR system improves the survival of cerebellar granule cells [103]. Interestingly, the levels of Trx80 were considerably decreased in AD brain tissue, including in areas with abundant inflammatory changes, indicating that Trx80 failure is a distinct feature of the disease [104]. On the other hand, Trx80, which lacks the C-terminal strand-helix of the Trx fold, induces the proliferation of peripheral blood mononuclear cells [105] and may restrict in vitro accumulation of Aβ as well as Aβ toxicity in SH-SY5Y cells, indicating that increasing Trx1 levels and activities could interact with the toxic effects of Aβ [104]. Moreover, a Tg mouse model overexpressing Aβ showed that TrxR1 activity was significantly decreased at 14 months of age, indicating loss of the Trx1 functions [106]. Trx1 functions in the CNS comprise antioxidant activities and modulate nerve growth factors and other signal transduction pathways [18,107]. Genetic mutations of Trx or TrxR may affect neuronal degeneration [108].
The Prxs system is an antioxidant enzymatic system of emerging significance in the pathophysiology of neuronal aberrations [9]. Gene overexpression, knockdown, and knockout approaches revealed the critical function of Prxs-mediated neuron protection against oxidative stress. Post-translational modifications of neuronal Prxs may cause impairments in their functions as a part of disease pathology. Therefore, members of the Trx superfamily of proteins, the Trx-Prx system, could constitute promising biomarker candidates for the early diagnosis of AD, reflecting their crucial involvement in the treatment and pathogenesis of the disease.
Numerous studies suggest that TrxRs are essential players in antioxidant defenses, cellular redox systems, as well as in growth control, and selenium metabolism [11,33,109]. Decreased selenium is a prominent feature of AD [81,110,111,112]. Selenium (Se), botshowsh as Se-containing compounds and selenoproteins, may be critical in preventing Alzheimer’s pathology [111]. Seleno-L-methionine mixed with vitamin E may have a beneficial role against toxicity from β-amyloid and oxidative stress in cell cultures [113,114]. Also, a mutant APP and presenilin AD rodent model shows the efficacy of organic Se in decreasing AD pathology, reducing the Aβ burden, and minimizing DNA and RNA oxidation [115]. Injections of tricyclodecan-9-ylxanthogenate (D609), a compound that mimics the antioxidant properties of GSH, may decrease Aβ toxicity and oxidative stress by elevating of GPx activity [116].
Furthermore, Trx protein contents are reduced in postmortem AD brains. Still, TrxR activity may be increased [99], which is probably a compensatory mechanism to attenuate activated oxidative stress pathways. Moreover, apoptosis signal-regulating kinase 1 (ASK1) has a vital role in the pathogenesis of neurodegenerative disorders, including AD [117], and Trx is bound directly to the N-terminal region of ASK1 [118] and induces dissociation of Trx from ASK1 in the oxidative stress state. Also, Aβ induces an early, strong, and transient oxidation of glutaredoxin-1 (Grx1) and Trx1.
Mitochondrial dysfunctions coupled with increased oxidative stress (resulting partly from oxidative damage to mitochondrial DNA) may play a significant role in the etiopathogenesis of various degenerative brain diseases, including AD [119,120,121]. Aging mitochondrial DNA [122] will sooner or later lead to enhanced ROS production in mitochondria [123], thereby causing an interaction between mitochondrial DNA aging and Hg inhibition of antioxidant defense enzymes, which will cause enhanced oxidative stress in brain cells. Moreover, the expression of Prx1 is enhanced in an animal model of AD, while this enzyme function is a negative regulator of the expression of the γ-secretase complex, which is central to the pathogenesis of AD [124].
5.3. Age-Related Decline in Memory Function
There is little doubt that Hg and other Se-antagonistic toxic metals, including Cd and Ag, may cause enhancement of mitochondrial DNA aging in the brain when the exposure is large enough to cause significant inhibition of TrxRs or impaired availability of selenide ions for the synthesis of selenocysteyl-tRNA and incorporation into the Fe-sulphur groups of enzyme complexes in the mitochondrial respiratory chain. Therefore, TrxRs should also be considered one of the prime targets for therapeutic intervention in well-established or suspected Hg poisoning cases.
Enhanced expression of Trx2 and Prx3 (a 2-Cys peroxiredoxin) was observed in the spinal cord and hippocampus of aged dogs [125], suggesting redox regulation of the expression of these proteins with higher intracellular oxidant stress. As described above, oxidative stress occurs in elderly individuals due to mitochondrial DNA aging leading to enhanced production of ROS in the mitochondria [123,126].
Increased oxidative and nitrosative stress in the brain is crucial as a cause of pathological aging processes that ultimately may accumulate in neurodegenerative brain disease. At the same time, the rate-controlling enzymes in the biosynthetic pathways of several important transmitter substances and hormones are inhibited by nitrosative and/or oxidative stress. One example is tryptophan hydroxylase [127,128,129], which is the rate-limiting enzyme in the serotonin biosynthesis pathway, with serotonin and melatonin as end products. Another example is tyrosine hydroxylase [130,131,132], which is a rate-limiting enzyme in the pathway leading to dopamine synthesis, with dopamine and noradrenaline as end products (and for adrenaline in the adrenal glands). A third example is glutamate decarboxylase [133,134,135,136,137], which produces GABA from glutamate, but with one of the glutamate decarboxylase isozymes being much more sensitive to oxidative stress than the other [138].
Figure 2 shows the Trx system’s role in AD pathophysiology. Research has shown that the decreases of Trx1, Trx80, and TrxR in AD brain tissue are distinct disease hallmarks. As a critical protective factor against oxidative stress decreased, Trx and TrxR levels are associated with the accumulation of Aβ and may affect neuronal degeneration. Furthermore, the oxidative state may induce Trx dissociation from ASK1, which, in turn, may induce neuronal death. Also, the Trx-Prx system can be used as a promising biomarker in diagnosing AD.
6. Parkinson’s Disease
6.1. Pathophysiology of Parkinson’s Disease
Parkinson’s disease (PD) is a common neurodegenerative disorder and the second most prevalent disease, after AD, affecting 0.3% of the general population and 1–3% of the population older than 65 years [139,140,141]. PD is identified by accumulation of α-synuclein within neurons [142], loss of motor control [143], and a progressive, chronic, and profoundoverexpression loss of neuromelanin-containing dopaminergic neurons in the midbrain of the substantia nigra with eosinophilic, intracytoplasmic, proteinaceous inclusion bodies termed Lewy bodies and dystrophic Lewy neurites in surviving neurons [144,145]. Lewy bodies are insoluble proteins, especially α-synuclein, which are produced in these neurons before cell loss [146] as a consequence of incorrect processing of proteins, malfunction of the ubiquitination system, and the unfolded protein response [147]. Most cases of PD are sporadic, although genetic and environmental factors [148] and more likely, a combination of both factors are associated with PD while also familial mutations may be involved [149]. Factors that may induce neurodegenerative processes in the substantia nigra include mitochondrial failure, adenosine triphosphate (ATP) depletion, aberrations in neurotrophic factors, elevated ROS, and excess glutamatergic transmission excitotoxicity, and increased quantities of iron associated with neuromelanin [150].
Some toxins such as 6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) are known to induce dopaminergic neuronal loss in Tg mouse models with null mutations of Parkin, DJ-1, and PINK1 or point mutations of genes located in different PARK loci that carry the same genetic mutations as patients suffering from familial PD [151,152]. Dopamine is used by substantia nigra cells to interact with neurons of the striatum [153] and, therefore, decreased contents of the nigral and, thus, striatal dopamine may cause PD symptoms. A large body of studies showed the effect of oxidative stress in the cascade of events inducing degeneration of dopaminergic neurons in PD [154,155]. Moreover, the oxidative deamination of dopamine and other monoamine transmitters by monoamine oxidase (MAO) is associated with the loss of dopamine neurons in the substantia nigra [156,157].
6.2. Trx and TrxR in Parkinson’s Disease
Animal and human MAO-B can produce the neurotoxin 1-methyl-4-phenylpyridinium ion [MPP+] from the MPTP that may induce a severe and irreversible PD-like syndrome [114]. Importantly, Trx1 expression may be decreased by MPP+ [158], and its oxidation may induce apoptosis by a caspase-12-dependent pathway and prevent the mitochondrial respiration complex I. Moreover, in vitro and in vivo, overexpression of Trx1 has cytoprotective and neuroprotective activities against MPTP [88,159,160]. The most abundant neuronal Prx, Prx2, has an essential role in dopaminergic neuron protection against PD-relevant toxin-induced cell death through modulation of Trx1-ASK-1 interactions and prevents the subsequent stimulation of ASK-1-dependent cell death pathways [161]. Furthermore, neurodegeneration is accompanied by loss of DJ-1, a multifunctional protein with antioxidant activity as well as a transcriptional regulator and stabilizer of nuclear factor erythroid 2-related factor 2 (Nrf-2) [162]. Its oxidative modification has been identified in the brain tissue of PD and AD patients [163]. Moreover, DJ-1, as a redox sensor, can co-immunoprecipitate with ASK-1 during exposure with H2O2 after dissociation of Trx1 [164,165]. DJ-1 can protect cells against oxidative stress through increased expression of Trx1 via the transcription factor Nrf-2. Therefore, DJ-1 and the Nrf-2-mediated increased Trx1 contents may protect cells against oxidative stress [166]. Furthermore, Mito-TRFS, the first synthetic turn-on probe for mitochondrial TrxR, may provide an accurate discrimination method to image TrxR2 function in living cells [167]. Also, Mito-TRFS showed that PD models are accompanied by severe decrements in TrxR2 activity, suggesting a mechanistic link between dysfunction of TrxR2 and PD etiology.
Selenium may have a critical role in PD by alleviating oxidative stress via selenoproteins [20,112,168]. The plasma levels of selenium may be decreased in PD patients due to higher utilization of selenium for brain selenoprotein production, likely used to attenuate further oxidative damage [169]. A study revealed that blocking the mitochondrial Trx/Prx system induces dopaminergic cells towards mitochondrial dysfunction, elevated steady-state H2O2, and cell death against toxicants implicated in PD. Also, the lentiviral knockdown of TrxR2 or pharmacological inhibition of TrxR induces dopaminergic cells to sub-toxic concentrations of PD toxicants paraquat (PQ) and 6-hydroxydopamine (6OHDA) [170].
TrxR deficiency may reduce the activity of all 2-Cys Prxs in the brain and may potentiate oxidative stress, mitochondrial dysfunction, and cell death in dopaminergic cells [170]. Therefore, TrxR deficiency is probably associated with a faster progression of PD and should be regarded as a new drug target to treat PD and its progression by enhancing TrxRs and other antioxidant defenses in the brain. Moreover, activated nitro-oxidative stress pathways in PD may also cause comorbid depression, characterized by activated immune and nitro-oxidative stress pathways [171].
Dopamine is an essential neurotransmitter in brain centers, activated during context learning and activities associated with reward and pleasure [172,173,174]. These centers are also activated by nicotine and some psychoactive substances, which may induce dependence, including legal and illegal drugs [175,176,177,178]. Despite having distinct pharmacological targets, these psychoactive agents may enhance dopamine release in the nucleus accumbens [178]. There can be little doubt that inhibition of dopamine synthesis in the brain, accompanied by lowered antioxidant defenses, may increase the likelihood of substituting decreased endogenous dopamine levels with the effects of exogenic sources, including alcohol, smoking, or illegal psychoactive drugs. However, this stimulation induced by drug abuse is much more problematic in intensity and duration than the stimulation induced by natural rewards because exogenic provision tends to affect feedback circuits, reducing endogenic production, producing renewed craving, and thus causing a vicious circle [178].
Figure 3 shows the involvement of Trx1 in the pathophysiological signal pathways in PD. Oxidative stress may induce dissociation of ASK-1 from Trx1 to stimulate ASK-1-dependent cell death, and Prx2 may distinctly protect against the corresponding effect. H2O2-exposed ASK-1 further co-immunoprecipitates with DJ-1, which mediates an increase of Trx1 against oxidative stress via the Nrf2 signal pathway. Moreover, the decrease and oxidation of Trx1 may induce caspase-12-dependent apoptosis.
7. Huntington’s Disease
7.1. Pathophysiology of Huntington’s Disease
Huntington’s disease (HD) is a progressive autosomal dominant neurodegenerative disorder [179,180] caused by a trinucleotide CAG repeats in exon-1 of the huntingtin gene (HTT) on the fourth chromosome (4p16.3) [181], resulting in the expression of a polyglutamine-expanded mutant Huntington protein (mHTT) at the amino-terminal end of the protein [182]. The onset of the disease is typically in early to mid-adult life with a range from childhood to advanced age and is defined by an excess of least 39–42 CAG repeats [183,184,185]. The full-length soluble mHTT undergoes enzymatic cleavage to produce soluble N-terminal mHTT polyglutamine involved fragments as monomers, soluble oligomers, and larger insoluble aggregates [186,187]. Soluble N-terminal mHTT fragments are probably the primary drivers of disease progression through accumulation in cells, aberrant interactions with numerous proteins, intracellular inclusion bodies, neuronal death, and possibly direct production of ROS [184,188,189].
The striatum exhibits marked variation in the severity of neurodegeneration involvement [190] with the loss of medium spiny neurons (MSN) in the basal ganglia [181]. In a later phase of the disease, the pathology may spread through the globus pallidus, thalamus, hypothalamus, subthalamic nucleus, substantia nigra, and cerebellum [151,184]. At the onset of disease, the indirect pathway of the basal ganglia could be affected by the inhibitory of dopamine D2-type receptors that extend to the globus pallidus externa (GPe) [172]. In the later stages of the disease, MSNs of the direct excitatory pathway that connects to the globus pallidus internal segment (GPi) are also lost, resulting in symptoms such as akinesia and dystonia [114].
7.2. Trx and TrxR in Huntington’s Disease
Trx1 is described as a cytoplasmic and nuclear thiol-disulfide oxidoreductase that has protective effects in acute and chronic models of neurodegeneration. Trx1 transgenic mice display elevated resistance to neuronal degeneration induced by transient focal ischemia [191]. The protective function of Trx1 in various models of neuronal degeneration indicates the critical role of signaling pathways in redox regulation and repair of oxidatively-modified thiols in various proteins.
