


From Gasotransmitter to Immunomodulator: The Emerging Role of Hydrogen Sulfide in Macrophage Biology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
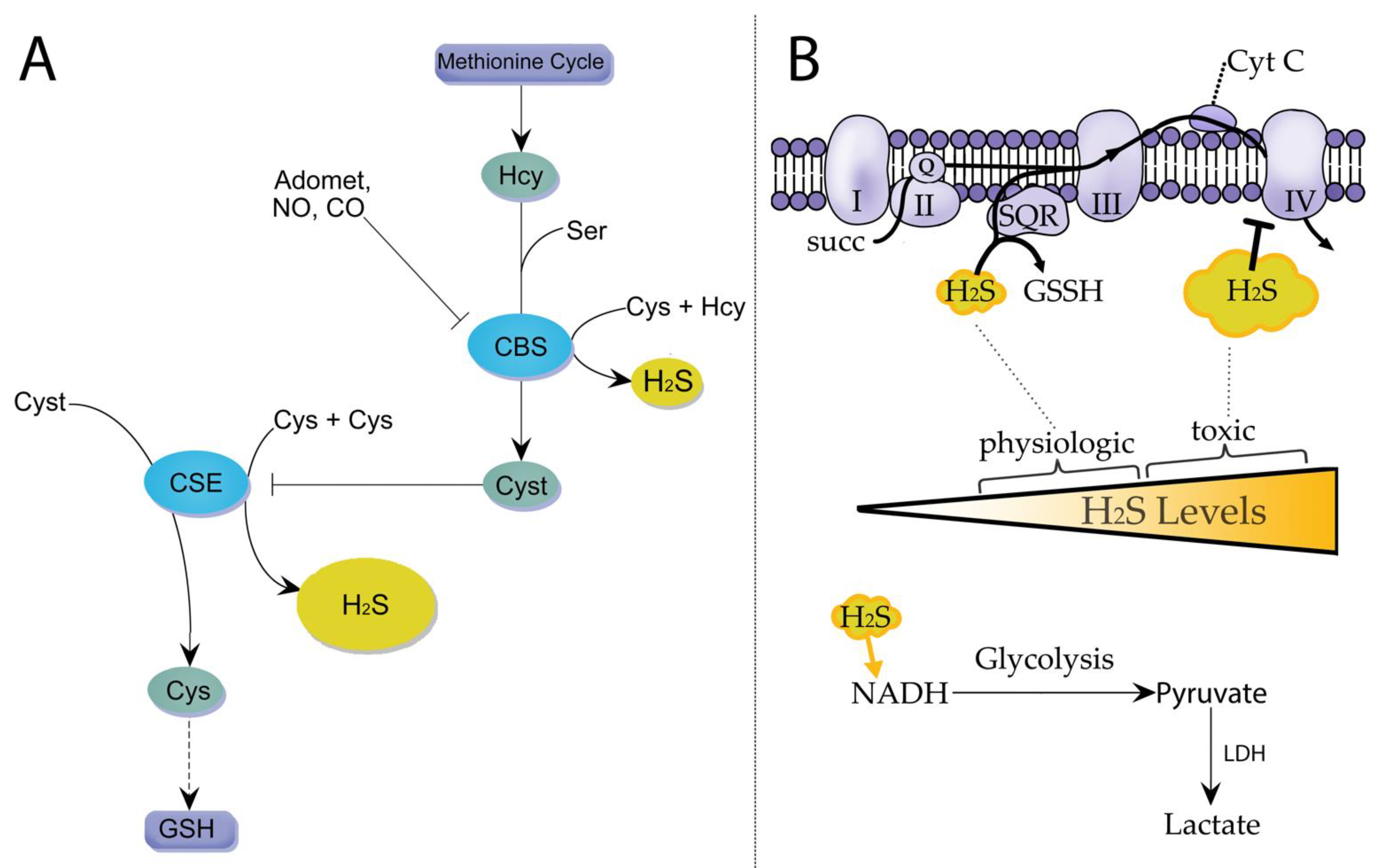
2. The Synthesis of Hydrogen Sulfide in Mammalian Cells
3. The Balancing Act: How Cells Control Cysteine and Hydrogen Sulfide Synthesis through the TSP Regulation
4. Persulfidation Is a Regulatory Mechanism for Cellular Processes
5. Hydrogen Sulfide and Redox Homeostasis
6. The Therapeutic Potential of Hydrogen Sulfide and Polysulfides
7. The Interplay between Metabolism and the Immune Response in Macrophages
8. The Complex Role of Hydrogen Sulfide in Modulating Cellular Energy Metabolism and Inflammatory Responses in Macrophages
9. Summary and Future Directions
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Martinez, F.O.; Gordon, S. The M1 and M2 Paradigm of Macrophage Activation: Time for Reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Herbst, S.; Schaible, U.E.; Schneider, B.E. Interferon Gamma Activated Macrophages Kill Mycobacteria by Nitric Oxide Induced Apoptosis. PLoS ONE 2011, 6, e19105. [Google Scholar] [CrossRef] [PubMed]
- Piedrafita, D.; Parsons, J.C.; Sandeman, R.M.; Wood, P.R.; Estuningsih, S.E.; Partoutomo, S.; Spithill, T.W. Antibody-Dependent Cell-Mediated Cytotoxicity to Newly Excysted Juvenile Fasciola Hepatica in Vitro Is Mediated by Reactive Nitrogen Intermediates. Parasite Immunol. 2001, 23, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.R.; McCrossan, M.; Selkirk, M.E. Cytostatic and Cytotoxic Effects of Activated Macrophages and Nitric Oxide Donors on Brugia Malayi. Infect. Immun. 1997, 65, 2732–2739. [Google Scholar] [CrossRef]
- Lavin, Y.; Mortha, A.; Rahman, A.; Merad, M. Regulation of Macrophage Development and Function in Peripheral Tissues. Nat. Rev. Immunol. 2015, 15, 731–744. [Google Scholar] [CrossRef]
- Sun, H.-J.; Wu, Z.-Y.; Nie, X.-W.; Bian, J.-S. Role of Hydrogen Sulfide and Polysulfides in Neurological Diseases: Focus on Protein S-Persulfidation. Curr. Neuropharmacol. 2021, 19, 868. [Google Scholar] [CrossRef]
- Wang, X.-Q.; Congyi, W.; Sun, F.; Luo, J.; Yue, T.; Wang, F.; Yang, C.; Zhang, S. The Hydrogen Sulfide Signaling in Macrophages: A Foe or Friend? Authorea Prepr. 2020. [Google Scholar] [CrossRef]
- Bhatia, M.; Gaddam, R.R. Hydrogen Sulfide in Inflammation: A Novel Mediator and Therapeutic Target. Antioxid Redox Signal 2021, 34, 1368–1377. [Google Scholar] [CrossRef]
- Fagone, P.; Mazzon, E.; Bramanti, P.; Bendtzen, K.; Nicoletti, F. Gasotransmitters and the Immune System: Mode of Action and Novel Therapeutic Targets. Eur. J. Pharmacol. 2018, 834, 92–102. [Google Scholar] [CrossRef]
- Palmieri, E.M.; McGinity, C.; Wink, D.A.; McVicar, D.W. Nitric Oxide in Macrophage Immunometabolism: Hiding in Plain Sight. Metabolites 2020, 10, 429. [Google Scholar] [CrossRef]
- Morse, D.; Choi, A.M.K. Heme Oxygenase-1: The “Emerging Molecule” Has Arrived. Am. J. Respir. Cell Mol. Biol. 2002, 27, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.D.; Snyder, S.H. Gasotransmitter Hydrogen Sulfide Signaling in Neuronal Health and Disease. Biochem. Pharmacol. 2018, 149, 101–109. [Google Scholar] [CrossRef] [PubMed]