Nevertheless, oxidative stress in HD is accompanied by elevated GPx levels [192]. The activity of GPx is significantly increased in the brain of HD patients, especially GPx1 in the cerebral cortex and striatum, as well as GPx6 in the striatum. Using quinolinic acid in a rat model of HD, the activity of GPx also increased to induce neurodegeneration [193]. Moreover, GSH and GR act as a backup of human TrxR1 to reduce Trx1, thus preventing cell death caused by aurothioglucose [62]. There is probably a GSH redox cycle dysregulation in HD with a decreased de novo synthesis of GSH in brain cells [194]. This emphasizes the role of a dysfunctional GSH status in HD and targets this system to prevent the onset of HD in relatives of patients genetically predisposed to develop the disease.
GSH is transported by a Na+-coupled transport system across the blood-brain barrier in various mammalian species [195,196,197,198,199], including humans [198] with KM values, which are so high [195,200] that they regulate the uptake rate of GSH into the brain. Moreover, Trx1 and the thioredoxin-related transmembrane protein 3 (TMX3) both decrease mHTT in cells, but there is no evidence of direct interaction with mHTT. Moreover, Trx1 and TMX3 decrease striatal neuronal atrophy, suggesting a modulatory role of Trx1 and TMX3 in mouse HD model systems [201]. Moreover, Trx1 is described as a negative regulator of ASK1 and, consequently, is a potential protectant against neurodegeneration [202].
Figure 4 shows the role of Trx1 and Gpx in the pathophysiology of HD. Trx1 protects against neuronal atrophy and neurodegeneration, thereby decreasing the expressions of mHTT and ASK1, respectively. Meanwhile, in HD patients, oxidative stress upregulates GPx1 and GPx6.
8. Brain Stroke
8.1. Pathophysiology of Brain Stroke
Brain stroke induces degeneration of brain tissue that leads to functional impairment with limited brain self-repair [203,204]. Disturbance in the brain blood supply and consequent brain dysfunction is a frequent cause of adult disabilities, which may result in more than half of the patients suffering from long-term disabilities. Brain stroke is the primary cause of adult-acquired disabilities in developing and developed countries, accounting for 9% of deaths worldwide [205,206,207]. The incidence and prevalence of brain stroke are anticipated to increase with the aging of the population [207]. According to the World Health Organization (WHO), annually, 15 million people suffer a stroke worldwide. A brain stroke is an acute-onset clinical syndrome that progresses following a vascular insult to the brain. Following vascular occlusion, a complex chain of molecular events induces irreversible tissue injuries such as failure of energy synthesis, loss of transmembrane ionic gradients dependent on active transport, cell depolarization, and excitotoxicity over the release of excitatory neurotransmitters. Moreover, ROS plays a crucial role in brain injury after ischemic stroke [208,209]. Current data suggest that a rapid increase in ROS production following an acute ischemic stroke due to impaired antioxidant defenses may lead to increased tissue damage [205].
8.2. Trx and TrxR in Brain Stroke
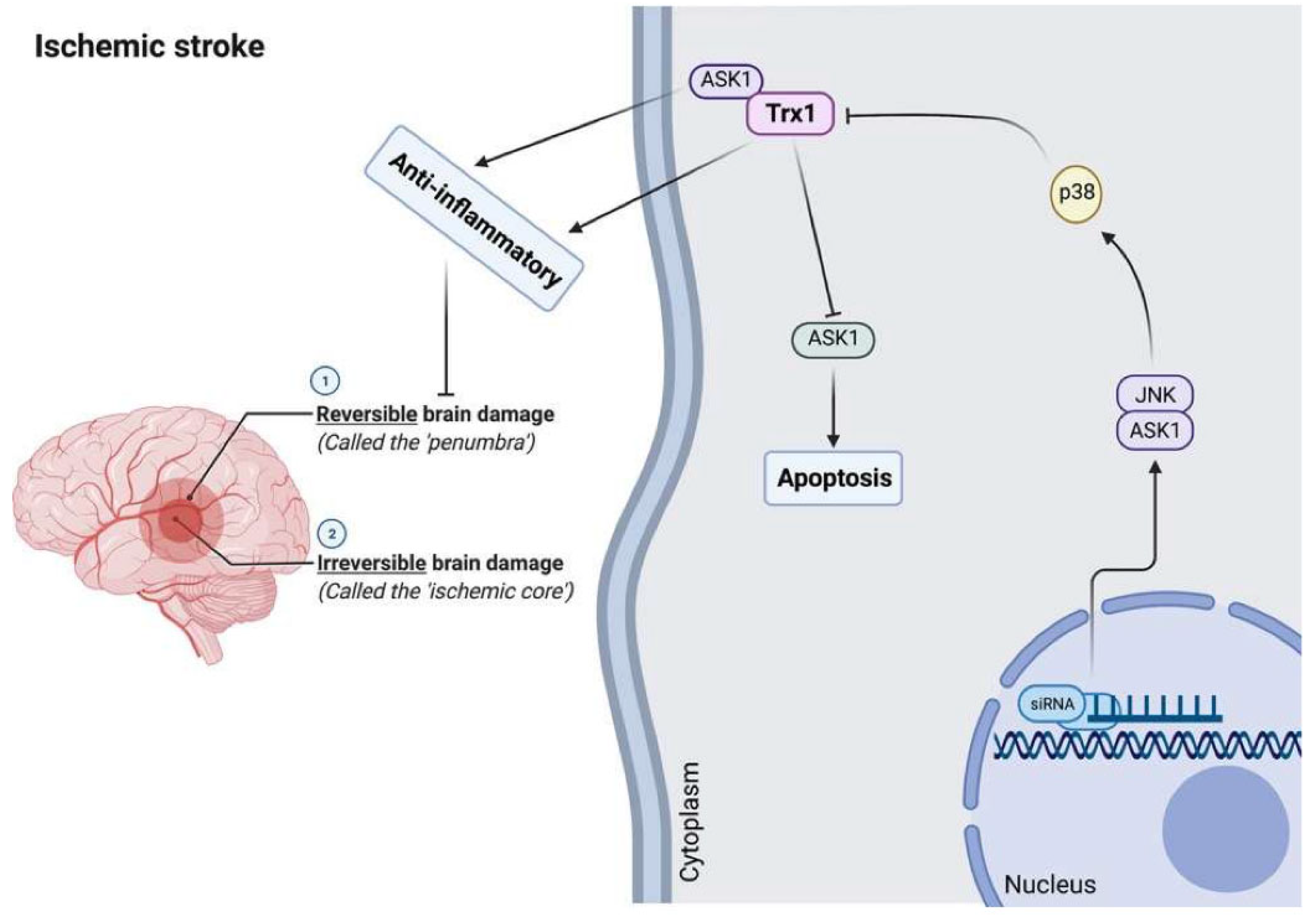
Trx has cellular antiapoptotic and anti-inflammatory activities by inhibiting and binding the pro-apoptotic protein ASK1, which stimulates the proapoptotic signaling pathways [202,210]. The Trx1 interacting protein (TXNIP), which may be induced by oxidative stress, is an endogenous inhibitor of Trx1 and is expressed in the brain [1,18,211,212]. Current data suggest that brain redox imbalances are actively involved in ischemic injury [213,214,215]. Furthermore, TXNIP inhibitors induce protection in rodent models of thromboembolic stroke [216] and ischemic-reperfusion injury [212,217]. Trx1 may promote recovery of cognitive functions and neurogenesis following cerebral ischemia in rodent models [218]. In mice, intraperitoneal administration of recombinant human Trx1 may decrease brain damage following ischemic stroke [219]. The neuroprotective activities against the consequences of cerebral ischemia injury may be explained by the anti-oxidative, anti-apoptotic, and anti-inflammatory properties of rhTrx1. Further, it may provide a novel therapeutic approach to decreasing neuronal apoptotic cell death induced by oxidative stress following acute ischemic stroke.
In a rodent stroke model based on middle cerebral artery occlusion injury, inhibition of Trx1 with siRNA induces neuronal apoptosis by stimulating the brain ASK1-JNK/p38 pathway [218,220,221]. Moreover, serum contents of Trx are associated with stroke risk, severity, and lesion volumes, and increased levels may be used as a diagnostic and prognostic biomarker of acute ischemic stroke in a Chinese sample [220,222]. In this respect, serum Trx levels ≥ 20.0 ng/mL are associated with a 9.482-fold increase in the risk of an unfavorable outcome [222].
Also, thioredoxin mimetic (TXM) tetrapeptides, derived from the canonical -CxxC-motif of the Trx1-active site, may reverse oxidative and inflammatory damage mimicking Trx1 activity as well as protect brain cognitive activity after weight-drop closed-head injury in a mouse model of Mild Traumatic Brain Injury (mTBI) [223].
Figure 5 shows the role of Trx1 in the pathophysiology of brain stroke. Although Trx1 may directly inhibit ASK1 and the corresponding ASK1-JNK/P38 pathway leading to anti-apoptosis and anti-inflammation, oxidative stress-induced TXNIP may inhibit its activity inducing stroke, which indicates that Trx1 may protect against ischemic stroke.
9. Multiple Sclerosis
9.1. Pathophysiology of Multiple Sclerosis
Multiple sclerosis (MS) is a chronic autoimmune and neuroinflammatory disorder characterized by foci of inflammatory demyelination in the optic nerves, spinal cord, and brain, as well as by axonal damage and neuronal degeneration [27,224,225,226]. It is one of the most common neurological disabilities in young adults, with a higher incidence in women. It is accompanied by lowered health-related quality of life, including social withdrawal and unemployment [227,228]. Besides genetic factors, also environmental factors may play a role in MS, including viral infections such as herpesviruses [229] and Human Endogenous Retrovirus (HERVs) [230,231]. Recent studies have demonstrated that oxidative stress plays a crucial role in the pathogenesis of MS. The primary pathologic aberrations in MS comprise axonal and neuronal damage and neuronal loss, leading to permanent neurologic disabilities in MS patients [19,27,232].
Astrocytes are primary regulators of brain oxidative homeostasis, and dysregulation of these cells probably leads to accumulated oxidative damage. On the other hand, the primary regulator of the antioxidant stress defense is Nrf2 [233], as well as peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α) [234]. Also, modified redox homeostasis and elevated oxidative stress are primarily a pathway of neurodegeneration and demyelination in the MS brain [235]. Integrated mechanisms regulated by vitagenes operated and encoded by the heat shock proteins (Hsp) Hsp32, Hsp70, Trx, and the sirtuin protein system in the brain are essential for neuronal survival during stressful conditions [27].
9.2. Trx and TrxR in Multiple Sclerosis
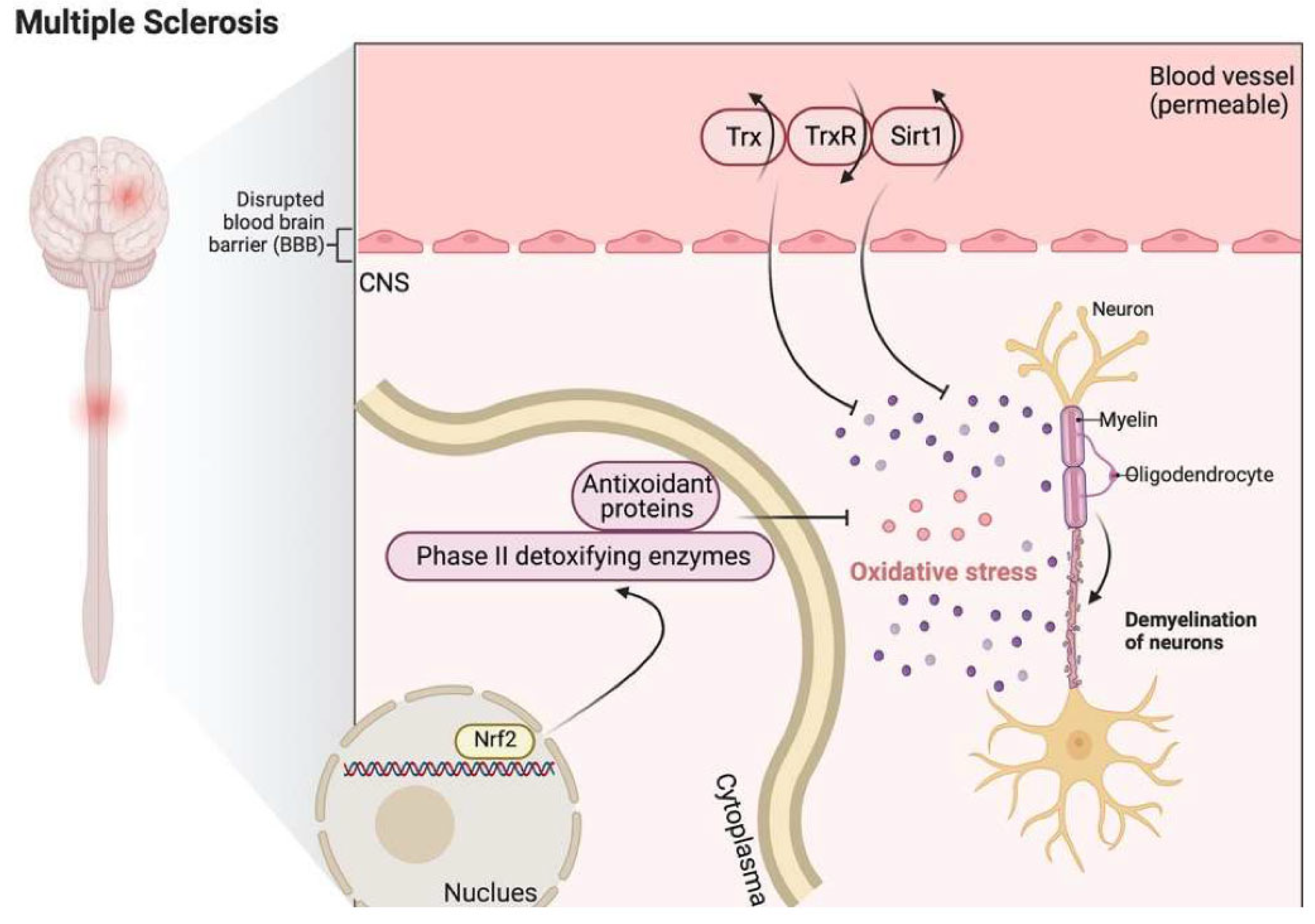
Numerous enzymes involved in antioxidant defense pathways are upregulated in active MS lesions, including Trx2 [234]. Moreover, in the blood and CSF of MS patients, elevated expression of Trx and sirtuin 1, together with a reduction in the expression of TrxR, were observed [27]. Moreover, Trx has a critical role in the reconstitution of Prx [236], which plays a vital role in preventing oxidative damage in MS lesions and increases astrocyte resilience against oxidative damage [224]. During anti-oxidative stress responses, upregulation of phase II detoxifying enzymes and antioxidant proteins may occur through Nrf2, which has an essential role in inducing the cellular pathways counteracting ROS [237,238]. This Nrf2-induced enzymatic machinery includes enzymes mediating glutathione (GSH) synthesis, the Trx enzyme system, and detoxifying enzymes like heme oxygenases (HO) or NAD(P)H: quinone oxidoreductase 1 (NQO1) [239].