- McBean, G.J.; Aslan, M.; Griffiths, H.R.; Torrão, R.C. Thiol Redox Homeostasis in Neurodegenerative Disease. Redox Biol. 2015, 5, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Sbodio, J.I.; Snyder, S.H.; Paul, B.D. Regulators of the Transsulfuration Pathway. Br. J. Pharmacol. 2019, 176, 583–593. [Google Scholar] [CrossRef]
- Carballal, S.; Vitvitsky, V.; Kumar, R.; Hanna, D.A.; Libiad, M.; Gupta, A.; Jones, J.W.; Banerjee, R. Hydrogen Sulfide Stimulates Lipid Biogenesis from Glutamine That Is Dependent on the Mitochondrial NAD(P)H Pool. J. Biol. Chem. 2021, 297, 100950. [Google Scholar] [CrossRef] [PubMed]
- Libiad, M.; Vitvitsky, V.; Bostelaar, T.; Bak, D.W.; Lee, H.J.; Sakamoto, N.; Fearon, E.; Lyssiotis, C.A.; Weerapana, E.; Banerjee, R. Hydrogen Sulfide Perturbs Mitochondrial Bioenergetics and Triggers Metabolic Reprogramming in Colon Cells. J. Biol. Chem. 2019, 294, 12077–12090. [Google Scholar] [CrossRef]
- Vitvitsky, V.; Kumar, R.; Libiad, M.; Maebius, A.; Landry, A.P.; Banerjee, R. The Mitochondrial NADH Pool Is Involved in Hydrogen Sulfide Signaling and Stimulation of Aerobic Glycolysis. J. Biol. Chem. 2021, 296, 100736. [Google Scholar] [CrossRef]
- Fu, M.; Zhang, W.; Wu, L.; Yang, G.; Li, H.; Wang, R. Hydrogen Sulfide (H2S) Metabolism in Mitochondria and Its Regulatory Role in Energy Production. Proc. Natl. Acad. Sci. USA 2012, 109, 2943–2948. [Google Scholar] [CrossRef]
- Sen, U.; Pushpakumar, S.B.; Amin, M.A.; Tyagi, S.C. Homocysteine in Renovascular Complications: Hydrogen Sulfide Is a Modulator and Plausible Anaerobic ATP Generator. Nitric Oxide 2014, 41, 27–37. [Google Scholar] [CrossRef]
- Freemerman, A.J.; Johnson, A.R.; Sacks, G.N.; Milner, J.J.; Kirk, E.L.; Troester, M.A.; Macintyre, A.N.; Goraksha-Hicks, P.; Rathmell, J.C.; Makowski, L. Metabolic Reprogramming of Macrophages: Glucose Transporter 1 (GLUT1)-Mediated Glucose Metabolism Drives a Proinflammatory Phenotype. J. Biol. Chem. 2014, 289, 7884–7896. [Google Scholar] [CrossRef]
- Cornwell, A.; Fedotova, S.; Cowan, S.; Badiei, A. Cystathionine γ-Lyase and Hydrogen Sulfide Modulates Glucose Transporter Glut1 Expression via NF-ΚB and PI3k/Akt in Macrophages during Inflammation. PLoS ONE 2022, 17, e0278910. [Google Scholar] [CrossRef] [PubMed]
- Badiei, A.; Gieseg, S.; Davies, S.; Othman, M.I.; Bhatia, M. LPS Up-Regulates Cystathionine γ -Lyase Gene Expression in Primary Human Macrophages via NF-ΚB/ERK Pathway. Inflamm. Allergy Drug Targets 2015, 14, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Gaddam, R.R.; Fraser, R.; Badiei, A.; Chambers, S.; Cogger, V.C.; le Couteur, D.G.; Ishii, I.; Bhatia, M. Cystathionine-Gamma-Lyase Gene Deletion Protects Mice against Inflammation and Liver Sieve Injury Following Polymicrobial Sepsis. PLoS ONE 2016, 11, e0160521. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.H.; Teng, X.; Hu, Z.J.; Tian, D.Y.; Jin, S.; Wu, Y.M. Hydrogen Sulfide Attenuated Sepsis-Induced Myocardial Dysfunction Through TLR4 Pathway and Endoplasmic Reticulum Stress. Front. Physiol. 2021, 12, 653601. [Google Scholar] [CrossRef]
- Li, J.; Li, M.; Li, L.; Ma, J.; Yao, C.; Yao, S. Hydrogen Sulfide Attenuates Ferroptosis and Stimulates Autophagy by Blocking MTOR Signaling in Sepsis-Induced Acute Lung Injury. Mol. Immunol. 2022, 141, 318–327. [Google Scholar] [CrossRef]
- Li, J.; Ma, J.; Li, M.; Tao, J.; Chen, J.; Yao, C.; Yao, S. GYY4137 Alleviates Sepsis-Induced Acute Lung Injury in Mice by Inhibiting the PDGFRβ/Akt/NF-ΚB/NLRP3 Pathway. Life Sci. 2021, 271, 119192. [Google Scholar] [CrossRef]
- Zhou, T.; Qian, H.; Zheng, N.; Lu, Q.; Han, Y. GYY4137 Ameliorates Sepsis-Induced Cardiomyopathy via NLRP3 Pathway. Biochim. Biophys Acta Mol. Basis. Dis. 2022, 1868. [Google Scholar] [CrossRef]
- Beatty, P.W.; Reed, D.J. Involvement of the Cystathionine Pathway in the Biosynthesis of Glutathione by Isolated Rat Hepatocytes. Arch. Biochem. Biophys. 1980, 204, 80–87. [Google Scholar] [CrossRef]
- Filipovic, M.R.; Zivanovic, J.; Alvarez, B.; Banerjee, R. Chemical Biology of H2S Signaling through Persulfidation. Chem. Rev. 2018, 118, 1253. [Google Scholar] [CrossRef]
- Nagahara, N.; Ito, T.; Kitamura, H.; Nishino, T. Tissue and Subcellular Distribution of Mercaptopyruvate Sulfurtransferase in the Rat: Confocal Laser Fluorescence and Immunoelectron Microscopic Studies Combined with Biochemical Analysis. Histochem. Cell Biol. 1998, 110, 243–250. [Google Scholar] [CrossRef]
- Mikami, Y.; Shibuya, N.; Kimura, Y.; Nagahara, N.; Yamada, M.; Kimura, H. Hydrogen Sulfide Protects the Retina from Light-Induced Degeneration by the Modulation of Ca2+ Influx. J. Biol. Chem. 2011, 286, 39379. [Google Scholar] [CrossRef]
- Chen, X.; Jhee, K.H.; Kruger, W.D. Production of the Neuromodulator H2S by Cystathionine Beta-Synthase via the Condensation of Cysteine and Homocysteine. J. Biol. Chem. 2004, 279, 52082–52086. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Padovani, D.; Leslie, R.A.; Chiku, T.; Banerjee, R. Relative Contributions of Cystathionine Beta-Synthase and Gamma-Cystathionase to H2S Biogenesis via Alternative Trans-Sulfuration Reactions. J. Biol. Chem. 2009, 284, 22457–22466. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Li, Q.; Du, H.P.; Wang, Y.L.; You, S.J.; Wang, F.; Xu, X.S.; Cheng, J.; Cao, Y.J.; Liu, C.F.; et al. Homocysteine Triggers Inflammatory Responses in Macrophages through Inhibiting CSE-H2S Signaling via DNA Hypermethylation of CSE Promoter. Int. J. Mol. Sci. 2015, 16, 12560–12577. [Google Scholar] [CrossRef] [PubMed]
- Bronowicka-Adamska, P.; Hutsch, T.; Gawrys-Kopczynska, M.; Maksymiuk, K.; Wróbel, M. Hydrogen Sulfide Formation in Experimental Model of Acute Pancreatitis. Acta Biochim. Pol. 2019, 66, 611–618. [Google Scholar] [CrossRef]
- Bronowicka-Adamska, P.; Jurkowska, H.; Gawda, A.; Skalska, P.; Nazimek, K.; Marcinkiewicz, J.; Wróbel, M. Expression and Activity of Hydrogen Sulfide Generating Enzymes in Murine Macrophages Stimulated with Lipopolysaccharide and Interferon-γ. Mol. Biol. Rep. 2019, 46, 2791–2798. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Vitvitsky, V.; Gendelman, H.E.; Banerjee, R. Monocyte Differentiation, Activation, and Mycobacterial Killing Are Linked to Transsulfuration-Dependent Redox Metabolism. J. Biol. Chem. 2006, 281, 38712–38720. [Google Scholar] [CrossRef]
- Taoka, S.; Lepore, B.W.; Kabil, Ö.; Ojha, S.; Ringe, D.; Banerjee, R. Human Cystathionine Beta-Synthase Is a Heme Sensor Protein. Evidence That the Redox Sensor Is Heme and Not the Vicinal Cysteines in the CXXC Motif Seen in the Crystal Structure of the Truncated Enzyme. Biochemistry 2002, 41, 10454–10461. [Google Scholar] [CrossRef]