Figure 6 shows that Trx and sirt1 are upregulated, and TrxR is downregulated in MS. Nrf2-induced GSH synthesis and Trx system activation are involved in anti-oxidative stress responses.
9.3. Effects of Metals on the Trx System
Gold (Au) compounds have a long history of use in medicine [240]. Mammalian TrxRs are effectively inhibited by goldthioglucose (aurothioglucose, ATG) and other clinically used drugs [20,241,242,243]. ATG was the first TrxR inhibitor-containing gold [244]. Auranofin is a gold-containing complex used to treat rheumatoid arthritis, which inhibits the activity of TrxR, causing mitochondrial dysfunction, oxidative stress, and mitophagy flux to lysosomes [245,246]. Both ATG and auranofin cannot inhibit GR and GPx, indicating the Sec selenol group of TrxR is the main target. Interestingly, platinum (Pt) and auranofin, which have anti-arthritis effects, may inhibit the selenoprotein TrxR [247]. Also, Palladium (Pd) and Au are remarkably more potent inhibitors of recombinant TrxR1 than Pt. Moreover, gold compounds are highly specific inhibitors of mitochondrial TrxRs, and their activities may induce other functions such as membrane permeability properties. On the other hand, metal ions (cadmium acetate) and metal complexes (cisplatin, zinc pyrithione and tributyltin) significantly inhibit TrxRs activity although with less potency than gold compounds (auranofin, triethylphosphine gold and aurothiomalate, [Au(2,2′-diethylendiamine)Cl]Cl2, [(Au(2-(1,1-dimethylbenzyl)-pyridine) (CH3COO)2], [Au(6-(1,1-dimethylbenzyl)-2,2′-bipyridine)(OH)](PF6), [Au(bipydmb-H)(2,6-xylidine)](PF6)) [20,240,248].
Mercury compounds (methylmercury (MeHg+) or inorganic mercury (Hg2+) interact with many essential enzymes implicated in antioxidant regulation, including selenoenzymes TrxRs and glutathione peroxidase (GPx) [249,250,251]. Mercury inhibits the Trx system, while the primary molecular mechanism of mercury toxicity is associated with the thiol-selenol in the C-terminal active site of TrxR [252]. Mercury poisoning may cause inhibition of glutamate decarboxylase activity and additionally may reduce dopamine and serotonin levels in the brain [253,254]. There is no doubt that too much Hg may cause harm to the brain [255]. Still, it is a challenging task to assess more quantitatively the consequences of Hg in a health context in comparison with other toxic metals, alcohol, unnatural Ah receptor agonists (including PCBs, PAHs, and various brominated flame retardants) [256] or mutagenic drugs, such as paracetamol [257]. Also, mercury, cadmium, and arsenic exert relatively little GSH oxidation but powerfully cause oxidation of both cytoplasmic Trx1 and mitochondrial Trx2 [258]. The magnitude of mercury, cadmium and arsenic effects was greater for the Trx2 than the Trx1.
As a derivative of Trx, thioredoxin-Albumin fusion (HAS-Trx) counteracted copper-induced neurotoxicity by suppressing ROS production [259]. It is also shown that both iron and copper potentiate GSH loss. Part of the effects of copper on GSH is related to its ability to reduce the activity of glutamate-cysteine ligase [260]. Moreover, Copper, iron, and nickel may induce GSH oxidation, resulting in lower oxidation of cytoplasmic Trx1 and mitochondrial Trx2 [30,242,261].
10. Conclusions
There is evidence that Trx and the TrxRs enzyme systems are essential for regulating the cellular redox system, energy metabolism, modulation of immune responses and antioxidant defenses, cell growth and survival, and selenium metabolism in the CNS suppressing neurodegenerative, neuro-oxidative, and neuroinflammatory processes. Many compounds targeting Trx/TrxR functions, such as various flavanoids, gold compounds, platinum compounds, arsenic trioxide, motexafin gadolinium, and nitrous compounds, gained interest in the functions of the Trx/TrxR system. Given the broad range of protective functions, it is evident that Trx and the TrxRs enzyme systems play a crucial role in health and disease, explaining that dysfunctions/overactivation of Trx and TrxRs are associated with various neurodegenerative, neuroinflammatory, or neuro-oxidative brain disorders. The present review indicates multiple aberrations in the Trx and TrxR and other redox systems in AD, PD, HD, stroke, and MS and their multiple reciprocal relationships with the neurodegenerative, neuro-inflammatory, and neuro-oxidative pathways involved in those disorders. Future research should examine whether combining Trx and TrxR and other oxidative stress biomarkers (for example, in machine learning models) could provide novel tool which reflect the severity of neurodegenerative, neuroinflammatory, or neuro-oxidative processes or may be employed as diagnostic or prognostic tools to help in the (differential) diagnosis of AD, PD, HD, stroke, and MS or their staging characteristics. Furthermore, aberrations in the Trx and TrxR systems are probably new drug targets to treat and prevent neurodegenerative, neuroinflammatory, neuro-oxidative stress processes and related brain disorders. Future research should delineate whether these Trx and TrxR targeting drugs may augment the current gold-standard treatment strategies used to treat those disorders.
Author Contributions
Conceptualization, G.B., L.Z., M.P., C.T.C., T.H., J.L. and M.M.; methodology, G.B., L.Z., M.P., C.T.C., T.H., J.L. and M.M.; writing—original draft preparation, G.B., L.Z., M.P., C.T.C., T.H., J.L. and M.M.; writing—review and editing, L.Z., G.B. and M.P.; visualization, L.Z. and J.L.; revision, L.Z., J.L., G.B. and M.P.; supervision, G.B.; funding acquisition, L.Z., J.L. and M.P. All authors have read and agreed to the published version of the manuscript.
Funding
L.Z. and J.L. are grateful for support from the National Natural Science Foundation of China (81903105). M.P. acknowledges Università degli Studi di Sassari (UNISS) for the financial support received within the program “Fondo di Ateneo per la ricerca 2020, FAR 2020.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nasoohi, S.; Ismael, S.; Ishrat, T. Thioredoxin-Interacting Protein (TXNIP) in Cerebrovascular and Neurodegenerative Diseases: Regulation and Implication. Mol. Neurobiol. 2018, 55, 7900–7920. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Dhillon, P.; Shorter, J. Neurodegenerative disease: Models, mechanisms, and a new hope. Dis. Model. Mech. 2017, 10, 499–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, B.; Sharma, D. Recent Patent Advances for Neurodegenerative Disorders and its Treatment. Recent Pat. Drug Deliv. Formul. 2017, 11, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Ruszkiewicz, J.; Albrecht, J. Changes in the mitochondrial antioxidant systems in neurodegenerative diseases and acute brain disorders. Neurochem. Int. 2015, 88, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Hattori, F.; Oikawa, S. Peroxiredoxins in the central nervous system. Subcell Biochem. 2007, 44, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Collin, F. Chemical Basis of Reactive Oxygen Species Reactivity and Involvement in Neurodegenerative Diseases. Int. J. Mol. Sci 2019, 20, 2407. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Lim, J.L.; Wilhelmus, M.M.; de Vries, H.E.; Drukarch, B.; Hoozemans, J.J.; van Horssen, J. Antioxidative defense mechanisms controlled by Nrf2: State-of-the-art and clinical perspectives in neurodegenerative diseases. Arch. Toxicol. 2014, 88, 1773–1786. [Google Scholar] [CrossRef]
- Mahmood, D.F.; Abderrazak, A.; El Hadri, K.; Simmet, T.; Rouis, M. The thioredoxin system as a therapeutic target in human health and disease. Antioxid. Redox Signal. 2013, 19, 1266–1303. [Google Scholar] [CrossRef]
- Bharti, V.; Tan, H.; Chow, D.; Wang, Y.; Nagakannan, P.; Eftekharpour, E.; Wang, J.F. Glucocorticoid Upregulates Thioredoxin-interacting Protein in Cultured Neuronal Cells. Neuroscience 2018, 384, 375–383. [Google Scholar] [CrossRef]
- Holmgren, A.; Lu, J. Thioredoxin and thioredoxin reductase: Current research with special reference to human disease. Biochem. Biophys. Res. Commun. 2010, 396, 120–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wadhwa, R.; Gupta, R.; Maurya, P.K. Oxidative Stress and Accelerated Aging in Neurodegenerative and Neuropsychiatric Disorder. Curr. Pharm. Des. 2018, 24, 4711–4725. [Google Scholar] [CrossRef] [PubMed]
- Hohn, A.; Weber, D.; Jung, T.; Ott, C.; Hugo, M.; Kochlik, B.; Kehm, R.; Konig, J.; Grune, T.; Castro, J.P. Happily (n)ever after: Aging in the context of oxidative stress, proteostasis loss and cellular senescence. Redox Biol. 2017, 11, 482–501. [Google Scholar] [CrossRef]
- Zhang, Y.; Unnikrishnan, A.; Deepa, S.S.; Liu, Y.; Li, Y.; Ikeno, Y.; Sosnowska, D.; van Remmen, H.; Richardson, A. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1(-/)(-) mice is correlated to increased cellular senescence. Redox Biol. 2017, 11, 30–37. [Google Scholar] [CrossRef] [Green Version]
- Nocella, C.; Cammisotto, V.; Pigozzi, F.; Borrione, P.; Fossati, C.; D’Amico, A.; Cangemi, R.; Peruzzi, M.; Gobbi, G.; Ettorre, E.; et al. Impairment between Oxidant and Antioxidant Systems: Short- and Long-term Implications for Athletes’ Health. Nutrients 2019, 11, 1353. [Google Scholar] [CrossRef] [Green Version]
- Chasapis, C.T.; Makridakis, M.; Damdimopoulos, A.E.; Zoidakis, J.; Lygirou, V.; Mavroidis, M.; Vlahou, A.; Miranda-Vizuete, A.; Spyrou, G.; Vlamis-Gardikas, A. Implications of the mitochondrial interactome of mammalian thioredoxin 2 for normal cellular function and disease. Free Radic. Biol. Med. 2019, 137, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Kahya, M.; Sood, P.; Devos, H.; Krishnan, S.; Hirsch, M.A.; Heyn, P. Falls Risk and Alzheimer Disease: A Patient Guide. Arch. Phys. Med. Rehabil. 2020, 101, 931–935. [Google Scholar] [CrossRef]
- Wang, C.Y.; Xu, Y.; Wang, X.; Guo, C.; Wang, T.; Wang, Z.Y. Dl-3-n-Butylphthalide Inhibits NLRP3 Inflammasome and Mitigates Alzheimer’s-Like Pathology via Nrf2-TXNIP-TrX Axis. Antioxid. Redox Signal. 2019, 30, 1411–1431. [Google Scholar] [CrossRef]
- Armon-Omer, A.; Waldman, C.; Simaan, N.; Neuman, H.; Tamir, S.; Shahien, R. New Insights on the Nutrition Status and Antioxidant Capacity in Multiple Sclerosis Patients. Nutrients 2019, 11, 427. [Google Scholar] [CrossRef] [Green Version]
- Bjørklund, G.; Zou, L.; Wang, J.; Chasapis, C.T.; Peana, M. Thioredoxin reductase as a pharmacological target. Pharmacol. Res. 2021, 174, 105854. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef] [Green Version]
- Bazinet, R.P.; Laye, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.Z.; He, L. The role of polyunsaturated fatty acids and GPR40 receptor in brain. Neuropharmacology 2017, 113, 639–651. [Google Scholar] [CrossRef] [PubMed]
- McNamara, R.K.; Asch, R.H.; Lindquist, D.M.; Krikorian, R. Role of polyunsaturated fatty acids in human brain structure and function across the lifespan: An update on neuroimaging findings. Prostaglandins Leukot. Essent. Fatty Acids 2018, 136, 23–34. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef]
- Niedzielska, E.; Smaga, I.; Gawlik, M.; Moniczewski, A.; Stankowicz, P.; Pera, J.; Filip, M. Oxidative Stress in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 4094–4125. [Google Scholar] [CrossRef] [Green Version]
- Pennisi, G.; Cornelius, C.; Cavallaro, M.M.; Salinaro, A.T.; Cambria, M.T.; Pennisi, M.; Bella, R.; Milone, P.; Ventimiglia, B.; Migliore, M.R.; et al. Redox regulation of cellular stress response in multiple sclerosis. Biochem. Pharmacol. 2011, 82, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Elmann, A.; Wang, C.K.; Vauzour, D. Polyphenols Targeting Brain Cells Longevity, Brain’s Redox Status, and Neurodegenerative Diseases. Oxid. Med. Cell Longev. 2018, 2018, 7402795. [Google Scholar] [CrossRef]
- Arner, E.S.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef]
- Ouyang, Y.; Peng, Y.; Li, J.; Holmgren, A.; Lu, J. Modulation of thiol-dependent redox system by metal ions via thioredoxin and glutaredoxin systems. Metallomics 2018, 10, 218–228. [Google Scholar] [CrossRef]
- Ren, X.; Zou, L.; Zhang, X.; Branco, V.; Wang, J.; Carvalho, C.; Holmgren, A.; Lu, J. Redox Signaling Mediated by Thioredoxin and Glutathione Systems in the Central Nervous System. Antioxid. Redox Signal. 2017, 27, 989–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmgren, A. Antioxidant function of thioredoxin and glutaredoxin systems. Antioxid. Redox Signal. 2000, 2, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Martin, S.G. The thioredoxin system: A key target in tumour and endothelial cells. Br. J. Radiol. 2008, 81, S57–S68. [Google Scholar] [CrossRef] [PubMed]
- Bas, A.; Gultekin, G.; Incir, S.; Bas, T.O.; Emul, M.; Duran, A. Level of serum thioredoxin and correlation with neurocognitive functions in patients with schizophrenia using clozapine and other atypical antipsychotics. Psychiatry Res. 2017, 247, 84–89. [Google Scholar] [CrossRef]