- Taoka, S.; Banerjee, R. Characterization of NO Binding to Human Cystathionine β-Synthase: Possible Implications of the Effects of CO and NO Binding to the Human Enzyme. J. Inorg. Biochem. 2001, 87, 245–251. [Google Scholar] [CrossRef]
- Ereño-Orbea, J.; Majtan, T.; Oyenarte, I.; Kraus, J.P.; Martínez-Cruza, L.A. Structural Basis of Regulation and Oligomerization of Human Cystathionine β-Synthase, the Central Enzyme of Transsulfuration. Proc. Natl. Acad. Sci. USA 2013, 110, E3790–E3799. [Google Scholar] [CrossRef]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS Domains Form Energy-Sensing Modules Whose Binding of Adenosine Ligands Is Disrupted by Disease Mutations. J. Clin. Investig. 2004, 113, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.B.; Colaço, H.G.; Sarti, P.; Leandro, P.; Giuffrè, A. S-Adenosyl-l-Methionine Modulates CO and NO• Binding to the Human H2S-Generating Enzyme Cystathionine β-Synthase. J. Biol. Chem. 2016, 291, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Vicente, J.B.; Malagrinò, F.; Arese, M.; Forte, E.; Sarti, P.; Giuffrè, A. Bioenergetic Relevance of Hydrogen Sulfide and the Interplay between Gasotransmitters at Human Cystathionine β-Synthase. Biochim. Biophys. Acta 2016, 1857, 1127–1138. [Google Scholar] [CrossRef]
- Kabil, O.; Yadav, V.; Banerjee, R. Heme-Dependent Metabolite Switching Regulates H2S Synthesis in Response to Endoplasmic Reticulum (ER) Stress. J. Biol. Chem. 2016, 291, 16418–16423. [Google Scholar] [CrossRef]
- Mosharov, E.; Cranford, M.R.; Banerjee, R. The Quantitatively Important Relationship between Homocysteine Metabolism and Glutathione Synthesis by the Transsulfuration Pathway and Its Regulation by Redox Changes. Biochemistry 2000, 39, 13005–13011. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, G.; Tang, G.; Wu, L.; Wang, R. Rat Pancreatic Level of Cystathionine γ-Lyase Is Regulated by Glucose Level via Specificity Protein 1 (SP1) Phosphorylation. Diabetologia 2011, 54, 2615–2625. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Pei, Y.; Teng, H.; Cao, Q.; Wang, R. Specificity Protein-1 as a Critical Regulator of Human Cystathionine Gamma-Lyase in Smooth Muscle Cells. J. Biol. Chem. 2011, 286, 26450–26460. [Google Scholar] [CrossRef]
- Badiei, A.; Muniraj, N.; Chambers, S.; Bhatia, M. Inhibition of Hydrogen Sulfide Production by Gene Silencing Attenuates Inflammatory Activity by Downregulation of NF-ΚB and MAP Kinase Activity in LPS-Activated RAW 264.7 Cells. Biomed Res. Int. 2014, 2014, 848570. [Google Scholar] [CrossRef]
- Zheng, Y.; Luo, N.; Mu, D.; Jiang, P.; Liu, R.; Sun, H.; Xiong, S.; Liu, X.; Wang, L.; Chu, Y. Lipopolysaccharide Regulates Biosynthesis of Cystathionine γ-Lyase and Hydrogen Sulfide through Toll-like Receptor-4/P38 and Toll-like Receptor-4/NF-ΚB Pathways in Macrophages. Vitr. Cell Dev. Biol. Anim. 2013, 49, 679–688. [Google Scholar] [CrossRef]
- Mishanina, T.V.; Libiad, M.; Banerjee, R. Biogenesis of Reactive Sulfur Species for Signaling by Hydrogen Sulfide Oxidation Pathways. Nat. Chem. Biol. 2015, 11, 457–464. [Google Scholar] [CrossRef]
- Gao, X.H.; Krokowski, D.; Guan, B.J.; Bederman, I.; Majumder, M.; Parisien, M.; Diatchenko, L.; Kabil, O.; Willard, B.; Banerjee, R.; et al. Quantitative H2S-Mediated Protein Sulfhydration Reveals Metabolic Reprogramming during the Integrated Stress Response. Elife 2015, 4, e10067. [Google Scholar] [CrossRef] [PubMed]