- Pekkari, K.; Holmgren, A. Truncated thioredoxin: Physiological functions and mechanism. Antioxid. Redox Signal. 2004, 6, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. Selenoproteins. J. Biol. Chem. 2009, 284, 723–727. [Google Scholar] [CrossRef] [Green Version]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef] [Green Version]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with Txnip. Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.; Holmgren, A. Thioredoxin system in cell death progression. Antioxid. Redox Signal. 2012, 17, 1738–1747. [Google Scholar] [CrossRef]
- Yu, Y.; Xing, K.; Badamas, R.; Kuszynski, C.A.; Wu, H.; Lou, M.F. Overexpression of thioredoxin-binding protein 2 increases oxidation sensitivity and apoptosis in human lens epithelial cells. Free Radic. Biol. Med. 2013, 57, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Lerner, A.G.; Upton, J.P.; Praveen, P.V.; Ghosh, R.; Nakagawa, Y.; Igbaria, A.; Shen, S.; Nguyen, V.; Backes, B.J.; Heiman, M.; et al. IRE1alpha induces thioredoxin-interacting protein to activate the NLRP3 inflammasome and promote programmed cell death under irremediable ER stress. Cell Metab. 2012, 16, 250–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oslowski, C.M.; Hara, T.; O’Sullivan-Murphy, B.; Kanekura, K.; Lu, S.; Hara, M.; Ishigaki, S.; Zhu, L.J.; Hayashi, E.; Hui, S.T.; et al. Thioredoxin-interacting protein mediates ER stress-induced beta cell death through initiation of the inflammasome. Cell Metab. 2012, 16, 265–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Shih, A.Y.; Johannssen, H.C.; Erb, H.; Li, P.; Murphy, T.H. Two-photon imaging of glutathione levels in intact brain indicates enhanced redox buffering in developing neurons and cells at the cerebrospinal fluid and blood-brain interface. J. Biol. Chem. 2006, 281, 17420–17431. [Google Scholar] [CrossRef] [Green Version]
- Ignowski, E.; Winter, A.N.; Duval, N.; Fleming, H.; Wallace, T.; Manning, E.; Koza, L.; Huber, K.; Serkova, N.J.; Linseman, D.A. The cysteine-rich whey protein supplement, Immunocal(R), preserves brain glutathione and improves cognitive, motor, and histopathological indices of traumatic brain injury in a mouse model of controlled cortical impact. Free Radic. Biol. Med. 2018, 124, 328–341. [Google Scholar] [CrossRef]
- Garcia-Ruiz, C.; Morales, A.; Ballesta, A.; Rodes, J.; Kaplowitz, N.; Fernandez-Checa, J.C. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J. Clin. Investig. 1994, 94, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Castagna, A.; Le Grazie, C.; Accordini, A.; Giulidori, P.; Cavalli, G.; Bottiglieri, T.; Lazzarin, A. Cerebrospinal fluid S-adenosylmethionine (SAMe) and glutathione concentrations in HIV infection: Effect of parenteral treatment with SAMe. Neurology 1995, 45, 1678–1683. [Google Scholar] [CrossRef]
- Zachara, B.A.; Pawluk, H.; Bloch-Boguslawska, E.; Sliwka, K.M.; Korenkiewicz, J.; Skok, Z.; Ryc, K. Tissue level, distribution, and total body selenium content in healthy and diseased humans in Poland. Arch. Environ. Health 2001, 56, 461–466. [Google Scholar] [CrossRef]
- Varikasuvu, S.R.; Prasad, V.S.; Kothapalli, J.; Manne, M. Brain Selenium in Alzheimer’s Disease (BRAIN SEAD Study): A Systematic Review and Meta-Analysis. Biol. Trace Elem. Res. 2019, 189, 361–369. [Google Scholar] [CrossRef]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid-beta cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers Dis. 2013, 36, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Branco, V.; Pimentel, J.; Brito, M.A.; Carvalho, C. Thioredoxin, glutathione and related molecules in tumors of the nervous system. Curr. Med. Chem. 2019, 27, 1878–1900. [Google Scholar] [CrossRef] [PubMed]
- Satrustegui, J.; Richter, C. The role of hydroperoxides as calcium release agents in rat brain mitochondria. Arch. Biochem. Biophys. 1984, 233, 736–740. [Google Scholar] [CrossRef]
- Rowe, J.M. The evolving paradigm of prognostic factors in AML: Introduction to the Acute Leukemia Forum 2010. Best Pract. Res. Clin. Haematol. 2010, 23, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.H.; Choi, J.H.; Song, J.M.; Lee, C.H.; Yoo, K.Y.; Hwang, I.K.; Kim, J.S.; Shin, H.C.; Won, M.H. Increase in Trx2/Prx3 redox system immunoreactivity in the spinal cord and hippocampus of aged dogs. Exp. Gerontol. 2011, 46, 946–952. [Google Scholar] [CrossRef]
- Angeles, D.C.; Gan, B.H.; Onstead, L.; Zhao, Y.; Lim, K.L.; Dachsel, J.; Melrose, H.; Farrer, M.; Wszolek, Z.K.; Dickson, D.W.; et al. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum. Mutat. 2011, 32, 1390–1397. [Google Scholar] [CrossRef]
- Goemaere, J.; Knoops, B. Peroxiredoxin distribution in the mouse brain with emphasis on neuronal populations affected in neurodegenerative disorders. J. Comp. Neurol. 2012, 520, 258–280. [Google Scholar] [CrossRef]
- Irwin, R.W.; Yao, J.; To, J.; Hamilton, R.T.; Cadenas, E.; Brinton, R.D. Selective oestrogen receptor modulators differentially potentiate brain mitochondrial function. J. Neuroendocrinol. 2012, 24, 236–248. [Google Scholar] [CrossRef]
- Pitts, A.; Dailey, K.; Newington, J.T.; Chien, A.; Arseneault, R.; Cann, T.; Thompson, L.M.; Cumming, R.C. Dithiol-based compounds maintain expression of antioxidant protein peroxiredoxin 1 that counteracts toxicity of mutant huntingtin. J. Biol. Chem. 2012, 287, 22717–22729. [Google Scholar] [CrossRef] [Green Version]
- Shim, S.Y.; Kim, H.S.; Kim, E.K.; Choi, J.H. Expression of peroxiredoxin 1, 2, and 6 in the rat brain during perinatal development and in response to dexamethasone. Free Radic. Res. 2012, 46, 231–239. [Google Scholar] [CrossRef]
- Zhao, L.; Morgan, T.E.; Mao, Z.; Lin, S.; Cadenas, E.; Finch, C.E.; Pike, C.J.; Mack, W.J.; Brinton, R.D. Continuous versus cyclic progesterone exposure differentially regulates hippocampal gene expression and functional profiles. PLoS ONE 2012, 7, e31267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, Y.; Zhang, X.; Ji, H.; Liu, H.; Li, S.; Li, L. Probucol and atorvastatin in combination protect rat brains in MCAO model: Upregulating Peroxiredoxin2, Foxo3a and Nrf2 expression. Neurosci. Lett. 2012, 509, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Lin, W.P. Quantitative analysis of multiple exocyclic DNA adducts in human salivary DNA by stable isotope dilution nanoflow liquid chromatography-nanospray ionization tandem mass spectrometry. Anal. Chem. 2011, 83, 8543–8551. [Google Scholar] [CrossRef] [PubMed]
- Voulgaridou, G.P.; Anestopoulos, I.; Franco, R.; Panayiotidis, M.I.; Pappa, A. DNA damage induced by endogenous aldehydes: Current state of knowledge. Mutat. Res. 2011, 711, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.E.; Park, Y.S. Role of lipid peroxidation-derived alpha, beta-unsaturated aldehydes in vascular dysfunction. Oxid Med. Cell Longev. 2013, 2013, 629028. [Google Scholar] [CrossRef] [Green Version]
- Yata, K.; Oikawa, S.; Sasaki, R.; Shindo, A.; Yang, R.; Murata, M.; Kanamaru, K.; Tomimoto, H. Astrocytic neuroprotection through induction of cytoprotective molecules; a proteomic analysis of mutant P301S tau-transgenic mouse. Brain Res. 2011, 1410, 12–23. [Google Scholar] [CrossRef]
- Power, J.H.; Asad, S.; Chataway, T.K.; Chegini, F.; Manavis, J.; Temlett, J.A.; Jensen, P.H.; Blumbergs, P.C.; Gai, W.P. Peroxiredoxin 6 in human brain: Molecular forms, cellular distribution and association with Alzheimer’s disease pathology. Acta Neuropathol. 2008, 115, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Drechsel, D.A.; Patel, M. Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J. Biol. Chem. 2010, 285, 27850–27858. [Google Scholar] [CrossRef] [Green Version]
- Kudin, A.P.; Augustynek, B.; Lehmann, A.K.; Kovacs, R.; Kunz, W.S. The contribution of thioredoxin-2 reductase and glutathione peroxidase to H2O2 detoxification of rat brain mitochondria. Biochim. Biophys. Acta 2012, 1817, 1901–1906. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Schuster, A.; Tang, C.; Yu, T.; Ortogero, N.; Bao, J.; Zheng, H.; Yan, W. Sperm-borne miRNAs and endo-siRNAs are important for fertilization and preimplantation embryonic development. Development 2016, 143, 635–647. [Google Scholar] [CrossRef]
- Lokken, A.A.; Ralston, A. The Genetic Regulation of Cell Fate during Preimplantation Mouse Development. Curr. Top. Dev. Biol. 2016, 120, 173–202. [Google Scholar] [CrossRef] [PubMed]
- Fessel, J. Alzheimer’s disease combination treatment. Neurobiol. Aging 2018, 63, 165. [Google Scholar] [CrossRef] [PubMed]
- Plassman, B.L.; Langa, K.M.; Fisher, G.G.; Heeringa, S.G.; Weir, D.R.; Ofstedal, M.B.; Burke, J.R.; Hurd, M.D.; Potter, G.G.; Rodgers, W.L.; et al. Prevalence of dementia in the United States: The aging, demographics, and memory study. Neuroepidemiology 2007, 29, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Abolhassani, N.; Leon, J.; Sheng, Z.; Oka, S.; Hamasaki, H.; Iwaki, T.; Nakabeppu, Y. Molecular pathophysiology of impaired glucose metabolism, mitochondrial dysfunction, and oxidative DNA damage in Alzheimer’s disease brain. Mech. Ageing Dev. 2017, 161, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Hebert, L.E.; Beckett, L.A.; Scherr, P.A.; Evans, D.A. Annual incidence of Alzheimer disease in the United States projected to the years 2000 through 2050. Alzheimer Dis. Assoc. Disord 2001, 15, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimers Dement. 2013, 9, 63–75.e62. [Google Scholar] [CrossRef]
- Alzheimer’s, A. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015, 11, 332–384. [Google Scholar] [CrossRef]
- Dye, R.V.; Miller, K.J.; Singer, E.J.; Levine, A.J. Hormone replacement therapy and risk for neurodegenerative diseases. Int. J. Alzheimers Dis. 2012, 2012, 258454. [Google Scholar] [CrossRef] [Green Version]
- Robinson, R.A.; Amin, B.; Guest, P.C. Multiplexing Biomarker Methods, Proteomics and Considerations for Alzheimer’s Disease. Adv. Exp. Med. Biol. 2017, 974, 21–48. [Google Scholar] [CrossRef]
- Gotz, J.; Ittner, A.; Ittner, L.M. Tau-targeted treatment strategies in Alzheimer’s disease. Br. J. Pharmacol. 2012, 165, 1246–1259. [Google Scholar] [CrossRef]
- Aaseth, J.; Skaug, M.A.; Cao, Y.; Andersen, O. Chelation in metal intoxication—Principles and paradigms. J. Trace Elem. Med. Biol. 2015, 31, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Cieslik, M.; Czapski, G.A.; Wojtowicz, S.; Wieczorek, I.; Wencel, P.L.; Strosznajder, R.P.; Jaber, V.; Lukiw, W.J.; Strosznajder, J.B. Alterations of Transcription of Genes Coding Anti-oxidative and Mitochondria-Related Proteins in Amyloid beta Toxicity: Relevance to Alzheimer’s Disease. Mol. Neurobiol. 2020, 57, 1374–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dickerson, B.C.; Brickhouse, M.; McGinnis, S.; Wolk, D.A. Alzheimer’s disease: The influence of age on clinical heterogeneity through the human brain connectome. Alzheimers Dement. 2017, 6, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Wiste, H.J.; Weigand, S.D.; Therneau, T.M.; Lowe, V.J.; Knopman, D.S.; Gunter, J.L.; Senjem, M.L.; Jones, D.T.; Kantarci, K.; et al. Defining imaging biomarker cut points for brain aging and Alzheimer’s disease. Alzheimers Dement. 2017, 13, 205–216. [Google Scholar] [CrossRef] [Green Version]
- Feger, B.J.; Thompson, J.W.; Dubois, L.G.; Kommaddi, R.P.; Foster, M.W.; Mishra, R.; Shenoy, S.K.; Shibata, Y.; Kidane, Y.H.; Moseley, M.A.; et al. Microgravity induces proteomics changes involved in endoplasmic reticulum stress and mitochondrial protection. Sci. Rep. 2016, 6, 34091. [Google Scholar] [CrossRef] [Green Version]
- Scheff, S.W.; Ansari, M.A.; Mufson, E.J. Oxidative stress and hippocampal synaptic protein levels in elderly cognitively intact individuals with Alzheimer’s disease pathology. Neurobiol. Aging 2016, 42, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, N.; Ghosh, R.; Mandal, S.C. Antioxidant protection: A promising therapeutic intervention in neurodegenerative disease. Free Radic. Res. 2011, 45, 888–905. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Ong, T.P.; Jacob-Filho, W.; Jaluul, O.; Freitas, M.I.; Cominetti, C.; Cozzolino, S.M. Glutathione peroxidase 1 Pro198Leu polymorphism in Brazilian Alzheimer’s disease patients: Relations to the enzyme activity and to selenium status. J. Nutrigenet. Nutr. 2012, 5, 72–80. [Google Scholar] [CrossRef]
- Bateman, R.J.; Siemers, E.R.; Mawuenyega, K.G.; Wen, G.; Browning, K.R.; Sigurdson, W.C.; Yarasheski, K.E.; Friedrich, S.W.; Demattos, R.B.; May, P.C.; et al. A gamma-secretase inhibitor decreases amyloid-beta production in the central nervous system. Ann. Neurol. 2009, 66, 48–54. [Google Scholar] [CrossRef] [Green Version]
- Chouraki, V.; Reitz, C.; Maury, F.; Bis, J.C.; Bellenguez, C.; Yu, L.; Jakobsdottir, J.; Mukherjee, S.; Adams, H.H.; Choi, S.H.; et al. Evaluation of a Genetic Risk Score to Improve Risk Prediction for Alzheimer’s Disease. J. Alzheimers Dis. 2016, 53, 921–932. [Google Scholar] [CrossRef]
- Corder, E.H.; Saunders, A.M.; Strittmatter, W.J.; Schmechel, D.E.; Gaskell, P.C.; Small, G.W.; Roses, A.D.; Haines, J.L.; Pericak-Vance, M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993, 261, 921–923. [Google Scholar] [CrossRef] [PubMed]
- Bertram, L.; McQueen, M.B.; Mullin, K.; Blacker, D.; Tanzi, R.E. Systematic meta-analyses of Alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 2007, 39, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Tran, L.; Emilsson, V.; Zhu, J. Characterization of Genetic Networks Associated with Alzheimer’s Disease. Methods Mol. Biol. 2016, 1303, 459–477. [Google Scholar] [CrossRef]
- Chouliaras, L.; Sierksma, A.S.; Kenis, G.; Prickaerts, J.; Lemmens, M.A.; Brasnjevic, I.; van Donkelaar, E.L.; Martinez-Martinez, P.; Losen, M.; de Baets, M.H.; et al. Gene-environment interaction research and transgenic mouse models of Alzheimer’s disease. Int. J. Alzheimers Dis. 2010, 2010, 859101. [Google Scholar] [CrossRef] [Green Version]
- Huse, J.T.; Doms, R.W. Neurotoxic traffic: Uncovering the mechanics of amyloid production in Alzheimer’s disease. Traffic 2001, 2, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Perluigi, M.; Sultana, R. Oxidative stress in Alzheimer’s disease brain: New insights from redox proteomics. Eur. J. Pharmacol. 2006, 545, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Stoothoff, W.H.; de Calignon, A.; Jones, P.B.; Hyman, B.T. Tau pathophysiology in neurodegeneration: A tangled issue. Trends Neurosci. 2009, 32, 150–159. [Google Scholar] [CrossRef]
- Selenius, M.; Rundlof, A.K.; Olm, E.; Fernandes, A.P.; Bjornstedt, M. Selenium and the selenoprotein thioredoxin reductase in the prevention, treatment and diagnostics of cancer. Antioxid. Redox Signal. 2010, 12, 867–880. [Google Scholar] [CrossRef]
- Lovell, M.A.; Xie, C.; Gabbita, S.P.; Markesbery, W.R. Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer’s disease brain. Free Radic. Biol. Med. 2000, 28, 418–427. [Google Scholar] [CrossRef]
- Takagi, Y.; Mitsui, A.; Nishiyama, A.; Nozaki, K.; Sono, H.; Gon, Y.; Hashimoto, N.; Yodoi, J. Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proc. Natl. Acad. Sci. USA 1999, 96, 4131–4136. [Google Scholar] [CrossRef]
- Akterin, S.; Cowburn, R.F.; Miranda-Vizuete, A.; Jimenez, A.; Bogdanovic, N.; Winblad, B.; Cedazo-Minguez, A. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer’s disease. Cell Death Differ. 2006, 13, 1454–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzawa, A. Thioredoxin and redox signaling: Roles of the thioredoxin system in control of cell fate. Arch. Biochem. Biophys. 2017, 617, 101–105. [Google Scholar] [CrossRef]
- Bobba, A.; Casalino, E.; Petragallo, V.A.; Atlante, A. Thioredoxin/thioredoxin reductase system involvement in cerebellar granule cell apoptosis. Apoptosis 2014, 19, 1497–1508. [Google Scholar] [CrossRef]
- Gil-Bea, F.; Akterin, S.; Persson, T.; Mateos, L.; Sandebring, A.; Avila-Carino, J.; Gutierrez-Rodriguez, A.; Sundstrom, E.; Holmgren, A.; Winblad, B.; et al. Thioredoxin-80 is a product of alpha-secretase cleavage that inhibits amyloid-beta aggregation and is decreased in Alzheimer’s disease brain. EMBO Mol. Med. 2012, 4, 1097–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Ma, H.; Zhang, L.; Cui, Y.; Liu, X.; Fang, J. A small molecule probe reveals declined mitochondrial thioredoxin reductase activity in a Parkinson’s disease model. Chem. Commun. 2016, 52, 2296–2299. [Google Scholar] [CrossRef] [PubMed]
- Lamoke, F.; Ripandelli, G.; Webster, S.; Montemari, A.; Maraschi, A.; Martin, P.; Marcus, D.M.; Liou, G.I.; Bartoli, M. Loss of thioredoxin function in retinas of mice overexpressing amyloid beta. Free Radic. Biol. Med. 2012, 53, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Masutani, H.; Bai, J.; Kim, Y.C.; Yodoi, J. Thioredoxin as a neurotrophic cofactor and an important regulator of neuroprotection. Mol. Neurobiol. 2004, 29, 229–242. [Google Scholar] [CrossRef]
- Seyfried, J.; Wullner, U. Inhibition of thioredoxin reductase induces apoptosis in neuronal cell lines: Role of glutathione and the MKK4/JNK pathway. Biochem. Biophys. Res. Commun. 2007, 359, 759–764. [Google Scholar] [CrossRef]
- Arner, E.S. Focus on mammalian thioredoxin reductases—Important selenoproteins with versatile functions. Biochim. Biophys. Acta 2009, 1790, 495–526. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Li, Q.X.; Fowler, C.J.; Masters, C.L.; Martins, R.N.; Ganio, K.; Lothian, A.; Mukherjee, S.; et al. Selenium Levels in Serum, Red Blood Cells, and Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Report from the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL). J. Alzheimers Dis. 2017, 57, 183–193. [Google Scholar] [CrossRef]
- Du, X.; Wang, C.; Liu, Q. Potential Roles of Selenium and Selenoproteins in the Prevention of Alzheimer’s Disease. Curr. Top. Med. Chem. 2016, 16, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Shanaida, M.; Lysiuk, R.; Antonyak, H.; Klishch, I.; Shanaida, V.; Peana, M. Selenium: An Antioxidant with a Critical Role in Anti-Aging. Molecules 2022, 27, 6613. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.; Markesbery, W.R.; Shao, C.; Lovell, M.A. Seleno-L-methionine protects against beta-amyloid and iron/hydrogen peroxide-mediated neuron death. Antioxid. Redox Signal. 2007, 9, 457–467. [Google Scholar] [CrossRef] [PubMed]
- Pillai, R.; Uyehara-Lock, J.H.; Bellinger, F.P. Selenium and selenoprotein function in brain disorders. IUBMB Life 2014, 66, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Lovell, M.A.; Xiong, S.; Lyubartseva, G.; Markesbery, W.R. Organoselenium (Sel-Plex diet) decreases amyloid burden and RNA and DNA oxidative damage in APP/PS1 mice. Free Radic. Biol. Med. 2009, 46, 1527–1533. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.A.; Joshi, G.; Huang, Q.; Opii, W.O.; Abdul, H.M.; Sultana, R.; Butterfield, D.A. In vivo administration of D609 leads to protection of subsequently isolated gerbil brain mitochondria subjected to in vitro oxidative stress induced by amyloid beta-peptide and other oxidative stressors: Relevance to Alzheimer’s disease and other oxidative stress-related neurodegenerative disorders. Free Radic. Biol. Med. 2006, 41, 1694–1703. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Park, K.A.; Lee, W.T.; Lee, J.E. Apoptosis signal regulating kinase 1 (ASK1): Potential as a therapeutic target for Alzheimer’s disease. Int. J. Mol. Sci. 2014, 15, 2119–2129. [Google Scholar] [CrossRef] [Green Version]
- Nishitoh, H.; Matsuzawa, A.; Tobiume, K.; Saegusa, K.; Takeda, K.; Inoue, K.; Hori, S.; Kakizuka, A.; Ichijo, H. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002, 16, 1345–1355. [Google Scholar] [CrossRef] [Green Version]
- Moran, C.; Kirk, C.; Powell, E. Spoken persuasive discourse abilities of adolescents with acquired brain injury. Lang. Speech Hear. Serv. Sch. 2012, 43, 264–275. [Google Scholar] [CrossRef]
- Santos, R.X.; Correia, S.C.; Zhu, X.; Smith, M.A.; Moreira, P.I.; Castellani, R.J.; Nunomura, A.; Perry, G. Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease. Antioxid. Redox Signal. 2013, 18, 2444–2457. [Google Scholar] [CrossRef]
- Bradley-Whitman, M.A.; Timmons, M.D.; Beckett, T.L.; Murphy, M.P.; Lynn, B.C.; Lovell, M.A. Nucleic acid oxidation: An early feature of Alzheimer’s disease. J. Neurochem. 2014, 128, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejczyk, M.; Zalewska, A.; Gerreth, A.K. Salivary Redox Biomarkers in Selected Neurodegenerative Diseases. J. Clin. Med. 2020, 9, 497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.J.; Song, D.S.; Yoo, J.S.; Hyung, K.E.; Lee, M.J.; Moon, Y.H.; Lee, I.H.; Go, B.S.; Park, S.Y.; Hwang, K.W. Protective functions of peroxiredoxin-1 against cytokine-induced MIN6 pancreatic beta-cell line death. Can. J. Physiol. Pharmacol. 2013, 91, 1037–1043. [Google Scholar] [CrossRef]
- Ahn, S.; Kearbey, J.D.; Li, C.M.; Duke, C.B., 3rd; Miller, D.D.; Dalton, J.T. Biotransformation of a novel antimitotic agent, I-387, by mouse, rat, dog, monkey, and human liver microsomes and in vivo pharmacokinetics in mice. Drug Metab. Dispos. 2011, 39, 636–643. [Google Scholar] [CrossRef] [Green Version]
- Barja, I.; Silvan, G.; Martinez-Fernandez, L.; Illera, J.C. Physiological stress responses, fecal marking behavior, and reproduction in wild European pine martens (Martes martes). J. Chem. Ecol. 2011, 37, 253–259. [Google Scholar] [CrossRef]
- Kuhn, D.M.; Arthur, R.E., Jr. Inactivation of tryptophan hydroxylase by nitric oxide: Enhancement by tetrahydrobiopterin. J. Neurochem. 1997, 68, 1495–1502. [Google Scholar] [CrossRef]
- Stone, D.M.; Johnson, M.; Hanson, G.R.; Gibb, J.W. Acute inactivation of tryptophan hydroxylase by amphetamine analogs involves the oxidation of sulfhydryl sites. Eur. J. Pharmacol. 1989, 172, 93–97. [Google Scholar] [CrossRef]
- Kuhn, D.M.; Ruskin, B.; Lovenberg, W. Tryptophan hydroxylase. The role of oxygen, iron, and sulfhydryl groups as determinants of stability and catalytic activity. J. Biol. Chem. 1980, 255, 4137–4143. [Google Scholar] [CrossRef]
- Kuhn, D.M.; Arthur, R.E., Jr.; Thomas, D.M.; Elferink, L.A. Tyrosine hydroxylase is inactivated by catechol-quinones and converted to a redox-cycling quinoprotein: Possible relevance to Parkinson’s disease. J. Neurochem. 1999, 73, 1309–1317. [Google Scholar] [CrossRef]
- Blanchard-Fillion, B.; Souza, J.M.; Friel, T.; Jiang, G.C.; Vrana, K.; Sharov, V.; Barron, L.; Schoneich, C.; Quijano, C.; Alvarez, B.; et al. Nitration and inactivation of tyrosine hydroxylase by peroxynitrite. J. Biol. Chem. 2001, 276, 46017–46023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadidi, M.; Geddes, T.J.; Kuhn, D.M. S-thiolation of tyrosine hydroxylase by reactive nitrogen species in the presence of cysteine or glutathione. Antioxid. Redox Signal. 2005, 7, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Haugaard, N.; Lee, N.H.; Kostrzewa, R.; Horn, R.S.; Haugaard, E.S. The role of sulfhydryl groups in oxidative phosphorylation and ion transport by rat liver mitochrondia. Biochim. Biophys. Acta 1969, 172, 198–204. [Google Scholar] [CrossRef]
- Wood, J.D. The role of gamma-aminobutyric acid in the mechanism of seizures. Prog. Neurobiol. 1975, 5, 77–95. [Google Scholar] [CrossRef]
- Bondarenko, T.I.; Makletsova, M.G.; Uskova, N.I. Intensity of label incorporation from [1,4-14C]putrescine into homocarnosine in the brain of rats of various ages. Ukr. Biokhim. Zh. 1986, 58, 75–77. [Google Scholar]
- Hori, S. Study on hyperbaric oxygen-induced convulsion with particular reference to gamma-aminobutyric acid in synaptosomes. J. Biochem. 1982, 91, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Krichevskaia, A.A.; Bondarenko, T.I.; Makletsova, M.G.; Mikhaleva, I.I. Protective effect of the neuropeptide homocarnosine in hyperbaric oxygenation. Vopr. Med. Khim. 1985, 31, 75–79. [Google Scholar]
- Li, X.; Gardner, E.L.; Xi, Z.X. The metabotropic glutamate receptor 7 (mGluR7) allosteric agonist AMN082 modulates nucleus accumbens GABA and glutamate, but not dopamine, in rats. Neuropharmacology 2008, 54, 542–551. [Google Scholar] [CrossRef] [Green Version]
- De Rijk, M.C.; Launer, L.J.; Berger, K.; Breteler, M.M.; Dartigues, J.F.; Baldereschi, M.; Fratiglioni, L.; Lobo, A.; Martinez-Lage, J.; Trenkwalder, C.; et al. Prevalence of Parkinson’s disease in Europe: A collaborative study of population-based cohorts. Neurologic Diseases in the Elderly Research Group. Neurology 2000, 54, S21–S23. [Google Scholar]
- Cook, C.; Stetler, C.; Petrucelli, L. Disruption of protein quality control in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009423. [Google Scholar] [CrossRef] [Green Version]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. Off. J. Mov. Disord. Soc. 2014, 29, 1583–1590. [Google Scholar] [CrossRef] [PubMed]
- Engelender, S.; Isacson, O. The Threshold Theory for Parkinson’s Disease. Trends Neurosci. 2017, 40, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S. Description of Parkinson’s disease as a clinical syndrome. Ann. N. Y. Acad. Sci. 2003, 991, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Forno, L.S.; DeLanney, L.E.; Irwin, I.; Langston, J.W. Electron microscopy of Lewy bodies in the amygdala-parahippocampal region. Comparison with inclusion bodies in the MPTP-treated squirrel monkey. Adv. Neurol. 1996, 69, 217–228. [Google Scholar]
- Chinta, S.J.; Andersen, J.K. Dopaminergic neurons. Int. J. Biochem. Cell Biol. 2005, 37, 942–946. [Google Scholar] [CrossRef]
- Galvin, K.A.; Oorschot, D.E. Postinjury magnesium sulfate treatment is not markedly neuroprotective for striatal medium spiny neurons after perinatal hypoxia/ischemia in the rat. Pediatr. Res. 1998, 44, 740–745. [Google Scholar] [CrossRef] [Green Version]
- Alladi, P.A.; Mahadevan, A.; Vijayalakshmi, K.; Muthane, U.; Shankar, S.K.; Raju, T.R. Ageing enhances alpha-synuclein, ubiquitin and endoplasmic reticular stress protein expression in the nigral neurons of Asian Indians. Neurochem. Int. 2010, 57, 530–539. [Google Scholar] [CrossRef]
- Imai, Y.; Venderova, K.; Lim, K.L. Animal models of Parkinson’s disease 2012. Parkinsons Dis. 2012, 2012, 729428. [Google Scholar] [CrossRef] [Green Version]
- Greenamyre, J.T.; Hastings, T.G. Biomedicine. Parkinson’s—Divergent causes, convergent mechanisms. Science 2004, 304, 1120–1122. [Google Scholar] [CrossRef]
- Gandhi, S.; Wood, N.W. Molecular pathogenesis of Parkinson’s disease. Hum. Mol. Genet. 2005, 14, 2749–2755. [Google Scholar] [CrossRef]