- Greiner, R.; Pálinkás, Z.; Bäsell, K.; Becher, D.; Antelmann, H.; Nagy, P.; Dick, T.P. Polysulfides Link H2S to Protein Thiol Oxidation. Antioxid Redox Signal 2013, 19, 1749. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Mikami, Y.; Osumi, K.; Tsugane, M.; Oka, J.I.; Kimura, H. Polysulfides Are Possible H2S-Derived Signaling Molecules in Rat Brain. FASEB J. 2013, 27, 2451–2457. [Google Scholar] [CrossRef]
- Zhao, Z.Z.; Wang, Z.; Li, G.H.; Wang, R.; Tan, J.M.; Cao, X.; Suo, R.; Jiang, Z.S. Hydrogen Sulfide Inhibits Macrophage-Derived Foam Cell Formation. Exp. Biol. Med. 2011, 236, 169–176. [Google Scholar] [CrossRef]
- Shimizu, T.; Ida, T.; Antelo, G.T.; Ihara, Y.; Fakhoury, J.N.; Masuda, S.; Giedroc, D.P.; Akaike, T.; Capdevila, D.A.; Masuda, T. Polysulfide Metabolizing Enzymes Influence SqrR-Mediated Sulfide-Induced Transcription by Impacting Intracellular Polysulfide Dynamics. PNAS Nexus 2023, 2, 5–6. [Google Scholar] [CrossRef]
- Akaike, T.; Ida, T.; Wei, F.Y.; Nishida, M.; Kumagai, Y.; Alam, M.M.; Ihara, H.; Sawa, T.; Matsunaga, T.; Kasamatsu, S.; et al. Cysteinyl-TRNA Synthetase Governs Cysteine Polysulfidation and Mitochondrial Bioenergetics. Nat. Commun. 2017, 8, 1177. [Google Scholar] [CrossRef]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive Cysteine Persulfides and S-Polythiolation Regulate Oxidative Stress and Redox Signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [PubMed]
- Combi, Z.; Potor, L.; Nagy, P.; Sikura, K.É.; Ditrói, T.; Jurányi, E.P.; Galambos, K.; Szerafin, T.; Gergely, P.; Whiteman, M.; et al. Hydrogen Sulfide as an Anti-Calcification Stratagem in Human Aortic Valve: Altered Biogenesis and Mitochondrial Metabolism of H2S Lead to H2S Deficiency in Calcific Aortic Valve Disease. Redox Biol. 2023, 60, 102629. [Google Scholar] [CrossRef]
- Kimura, H. Signalling by Hydrogen Sulfide and Polysulfides via Protein S-Sulfuration. Br. J. Pharmacol. 2020, 177, 720–733. [Google Scholar] [CrossRef]
- Zivanovic, J.; Kouroussis, E.; Kohl, J.B.; Adhikari, B.; Bursac, B.; Schott-Roux, S.; Petrovic, D.; Miljkovic, J.L.; Thomas-Lopez, D.; Jung, Y.; et al. Selective Persulfide Detection Reveals Evolutionarily Conserved Anti-Aging Effects of S-Sulfhydration. Cell Metab. 2019, 30, 1152. [Google Scholar] [CrossRef]
- Trummer, M.; Galardon, E.; Mayer, B.; Steiner, G.; Stamm, T.; Kloesch, B. Polysulfides Derived from the Hydrogen Sulfide and Persulfide Donor P* Inhibit IL-1β-Mediated Inducible Nitric Oxide Synthase Signaling in ATDC5 Cells: Are CCAAT/Enhancer-Binding Proteins β and δ Involved in the Anti-Inflammatory Effects of Hydrogen Sulfide and Polysulfides? Nitric. Oxide 2022, 129, 41–52. [Google Scholar] [CrossRef]
- Lee, Z.W.; Low, Y.L.; Huang, S.; Wang, T.; Deng, L.W. The Cystathionine γ-Lyase/Hydrogen Sulfide System Maintains Cellular Glutathione Status. Biochem. J. 2014, 460, 425–435. [Google Scholar] [CrossRef] [PubMed]
- Ribas, V.; García-Ruiz, C.; Fernández-Checa, J.C. Glutathione and Mitochondria. Front. Pharmacol. 2014, 5 JUL, 151. [Google Scholar] [CrossRef]
- Lingappan, K. NF-ΚB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81. [Google Scholar] [CrossRef]