- Terzioglu, M.; Galter, D. Parkinson’s disease: Genetic versus toxin-induced rodent models. FEBS J. 2008, 275, 1384–1391. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.E.; Mouradian, M.M. Cytoprotective mechanisms of DJ-1 against oxidative stress through modulating ERK1/2 and ASK1 signal transduction. Redox Biol. 2018, 14, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.J.; Sailor, K.A.; Mahmood, Q.A.; Chavali, N.; Christian, K.M.; Song, H.; Ming, G.L. Seamless reconstruction of intact adult-born neurons by serial end-block imaging reveals complex axonal guidance and development in the adult hippocampus. J. Neurosci. 2013, 33, 11400–11411. [Google Scholar] [CrossRef] [Green Version]
- Brezova, V.; Dvoranova, D.; Zubor, V.; Breza, M.; Mazur, M.; Valko, M. Photochemical properties of camptothecin in the presence of copper(II) ions: The role of radicals as prospective species in photodynamic therapy. Mol. Biotechnol. 2007, 37, 48–51. [Google Scholar] [CrossRef]
- Bjørklund, G.; Peana, M.; Maes, M.; Dadar, M.; Severin, B. The glutathione system in Parkinson’s disease and its progression. Neurosci. Biobehav. Rev. 2021, 120, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Naoi, M.; Maruyama, W. Monoamine oxidase inhibitors as neuroprotective agents in age-dependent neurodegenerative disorders. Curr. Pharm. Des. 2010, 16, 2799–2817. [Google Scholar] [CrossRef] [PubMed]
- Naoi, M.; Maruyama, W.; Inaba-Hasegawa, K.; Akao, Y. Type A monoamine oxidase regulates life and death of neurons in neurodegeneration and neuroprotection. Int. Rev. Neurobiol. 2011, 100, 85–106. [Google Scholar] [CrossRef]
- Xia, L.; Nordman, T.; Olsson, J.M.; Damdimopoulos, A.; Bjorkhem-Bergman, L.; Nalvarte, I.; Eriksson, L.C.; Arner, E.S.; Spyrou, G.; Bjornstedt, M. The mammalian cytosolic selenoenzyme thioredoxin reductase reduces ubiquinone. A novel mechanism for defense against oxidative stress. J. Biol. Chem. 2003, 278, 2141–2146. [Google Scholar] [CrossRef] [Green Version]
- Blackinton, J.; Kumaran, R.; van der Brug, M.P.; Ahmad, R.; Olson, L.; Galter, D.; Lees, A.; Bandopadhyay, R.; Cookson, M.R. Post-transcriptional regulation of mRNA associated with DJ-1 in sporadic Parkinson disease. Neurosci. Lett. 2009, 452, 8–11. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.S.; Jia, J.J.; Kwon, Y.; Wang, S.D.; Bai, J. The role of thioredoxin-1 in suppression of endoplasmic reticulum stress in Parkinson disease. Free Radic. Biol. Med. 2014, 67, 10–18. [Google Scholar] [CrossRef]
- Hu, X.; Weng, Z.; Chu, C.T.; Zhang, L.; Cao, G.; Gao, Y.; Signore, A.; Zhu, J.; Hastings, T.; Greenamyre, J.T.; et al. Peroxiredoxin-2 protects against 6-hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J. Neurosci. 2011, 31, 247–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, J.; Sullards, M.C.; Olzmann, J.A.; Rees, H.D.; Weintraub, S.T.; Bostwick, D.E.; Gearing, M.; Levey, A.I.; Chin, L.S.; Li, L. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J. Biol. Chem. 2006, 281, 10816–10824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorner, K.; Holtorf, E.; Waak, J.; Pham, T.T.; Vogt-Weisenhorn, D.M.; Wurst, W.; Haass, C.; Kahle, P.J. Structural determinants of the C-terminal helix-kink-helix motif essential for protein stability and survival promoting activity of DJ-1. J. Biol. Chem. 2007, 282, 13680–13691. [Google Scholar] [CrossRef] [PubMed]
- Bahmed, K.; Boukhenouna, S.; Karim, L.; Andrews, T.; Lin, J.; Powers, R.; Wilson, M.A.; Lin, C.R.; Messier, E.; Reisdorph, N.; et al. The effect of cysteine oxidation on DJ-1 cytoprotective function in human alveolar type II cells. Cell Death Dis. 2019, 10, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, J.Y.; Lee, K.W.; Woo, J.M.; Junn, E.; Mouradian, M.M. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum. Mol. Genet. 2012, 21, 3013–3024. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.Y.; Liu, I.C.; Lin, T.Y. Truncated Escherichia coli thioredoxin induces proliferation of human blood mononuclear cells and production of reactive oxygen species as well as proinflammatory cytokines. Cell Biochem. Funct. 2016, 34, 226–232. [Google Scholar] [CrossRef]
- Chen, J.; Berry, M.J. Selenium and selenoproteins in the brain and brain diseases. J. Neurochem. 2003, 86, 1–12. [Google Scholar] [CrossRef]
- Shahar, A.; Patel, K.V.; Semba, R.D.; Bandinelli, S.; Shahar, D.R.; Ferrucci, L.; Guralnik, J.M. Plasma selenium is positively related to performance in neurological tasks assessing coordination and motor speed. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 1909–1915. [Google Scholar] [CrossRef] [Green Version]
- Lopert, P.; Day, B.J.; Patel, M. Thioredoxin reductase deficiency potentiates oxidative stress, mitochondrial dysfunction and cell death in dopaminergic cells. PLoS ONE 2012, 7, e50683. [Google Scholar] [CrossRef] [Green Version]
- Maes, M.; Moraes, J.B.; Congio, A.; Bonifacio, K.L.; Barbosa, D.S.; Vargas, H.O.; Michelin, A.P.; Carvalho, A.F.; Nunes, S.O.V. Development of a Novel Staging Model for Affective Disorders Using Partial Least Squares Bootstrapping: Effects of Lipid-Associated Antioxidant Defenses and Neuro-Oxidative Stress. Mol. Neurobiol. 2019, 56, 6626–6644. [Google Scholar] [CrossRef] [PubMed]
- Salimpoor, V.N.; Benovoy, M.; Larcher, K.; Dagher, A.; Zatorre, R.J. Anatomically distinct dopamine release during anticipation and experience of peak emotion to music. Nat. Neurosci. 2011, 14, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Baik, J.H. Dopamine signaling in reward-related behaviors. Front. Neural Circuits 2013, 7, 152. [Google Scholar] [CrossRef] [Green Version]
- Golden, J.P.; Demaro, J.A., 3rd; Knoten, A.; Hoshi, M.; Pehek, E.; Johnson, E.M., Jr.; Gereau, R.W.t.; Jain, S. Dopamine-dependent compensation maintains motor behavior in mice with developmental ablation of dopaminergic neurons. J. Neurosci. 2013, 33, 17095–17107. [Google Scholar] [CrossRef]
- Toates, F. Application of a multilevel model of behavioural control to understanding emotion. Behav. Process. 2002, 60, 99–114. [Google Scholar] [CrossRef]
- Brebner, K.; Wong, T.P.; Liu, L.; Liu, Y.; Campsall, P.; Gray, S.; Phelps, L.; Phillips, A.G.; Wang, Y.T. Nucleus accumbens long-term depression and the expression of behavioral sensitization. Science 2005, 310, 1340–1343. [Google Scholar] [CrossRef] [PubMed]
- Kutlu, A.; Karabacak, E.; Aydin, E.; Ozturk, S.; Bozkurt, B. A patient with steroids and antihistaminic drug allergy and newly occurred chronic urticaria angioedema: What about omalizumab? Hum. Exp. Toxicol. 2014, 33, 882–885. [Google Scholar] [CrossRef]
- Scuvee-Moreau, J. Neurobiology of addiction. Rev. Med. Liege 2013, 68, 211–217. [Google Scholar]
- Eisenstein, M. CRISPR takes on Huntington’s disease. Nature 2018, 557, S42–S43. [Google Scholar] [CrossRef] [Green Version]
- Nowogrodzki, A. Huntington’s disease: 4 big questions. Nature 2018, 557, S48. [Google Scholar] [CrossRef] [Green Version]
- Raymond, L.A.; Andre, V.M.; Cepeda, C.; Gladding, C.M.; Milnerwood, A.J.; Levine, M.S. Pathophysiology of Huntington’s disease: Time-dependent alterations in synaptic and receptor function. Neuroscience 2011, 198, 252–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fox, J.; Lu, Z.; Barrows, L. Thiol-disulfide Oxidoreductases TRX1 and TMX3 Decrease Neuronal Atrophy in a Lentiviral Mouse Model of Huntington’s Disease. PLoS Curr. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Galvan, A.; Wichmann, T. GABAergic circuits in the basal ganglia and movement disorders. Prog. Brain Res. 2007, 160, 287–312. [Google Scholar] [CrossRef] [PubMed]
- Hickey, M.A.; Chesselet, M.F. Apoptosis in Huntington’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2003, 27, 255–265. [Google Scholar] [CrossRef]
- Levine, M.S.; Cepeda, C.; Hickey, M.A.; Fleming, S.M.; Chesselet, M.F. Genetic mouse models of Huntington’s and Parkinson’s diseases: Illuminating but imperfect. Trends Neurosci. 2004, 27, 691–697. [Google Scholar] [CrossRef]
- Gafni, J.; Hermel, E.; Young, J.E.; Wellington, C.L.; Hayden, M.R.; Ellerby, L.M. Inhibition of calpain cleavage of huntingtin reduces toxicity: Accumulation of calpain/caspase fragments in the nucleus. J. Biol. Chem. 2004, 279, 20211–20220. [Google Scholar] [CrossRef] [Green Version]
- Fox, J.H.; Connor, T.; Stiles, M.; Kama, J.; Lu, Z.; Dorsey, K.; Lieberman, G.; Sapp, E.; Cherny, R.A.; Banks, M.; et al. Cysteine oxidation within N-terminal mutant huntingtin promotes oligomerization and delays clearance of soluble protein. J. Biol. Chem. 2011, 286, 18320–18330. [Google Scholar] [CrossRef] [Green Version]
- Arrasate, M.; Mitra, S.; Schweitzer, E.S.; Segal, M.R.; Finkbeiner, S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 2004, 431, 805–810. [Google Scholar] [CrossRef] [Green Version]
- Dunah, A.W.; Jeong, H.; Griffin, A.; Kim, Y.M.; Standaert, D.G.; Hersch, S.M.; Mouradian, M.M.; Young, A.B.; Tanese, N.; Krainc, D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science 2002, 296, 2238–2243. [Google Scholar] [CrossRef]
- Vonsattel, J.P.; Myers, R.H.; Stevens, T.J.; Ferrante, R.J.; Bird, E.D.; Richardson, E.P., Jr. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985, 44, 559–577. [Google Scholar] [CrossRef]
- Zhou, F.; Gomi, M.; Fujimoto, M.; Hayase, M.; Marumo, T.; Masutani, H.; Yodoi, J.; Hashimoto, N.; Nozaki, K.; Takagi, Y. Attenuation of neuronal degeneration in thioredoxin-1 overexpressing mice after mild focal ischemia. Brain Res. 2009, 1272, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sreekala, S.; Indira, M. Impact of co administration of selenium and quinolinic acid in the rat’s brain. Brain Res. 2009, 1281, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.; Rosenstock, T.R.; Cunha-Oliveira, T.; Ferreira, I.L.; Oliveira, C.R.; Rego, A.C. Glutathione redox cycle dysregulation in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2012, 53, 1857–1867. [Google Scholar] [CrossRef] [PubMed]
- Kannan, R.; Kuhlenkamp, J.F.; Ookhtens, M.; Kaplowitz, N. Transport of glutathione at blood-brain barrier of the rat: Inhibition by glutathione analogs and age-dependence. J. Pharmacol. Exp. Ther. 1992, 263, 964–970. [Google Scholar]
- Mackic, J.B.; Jinagouda, S.; McComb, J.G.; Weiss, M.H.; Kannan, R.; Kaplowitz, N.; Zlokovic, B.V. Transport of circulating reduced glutathione at the basolateral side of the anterior lens epithelium: Physiologic importance and manipulations. Exp. Eye Res. 1996, 62, 29–37. [Google Scholar] [CrossRef]
- Kannan, R.; Mackic, J.B.; Zlokovic, B.V. Corneal transport of circulating glutathione in normal and galactosemic guinea pigs. Cornea 1999, 18, 321–327. [Google Scholar] [CrossRef]
- Kannan, R.; Chakrabarti, R.; Tang, D.; Kim, K.J.; Kaplowitz, N. GSH transport in human cerebrovascular endothelial cells and human astrocytes: Evidence for luminal localization of Na+-dependent GSH transport in HCEC. Brain Res. 2000, 852, 374–382. [Google Scholar] [CrossRef]
- Zlokovic, B.V.; Mackic, J.B.; McComb, J.G.; Weiss, M.H.; Kaplowitz, N.; Kannan, R. Evidence for transcapillary transport of reduced glutathione in vascular perfused guinea-pig brain. Biochem. Biophys. Res. Commun. 1994, 201, 402–408. [Google Scholar] [CrossRef]
- Kannan, R.; Yi, J.R.; Tang, D.; Zlokovic, B.V.; Kaplowitz, N. Identification of a novel, sodium-dependent, reduced glutathione transporter in the rat lens epithelium. Investig. Ophthalmol. Vis. Sci. 1996, 37, 2269–2275. [Google Scholar]
- De Almeida, L.P.; Ross, C.A.; Zala, D.; Aebischer, P.; Deglon, N. Lentiviral-mediated delivery of mutant huntingtin in the striatum of rats induces a selective neuropathology modulated by polyglutamine repeat size, huntingtin expression levels, and protein length. J. Neurosci. 2002, 22, 3473–3483. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M.; Nishitoh, H.; Fujii, M.; Takeda, K.; Tobiume, K.; Sawada, Y.; Kawabata, M.; Miyazono, K.; Ichijo, H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998, 17, 2596–2606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterson, D.A. Umbilical cord blood cells and brain stroke injury: Bringing in fresh blood to address an old problem. J. Clin. Investig. 2004, 114, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Nih, L.R.; Gojgini, S.; Carmichael, S.T.; Segura, T. Dual-function injectable angiogenic biomaterial for the repair of brain tissue following stroke. Nat. Mater. 2018, 17, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, R.; Fernandez-Gajardo, R.; Gutierrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeutic opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714. [Google Scholar] [CrossRef]
- Norrving, B.; Kissela, B. The global burden of stroke and need for a continuum of care. Neurology 2013, 80, S5–S12. [Google Scholar] [CrossRef]
- Kalladka, D.; Muir, K.W. Brain repair: Cell therapy in stroke. Stem Cells Cloning 2014, 7, 31–44. [Google Scholar] [CrossRef]
- Tsuda, K. Letter by Tsuda Regarding Article, “Oxidative Stress Biomarkers of Brain Damage: Hyperacute Plasma F2-Isoprostane Predicts Infarct Growth in Stroke”. Stroke 2018, 49, e263. [Google Scholar] [CrossRef]
- Lorenzano, S.; Rost, N.S.; Khan, M.; Li, H.; Lima, F.O.; Maas, M.B.; Green, R.E.; Thankachan, T.K.; Dipietro, A.J.; Arai, K.; et al. Oxidative Stress Biomarkers of Brain Damage: Hyperacute Plasma F2-Isoprostane Predicts Infarct Growth in Stroke. Stroke 2018, 49, 630–637. [Google Scholar] [CrossRef]
- Watson, W.H.; Yang, X.; Choi, Y.E.; Jones, D.P.; Kehrer, J.P. Thioredoxin and its role in toxicology. Toxicol. Sci. 2004, 78, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Che, X.; Zhang, H.; Tan, G.; Liu, L.; Jiang, D.; Zhao, J.; Xiang, X.; Sun, X.; He, Z. Thioredoxin-Interacting Protein Mediates Apoptosis in Early Brain Injury after Subarachnoid Haemorrhage. Int. J. Mol. Sci. 2017, 18, 854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, G.S.; Jung, J.E.; Narasimhan, P.; Sakata, H.; Chan, P.H. Induction of thioredoxin-interacting protein is mediated by oxidative stress, calcium, and glucose after brain injury in mice. Neurobiol. Dis. 2012, 46, 440–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, W.H.; Lin, H.S.; Chen, C.I.; Chou, M.S.; Liou, C.W.; Chen, S.S.; Stroke Center, C.K. The medicolegal issue of tissue plasminogen activator in ischemic stroke: A review of judiciary decrees in Taiwan. Acta Neurol. Taiwan 2011, 20, 163–171. [Google Scholar] [PubMed]
- Hermann, D.M.; Matter, C.M. Tissue plasminogen activator-induced reperfusion injury after stroke revisited. Circulation 2007, 116, 363–365. [Google Scholar] [CrossRef] [Green Version]
- Kelly, P.J.; Morrow, J.D.; Ning, M.; Koroshetz, W.; Lo, E.H.; Terry, E.; Milne, G.L.; Hubbard, J.; Lee, H.; Stevenson, E.; et al. Oxidative stress and matrix metalloproteinase-9 in acute ischemic stroke: The Biomarker Evaluation for Antioxidant Therapies in Stroke (BEAT-Stroke) study. Stroke 2008, 39, 100–104. [Google Scholar] [CrossRef] [Green Version]
- Ishrat, T.; Mohamed, I.N.; Pillai, B.; Soliman, S.; Fouda, A.Y.; Ergul, A.; El-Remessy, A.B.; Fagan, S.C. Erratum to: Thioredoxin-interacting protein: A novel target for neuroprotection in experimental thromboembolic stroke in mice. Mol. Neurobiol. 2015, 51, 779–780. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Ismael, S.; Nasoohi, S.; Sakata, K.; Liao, F.F.; McDonald, M.P.; Ishrat, T. Thioredoxin-Interacting Protein (TXNIP) Associated NLRP3 Inflammasome Activation in Human Alzheimer’s Disease Brain. J. Alzheimers Dis. 2019, 68, 255–265. [Google Scholar] [CrossRef]
- Tian, L.; Nie, H.; Zhang, Y.; Chen, Y.; Peng, Z.; Cai, M.; Wei, H.; Qin, P.; Dong, H.; Xiong, L. Recombinant human thioredoxin-1 promotes neurogenesis and facilitates cognitive recovery following cerebral ischemia in mice. Neuropharmacology 2014, 77, 453–464. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tian, S.; Wang, J.; Han, F.; Zhao, L.; Wang, R.; Ning, W.; Chen, W.; Qu, Y. Intraperitoneal administration of thioredoxin decreases brain damage from ischemic stroke. Brain Res. 2015, 1615, 89–97. [Google Scholar] [CrossRef]
- Wu, X.; Li, L.; Zhang, L.; Wu, J.; Zhou, Y.; Zhou, Y.; Zhao, Y.; Zhao, J. Inhibition of thioredoxin-1 with siRNA exacerbates apoptosis by activating the ASK1-JNK/p38 pathway in brain of a stroke model rats. Brain Res. 2015, 1599, 20–31. [Google Scholar] [CrossRef]
- Li, L.; Zhu, K.; Liu, Y.; Wu, X.; Wu, J.; Zhao, Y.; Zhao, J. Targeting thioredoxin-1 with siRNA exacerbates oxidative stress injury after cerebral ischemia/reperfusion in rats. Neuroscience 2015, 284, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Qi, A.Q.; Li, Y.; Liu, Q.; Si, J.Z.; Tang, X.M.; Zhang, Z.Q.; Qi, Q.D.; Chen, W.B. Thioredoxin is a novel diagnostic and prognostic marker in patients with ischemic stroke. Free Radic. Biol. Med. 2015, 80, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Baratz-Goldstein, R.; Deselms, H.; Heim, L.R.; Khomski, L.; Hoffer, B.J.; Atlas, D.; Pick, C.G. Thioredoxin-Mimetic-Peptides Protect Cognitive Function after Mild Traumatic Brain Injury (mTBI). PLoS ONE 2016, 11, e0157064. [Google Scholar] [CrossRef] [PubMed]
- Voigt, D.; Scheidt, U.; Derfuss, T.; Bruck, W.; Junker, A. Expression of the Antioxidative Enzyme Peroxiredoxin 2 in Multiple Sclerosis Lesions in Relation to Inflammation. Int. J. Mol. Sci. 2017, 18, 760. [Google Scholar] [CrossRef] [Green Version]
- De Bock, L.; Somers, K.; Fraussen, J.; Hendriks, J.J.; van Horssen, J.; Rouwette, M.; Hellings, N.; Villar, L.M.; Alvarez-Cermeno, J.C.; Espino, M.; et al. Sperm-associated antigen 16 is a novel target of the humoral autoimmune response in multiple sclerosis. J. Immunol. 2014, 193, 2147–2156. [Google Scholar] [CrossRef] [Green Version]
- Edan, G. Multiple Sclerosis over the last 25 years: An introduction. Rev. Neurol. 2018, 174, 353–354. [Google Scholar] [CrossRef]
- Wendebourg, M.J.; Heesen, C.; Finlayson, M.; Meyer, B.; Pottgen, J.; Kopke, S. Patient education for people with multiple sclerosis-associated fatigue: A systematic review. PLoS ONE 2017, 12, e0173025. [Google Scholar] [CrossRef]
- Bar-Or, A. Multiple sclerosis and related disorders: Evolving pathophysiologic insights. Lancet Neurol. 2016, 15, 9–11. [Google Scholar] [CrossRef]
- Ascherio, A.; Munger, K.L. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann. Neurol. 2007, 61, 504–513. [Google Scholar] [CrossRef]
- Perron, H.; Bernard, C.; Bertrand, J.B.; Lang, A.B.; Popa, I.; Sanhadji, K.; Portoukalian, J. Endogenous retroviral genes, Herpesviruses and gender in Multiple Sclerosis. J. Neurol. Sci. 2009, 286, 65–72. [Google Scholar] [CrossRef]
- Morandi, E.; Tanasescu, R.; Tarlinton, R.E.; Constantinescu, C.S.; Zhang, W.; Tench, C.; Gran, B. The association between human endogenous retroviruses and multiple sclerosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0172415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Horssen, J.; Witte, M.E.; Schreibelt, G.; de Vries, H.E. Radical changes in multiple sclerosis pathogenesis. Biochim. Biophys. Acta 2011, 1812, 141–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draheim, T.; Liessem, A.; Scheld, M.; Wilms, F.; Weissflog, M.; Denecke, B.; Kensler, T.W.; Zendedel, A.; Beyer, C.; Kipp, M.; et al. Activation of the astrocytic Nrf2/ARE system ameliorates the formation of demyelinating lesions in a multiple sclerosis animal model. Glia 2016, 64, 2219–2230. [Google Scholar] [CrossRef] [PubMed]
- Nijland, P.G.; Witte, M.E.; van het Hof, B.; van der Pol, S.; Bauer, J.; Lassmann, H.; van der Valk, P.; de Vries, H.E.; van Horssen, J. Astroglial PGC-1alpha increases mitochondrial antioxidant capacity and suppresses inflammation: Implications for multiple sclerosis. Acta Neuropathol. Commun. 2014, 2, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lassmann, H.; van Horssen, J. Oxidative stress and its impact on neurons and glia in multiple sclerosis lesions. Biochim. Biophys. Acta 2016, 1862, 506–510. [Google Scholar] [CrossRef]
- Perkins, R.B.; Christiansen, C.L. The human papillomavirus vaccination is not associated with risk of multiple sclerosis or other demyelinating diseases. Evid. Based Med. 2015, 20, 116. [Google Scholar] [CrossRef]
- Pellegrini, G.G.; Cregor, M.; McAndrews, K.; Morales, C.C.; McCabe, L.D.; McCabe, G.P.; Peacock, M.; Burr, D.; Weaver, C.; Bellido, T. Nrf2 regulates mass accrual and the antioxidant endogenous response in bone differently depending on the sex and age. PLoS ONE 2017, 12, e0171161. [Google Scholar] [CrossRef]
- Choi, I.Y.; Lee, P.; Adany, P.; Hughes, A.J.; Belliston, S.; Denney, D.R.; Lynch, S.G. In vivo evidence of oxidative stress in brains of patients with progressive multiple sclerosis. Mult. Scler. 2018, 24, 1029–1038. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67. [Google Scholar] [CrossRef]
- Medici, S.; Peana, M.; Nurchi, V.M.; Lachowicz, J.I.; Crisponi, G.; Zoroddu, M.A. Noble metals in medicine: Latest advances. Coord. Chem. Rev. 2015, 284, 329–350. [Google Scholar] [CrossRef]
- Zhong, L.; Arner, E.S.; Ljung, J.; Aslund, F.; Holmgren, A. Rat and calf thioredoxin reductase are homologous to glutathione reductase with a carboxyl-terminal elongation containing a conserved catalytically active penultimate selenocysteine residue. J. Biol. Chem. 1998, 273, 8581–8591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, L.; Lu, J.; Wang, J.; Ren, X.; Zhang, L.; Gao, Y.; Rottenberg, M.E.; Holmgren, A. Synergistic antibacterial effect of silver and ebselen against multidrug-resistant Gram-negative bacterial infections. EMBO Mol. Med. 2017, 9, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Zhou, J.; Wang, P.; Li, T.; Zhao, Y.; Ren, X.; Lu, J.; Wang, J.; Holmgren, A.; Zou, L. Topical Therapeutic Efficacy of Ebselen Against Multidrug-Resistant Staphylococcus aureus LT-1 Targeting Thioredoxin Reductase. Front. Microbiol. 2019, 10, 3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunigan-Russell, K.; Lin, V.; Silverberg, M.; Wall, S.B.; Li, R.; Gotham, J.; Nicola, T.; Sridharan, A.; Snowball, J.; Delaney, C.; et al. Aurothioglucose Enhances Pro-Angiogenic Pathway Activation in Lungs from Room Air and Hyperoxia-exposed Newborn Mice. Am. J. Physiol. Lung Cell Mol. Physiol. 2020, 318, L1165–L1171. [Google Scholar] [CrossRef] [PubMed]
- Onodera, T.; Momose, I.; Kawada, M. Potential Anticancer Activity of Auranofin. Chem. Pharm. Bull. 2019, 67, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Yumnamcha, T.; Devi, T.S.; Singh, L.P. Auranofin Mediates Mitochondrial Dysregulation and Inflammatory Cell Death in Human Retinal Pigment Epithelial Cells: Implications of Retinal Neurodegenerative Diseases. Front. Neurosci. 2019, 13, 1065. [Google Scholar] [CrossRef]
- Prast-Nielsen, S.; Cebula, M.; Pader, I.; Arner, E.S. Noble metal targeting of thioredoxin reductase—Covalent complexes with thioredoxin and thioredoxin-related protein of 14 kDa triggered by cisplatin. Free Radic. Biol. Med. 2010, 49, 1765–1778. [Google Scholar] [CrossRef]
- Rigobello, M.P.; Messori, L.; Marcon, G.; Agostina Cinellu, M.; Bragadin, M.; Folda, A.; Scutari, G.; Bindoli, A. Gold complexes inhibit mitochondrial thioredoxin reductase: Consequences on mitochondrial functions. J. Inorg. Biochem. 2004, 98, 1634–1641. [Google Scholar] [CrossRef]
- Branco, V.; Canario, J.; Lu, J.; Holmgren, A.; Carvalho, C. Mercury and selenium interaction in vivo: Effects on thioredoxin reductase and glutathione peroxidase. Free Radic. Biol. Med. 2012, 52, 781–793. [Google Scholar] [CrossRef]
- Spiller, H.A. Rethinking mercury: The role of selenium in the pathophysiology of mercury toxicity. Clin. Toxicol. 2018, 56, 313–326. [Google Scholar] [CrossRef]
- Farina, M.; Aschner, M. Glutathione antioxidant system and methylmercury-induced neurotoxicity: An intriguing interplay. Biochim. Biophys. Acta Gen. Subj. 2019, 1863, 129285. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, C.M.; Chew, E.H.; Hashemy, S.I.; Lu, J.; Holmgren, A. Inhibition of the human thioredoxin system. A molecular mechanism of mercury toxicity. J. Biol. Chem. 2008, 283, 11913–11923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, C.L.; Jang, T.H.; Wang, L.H. Effects of mercury on serotonin concentration in the brain of tilapia, Oreochromis mossambicus. Neurosci. Lett. 1995, 184, 208–211. [Google Scholar] [CrossRef]
- Zhou, T.; Rademacher, D.J.; Steinpreis, R.E.; Weis, J.S. Neurotransmitter levels in two populations of larval Fundulus heteroclitus after methylmercury exposure. Comp Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1999, 124, 287–294. [Google Scholar] [CrossRef]
- Bjørklund, G.; Antonyak, H.; Polishchuk, A.; Semenova, Y.; Lesiv, M.; Lysiuk, R.; Peana, M. Effect of methylmercury on fetal neurobehavioral development: An overview of the possible mechanisms of toxicity and the neuroprotective effect of phytochemicals. Arch. Toxicol. 2022, 96, 3175–3199. [Google Scholar] [CrossRef] [PubMed]
- Hattori, K.; Sukenobu, N.; Sasaki, T.; Takasuga, S.; Hayashi, T.; Kasai, R.; Yamasaki, K.; Hazeki, O. Activation of insulin receptors by lagerstroemin. J. Pharmacol. Sci. 2003, 93, 69–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bessems, J.G.; Vermeulen, N.P. Paracetamol (acetaminophen)-induced toxicity: Molecular and biochemical mechanisms, analogues and protective approaches. Crit. Rev. Toxicol. 2001, 31, 55–138. [Google Scholar] [CrossRef]
- Hansen, J.M.; Zhang, H.; Jones, D.P. Differential oxidation of thioredoxin-1, thioredoxin-2, and glutathione by metal ions. Free Radic. Biol. Med. 2006, 40, 138–145. [Google Scholar] [CrossRef]
- Tanaka, K.I.; Shimoda, M.; Chuang, V.T.G.; Nishida, K.; Kawahara, M.; Ishida, T.; Otagiri, M.; Maruyama, T.; Ishima, Y. Thioredoxin-albumin fusion protein prevents copper enhanced zinc-induced neurotoxicity via its antioxidative activity. Int. J. Pharm. 2018, 535, 140–147. [Google Scholar] [CrossRef]
- Maher, P. Potentiation of glutathione loss and nerve cell death by the transition metals iron and copper: Implications for age-related neurodegenerative diseases. Free Radic. Biol. Med. 2018, 115, 92–104. [Google Scholar] [CrossRef]
- Zou, L.; Wang, J.; Gao, Y.; Ren, X.; Rottenberg, M.E.; Lu, J.; Holmgren, A. Synergistic antibacterial activity of silver with antibiotics correlating with the upregulation of the ROS production. Sci. Rep. 2018, 8, 11131. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
PRISMA 2009 Flow Diagram for the review detailing the database searches, the number of abstracts screened and the full texts retrieved.