- Das, K.C.; Lewis-Molock, Y.; White, C.W. Activation of NF-Kappa B and Elevation of MnSOD Gene Expression by Thiol Reducing Agents in Lung Adenocarcinoma (A549) Cells. Am. J. Physiol. 1995, 269, L588–L602. [Google Scholar] [CrossRef] [PubMed]
- Rojo, A.I.; Salinas, M.; Martín, D.; Perona, R.; Cuadrado, A. Regulation of Cu/Zn-Superoxide Dismutase Expression via the Phosphatidylinositol 3 Kinase/Akt Pathway and Nuclear Factor-KappaB. J. Neurosci. 2004, 24, 7324–7334. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, J.; Jenner, R.G.; Murray, H.L.; Gerber, G.K.; Gifford, D.K.; Young, R.A. Coordinated Binding of NF-KappaB Family Members in the Response of Human Cells to Lipopolysaccharide. Proc. Natl. Acad. Sci. USA 2006, 103, 5899–5904. [Google Scholar] [CrossRef]
- Pompella, A.; Visvikis, A.; Paolicchi, A.; de Tata, V.; Casini, A.F. The Changing Faces of Glutathione, a Cellular Protagonist. Biochem. Pharmacol. 2003, 66, 1499–1503. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Goto, Y.I.; Kimura, H. Hydrogen Sulfide Increases Glutathione Production and Suppresses Oxidative Stress in Mitochondria. Antioxid Redox Signal 2010, 12, 1–13. [Google Scholar] [CrossRef]
- Kimura, H. Signaling Molecules: Hydrogen Sulfide and Polysulfide. Antioxid Redox Signal 2015, 22, 362. [Google Scholar] [CrossRef]
- Kimura, Y.; Kimura, H. Hydrogen Sulfide Protects Neurons from Oxidative Stress. FASEB J. 2004, 18, 1165–1167. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Luo, J.H.; Yue, T.T.; Wang, F.X.; Yang, C.L.; Zhang, S.; Wang, X.Q.; Wang, C.Y. The Role of Hydrogen Sulphide Signalling in Macrophage Activation. Immunology 2021, 162, 3. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.H.; Liu, F.; Chen, Y.; Zhu, Y.C. Hydrogen Sulfide Decreases the Levels of ROS by Inhibiting Mitochondrial Complex IV and Increasing SOD Activities in Cardiomyocytes under Ischemia/Reperfusion. Biochem. Biophys. Res. Commun. 2012, 421, 164–169. [Google Scholar] [CrossRef]
- Zhang, H.; Du, J.; Huang, Y.; Tang, C.; Jin, H. Hydrogen Sulfide Regulates Macrophage Function in Cardiovascular Diseases. Antioxid Redox Signal 2023, 38, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Vitvitsky, V.; Kabil, O.; Banerjee, R. High Turnover Rates for Hydrogen Sulfide Allow for Rapid Regulation of Its Tissue Concentrations. Antioxid Redox Signal 2012, 17, 22–31. [Google Scholar] [CrossRef]
- Goubern, M.; Andriamihaja, M.; Nübel, T.; Blachier, F.; Bouillaud, F. Sulfide, the First Inorganic Substrate for Human Cells. FASEB J. 2007, 21, 1699–1706. [Google Scholar] [CrossRef] [PubMed]
- Lagoutte, E.; Mimoun, S.; Andriamihaja, M.; Chaumontet, C.; Blachier, F.; Bouillaud, F. Oxidation of Hydrogen Sulfide Remains a Priority in Mammalian Cells and Causes Reverse Electron Transfer in Colonocytes. Biochim. Biophys. Acta 2010, 1797, 1500–1511. [Google Scholar] [CrossRef]
- Kumar, R.; Landry, A.P.; Guha, A.; Vitvitsky, V.; Lee, H.J.; Seike, K.; Reddy, P.; Lyssiotis, C.A.; Banerjee, R. A Redox Cycle with Complex II Promotes Sulfide Quinone Oxidoreductase Dependent H2S Oxidation. bioRxiv 2021. bioRxiv:2021.09.08.459449. [Google Scholar] [CrossRef]
- Furne, J.; Saeed, A.; Levitt, M.D. Whole Tissue Hydrogen Sulfide Concentrations Are Orders of Magnitude Lower than Presently Accepted Values. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1479–R1485. [Google Scholar] [CrossRef]
- Levitt, M.D.; Abdel-Rehim, M.S.; Furne, J. Free and Acid-Labile Hydrogen Sulfide Concentrations in Mouse Tissues: Anomalously High Free Hydrogen Sulfide in Aortic Tissue. Antioxid Redox Signal 2011, 15, 373–378. [Google Scholar] [CrossRef]
- Zhou, T.; Liu, W.; Lai, H.; Liu, Y.; Su, W.; Xu, Z. Hydrogen Sulfide Promotes Osteogenesis by Modulating Macrophage Polarization. Int. Immunopharmacol. 2023, 115, 109564. [Google Scholar] [CrossRef] [PubMed]
- Geng, W.; Liu, X.; Tao, B.; He, Y.; Li, K.; Gao, P.; Feng, Q.; Zhao, P.; Luo, Z.; Cai, K. Nitric Oxide Scavenging and Hydrogen Sulfide Production Synergistically Treat Rheumatoid Arthritis. Adv. Healthc. Mater 2023, 12, 2202380. [Google Scholar] [CrossRef] [PubMed]
- Mulero, M.C.; Huxford, T.; Ghosh, G. NF-ΚB, IκB, and IKK: Integral Components of Immune System Signaling. Adv. Exp. Med. Biol. 2019, 1172, 207–226. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.C.; Yeh, W.C.; Ohashi, P.S. LPS/TLR4 Signal Transduction Pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Everts, B.; Amiel, E.; van der Windt, G.J.W.; Freitas, T.C.; Chott, R.; Yarasheski, K.E.; Pearce, E.L.; Pearce, E.J. Commitment to Glycolysis Sustains Survival of NO-Producing Inflammatory Dendritic Cells. Blood 2012, 120, 1422–1431. [Google Scholar] [CrossRef]
- Shearer, J.D.; Richards, J.R.; Mills, C.D.; Caldwell, M.D. Differential Regulation of Macrophage Arginine Metabolism: A Proposed Role in Wound Healing. Am. J. Physiol. 1997, 272, E181–E190. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network Integration of Parallel Metabolic and Transcriptional Data Reveals Metabolic Modules That Regulate Macrophage Polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef]
- Kelly, B.; O’Neill, L.A.J. Metabolic Reprogramming in Macrophages and Dendritic Cells in Innate Immunity. Cell Res. 2015, 25, 771–784. [Google Scholar] [CrossRef]
- Tannahill, G.M.; Curtis, A.M.; Adamik, J.; Palsson-Mcdermott, E.M.; McGettrick, A.F.; Goel, G.; Frezza, C.; Bernard, N.J.; Kelly, B.; Foley, N.H.; et al. Succinate Is an Inflammatory Signal That Induces IL-1β through HIF-1α. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.C.; Thompson, C.B.; Gottlieb, E. Succinate Links TCA Cycle Dysfunction to Oncogenesis by Inhibiting HIF-α Prolyl Hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef]
- Cramer, T.; Yamanishi, Y.; Clausen, B.E.; Förster, I.; Pawlinski, R.; Mackman, N.; Haase, V.H.; Jaenisch, R.; Corr, M.; Nizet, V.; et al. HIF-1α Is Essential for Myeloid Cell-Mediated Inflammation. Cell 2003, 112, 645–657. [Google Scholar] [CrossRef] [PubMed]
- Rius, J.; Guma, M.; Schachtrup, C.; Akassoglou, K.; Zinkernagel, A.S.; Nizet, V.; Johnson, R.S.; Haddad, G.G.; Karin, M. NF-KappaB Links Innate Immunity to the Hypoxic Response through Transcriptional Regulation of HIF-1alpha. Nature 2008, 453, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Obaid, M.; Udden, S.M.N.; Alluri, P.; Mandal, S.S. LncRNA HOTAIR Regulates Glucose Transporter Glut1 Expression and Glucose Uptake in Macrophages during Inflammation. Sci. Rep. 2021, 11, 232. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, F.R.; Kelher, M.R.; Moore, E.E.; McLaughlin, N.J.D.; Banerjee, A.; Silliman, C.C. Structural Organization of the Neutrophil NADPH Oxidase: Phosphorylation and Translocation during Priming and Activation. J. Leukoc. Biol. 2005, 78, 1025–1042. [Google Scholar] [CrossRef] [PubMed]