Figure 1.
PRISMA 2009 Flow Diagram for the review detailing the database searches, the number of abstracts screened and the full texts retrieved.
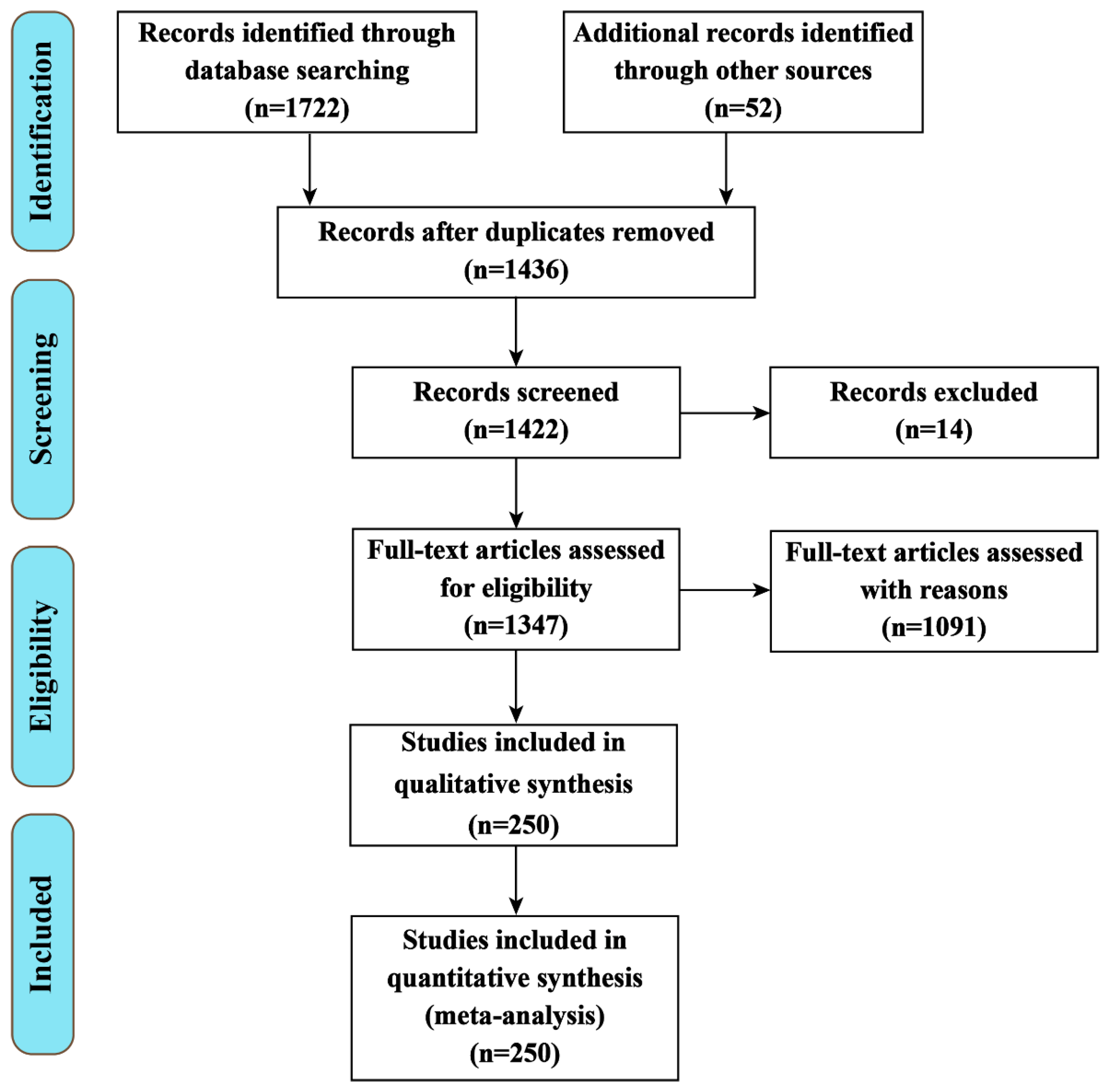
Figure 2.
The role of the Trx system in the pathophysiology of Alzheimer’s disease (AD). The combination of Trx with ASK1 may play a role in the neurodegeneration and survival of cerebellar cells. The increased expression of TrxR, Trx1 and Trx80 decreases the accumulation of Aβ in AD patients. Prx and Trx can be used to diagnose AD in lab instead of clinical application. Trx: thioredoxin; TrxR: thioredoxin reductase; Prx: peroxiredoxin; ASK1: apoptosis signal-regulating kinase 1; Aβ: amyloid β.
Figure 2.
The role of the Trx system in the pathophysiology of Alzheimer’s disease (AD). The combination of Trx with ASK1 may play a role in the neurodegeneration and survival of cerebellar cells. The increased expression of TrxR, Trx1 and Trx80 decreases the accumulation of Aβ in AD patients. Prx and Trx can be used to diagnose AD in lab instead of clinical application. Trx: thioredoxin; TrxR: thioredoxin reductase; Prx: peroxiredoxin; ASK1: apoptosis signal-regulating kinase 1; Aβ: amyloid β.

Figure 3.
The involvement of Trx1 in pathophysiological signal pathways of Parkinson’s disease (PD). Trx1 participates in a variety of biological pathways of PD. During oxidative conditions, Trx1 stimulates apoptosis signaling caspase 12, which antagonizes the activity of mitochondrial complex I. Trx1 might also suppress MPTP in cytoprotective and neuroprotective mechanisms. When exposed to H2O2 and dissociated from Trx1, ASK1 can co-immunoprecipitate with DJ-1. Meanwhile, DJ-1 induces expression of Trx1 via transcription factor Nrf-2 to protect cells against oxidative stress. Also, Prx has a protective role via Trx1-ASK1 against dopaminergic neuron cell death. Nrf-2: nuclear factor erythroid 2-related factor 2; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; Trx: thioredoxin; TrxR: thioredoxin reductase; Prx: peroxiredoxin; ASK1: apoptosis signal-regulating kinase 1; Aβ: amyloid β.
Figure 3.
The involvement of Trx1 in pathophysiological signal pathways of Parkinson’s disease (PD). Trx1 participates in a variety of biological pathways of PD. During oxidative conditions, Trx1 stimulates apoptosis signaling caspase 12, which antagonizes the activity of mitochondrial complex I. Trx1 might also suppress MPTP in cytoprotective and neuroprotective mechanisms. When exposed to H2O2 and dissociated from Trx1, ASK1 can co-immunoprecipitate with DJ-1. Meanwhile, DJ-1 induces expression of Trx1 via transcription factor Nrf-2 to protect cells against oxidative stress. Also, Prx has a protective role via Trx1-ASK1 against dopaminergic neuron cell death. Nrf-2: nuclear factor erythroid 2-related factor 2; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; Trx: thioredoxin; TrxR: thioredoxin reductase; Prx: peroxiredoxin; ASK1: apoptosis signal-regulating kinase 1; Aβ: amyloid β.

Figure 4.
The role of Trx1 and Gpx in the pathophysiology of Huntington’s disease (HD). Trx1 and GPx participate in various biological pathways in relation to HD. The neuron protection activity of Trx1 is achieved via a decrease of both mHTT and ASK1. Moreover, high expression levels of GPx1/6 also play a crucial role against oxidative stress in HD patients. Trx: thioredoxin reductase; TMX3: thioredoxin-related transmembrane protein 3; ASK1: apoptosis signal-regulating kinase 1; GPx: glutathione peroxidase; mHTT: mutant Huntington protein.
Figure 4.
The role of Trx1 and Gpx in the pathophysiology of Huntington’s disease (HD). Trx1 and GPx participate in various biological pathways in relation to HD. The neuron protection activity of Trx1 is achieved via a decrease of both mHTT and ASK1. Moreover, high expression levels of GPx1/6 also play a crucial role against oxidative stress in HD patients. Trx: thioredoxin reductase; TMX3: thioredoxin-related transmembrane protein 3; ASK1: apoptosis signal-regulating kinase 1; GPx: glutathione peroxidase; mHTT: mutant Huntington protein.

Figure 5.
The role of Trx1 in the pathophysiology of ischemic stroke. Trx1 participates in the pathophysiology of brain stroke. Although Trx1 could directly combine with ASK1 to decrease its activity, TXNIP may inhibit Trx1 activity from inducing brain ischemic stroke under oxidative stress conditions. Inhibition of Trx1 with siRNA induces neuronal apoptosis by stimulating the brain ASK1-JNK/p38 signaling pathway.Trx: thioredoxin; TXNIP: Trx1 interacting protein; ASK1: apoptosis signal-regulating kinase 1.
Figure 5.
The role of Trx1 in the pathophysiology of ischemic stroke. Trx1 participates in the pathophysiology of brain stroke. Although Trx1 could directly combine with ASK1 to decrease its activity, TXNIP may inhibit Trx1 activity from inducing brain ischemic stroke under oxidative stress conditions. Inhibition of Trx1 with siRNA induces neuronal apoptosis by stimulating the brain ASK1-JNK/p38 signaling pathway.Trx: thioredoxin; TXNIP: Trx1 interacting protein; ASK1: apoptosis signal-regulating kinase 1.

Figure 6.
The role of Trx in the pathophysiology of multiple sclerosis (MS). In MS, Trx and sirt1 were upregulated, and TrxR was downregulated. Meanwhile, the activation of the Nrf2 pathway might play a protecting role against oxidative stress by upregulating the expression levels of GSH and Trx. Trx: thioredoxin; TrxR: thioredoxin reductase; Sirt1: sirtuin 1; Nrf2: Nuclear factor (erythroid-derived 2)-like 2.
Figure 6.
The role of Trx in the pathophysiology of multiple sclerosis (MS). In MS, Trx and sirt1 were upregulated, and TrxR was downregulated. Meanwhile, the activation of the Nrf2 pathway might play a protecting role against oxidative stress by upregulating the expression levels of GSH and Trx. Trx: thioredoxin; TrxR: thioredoxin reductase; Sirt1: sirtuin 1; Nrf2: Nuclear factor (erythroid-derived 2)-like 2.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bjørklund, G.; Zou, L.; Peana, M.; Chasapis, C.T.; Hangan, T.; Lu, J.; Maes, M. The Role of the Thioredoxin System in Brain Diseases. Antioxidants 2022, 11, 2161. https://doi.org/10.3390/antiox11112161
AMA Style
Bjørklund G, Zou L, Peana M, Chasapis CT, Hangan T, Lu J, Maes M. The Role of the Thioredoxin System in Brain Diseases. Antioxidants. 2022; 11(11):2161. https://doi.org/10.3390/antiox11112161
Chicago/Turabian StyleBjørklund, Geir, Lili Zou, Massimiliano Peana, Christos T. Chasapis, Tony Hangan, Jun Lu, and Michael Maes. 2022. "The Role of the Thioredoxin System in Brain Diseases" Antioxidants 11, no. 11: 2161. https://doi.org/10.3390/antiox11112161
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.