- Kane, L.P.; Shapiro, V.S.; Stokoe, D.; Weiss, A. Induction of NF-KappaB by the Akt/PKB Kinase. Curr. Biol. 1999, 9, 601–604. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt Signaling Pathway in Macrophage Activation and M1/M2 Polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef]
- Luyendyk, J.P.; Schabbauer, G.A.; Tencati, M.; Holscher, T.; Pawlinski, R.; Mackman, N. Genetic Analysis of the Role of the PI3K-Akt Pathway in Lipopolysaccharide-Induced Cytokine and Tissue Factor Gene Expression in Monocytes/Macrophages. J. Immunol. 2008, 180, 4218. [Google Scholar] [CrossRef]
- Fukao, T.; Koyasu, S. PI3K and Negative Regulation of TLR Signaling. Trends Immunol. 2003, 24, 358–363. [Google Scholar] [CrossRef]
- Cheng, S.C.; Quintin, J.; Cramer, R.A.; Shepardson, K.M.; Saeed, S.; Kumar, V.; Giamarellos-Bourboulis, E.J.; Martens, J.H.A.; Rao, N.A.; Aghajanirefah, A.; et al. MTOR- and HIF-1α-Mediated Aerobic Glycolysis as Metabolic Basis for Trained Immunity. Science 2014, 345, 1250684. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Aksoylar, H.I.; Horng, T. Control of Macrophage Metabolism and Activation by MTOR and Akt Signaling. Semin. Immunol. 2015, 27, 286. [Google Scholar] [CrossRef]
- Joshi, S.; Singh, A.R.; Zulcic, M.; Durden, D.L. A Macrophage-Dominant PI3K Isoform Controls Hypoxia-Induced HIF1α and HIF2α Stability and Tumor Growth, Angiogenesis, and Metastasis. Mol. Cancer Res. 2014, 12, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wang, Y.; Li, Y.; Yu, Q.; Jin, X.; Wang, X.; Jia, A.; Hu, Y.; Han, L.; Wang, J.; et al. HIF1α-Dependent Glycolysis Promotes Macrophage Functional Activities in Protecting against Bacterial and Fungal Infection. Sci. Rep. 2018, 8, 3603. [Google Scholar] [CrossRef] [PubMed]
- Landry, A.P.; Ballou, D.P.; Banerjee, R. Hydrogen Sulfide Oxidation by Sulfide Quinone Oxidoreductase. Chembiochem 2021, 22, 949–960. [Google Scholar] [CrossRef]
- Lohninger, L.; Tomasova, L.; Praschberger, M.; Hintersteininger, M.; Erker, T.; Gmeiner, B.M.K.; Laggner, H. Hydrogen Sulphide Induces HIF-1α and Nrf2 in THP-1 Macrophages. Biochimie 2015, 112, 187–195. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornwell, A.; Badiei, A. From Gasotransmitter to Immunomodulator: The Emerging Role of Hydrogen Sulfide in Macrophage Biology. Antioxidants 2023, 12, 935. https://doi.org/10.3390/antiox12040935
Cornwell A, Badiei A. From Gasotransmitter to Immunomodulator: The Emerging Role of Hydrogen Sulfide in Macrophage Biology. Antioxidants. 2023; 12(4):935. https://doi.org/10.3390/antiox12040935
Chicago/Turabian StyleCornwell, Alex, and Alireza Badiei. 2023. "From Gasotransmitter to Immunomodulator: The Emerging Role of Hydrogen Sulfide in Macrophage Biology" Antioxidants 12, no. 4: 935. https://doi.org/10.3390/antiox12040935
APA StyleCornwell, A., & Badiei, A. (2023). From Gasotransmitter to Immunomodulator: The Emerging Role of Hydrogen Sulfide in Macrophage Biology. Antioxidants, 12(4), 935. https://doi.org/10.3390/antiox12040935