
Serelaxin Protects H9c2 Cardiac Myoblasts against Hypoxia and Reoxygenation-Induced Damage through Activation of AMP Kinase/Sirtuin1: Further Insight into the Molecular Mechanisms of the Cardioprotection of This Hormone




Abstract
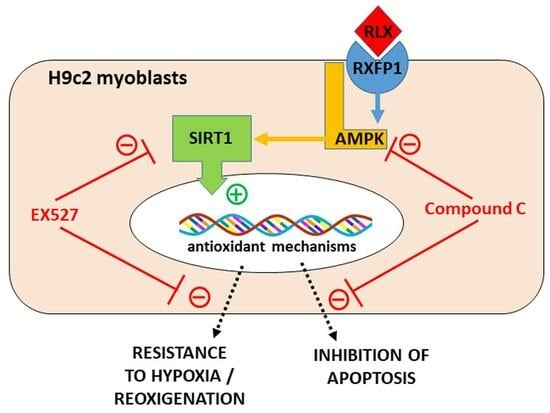
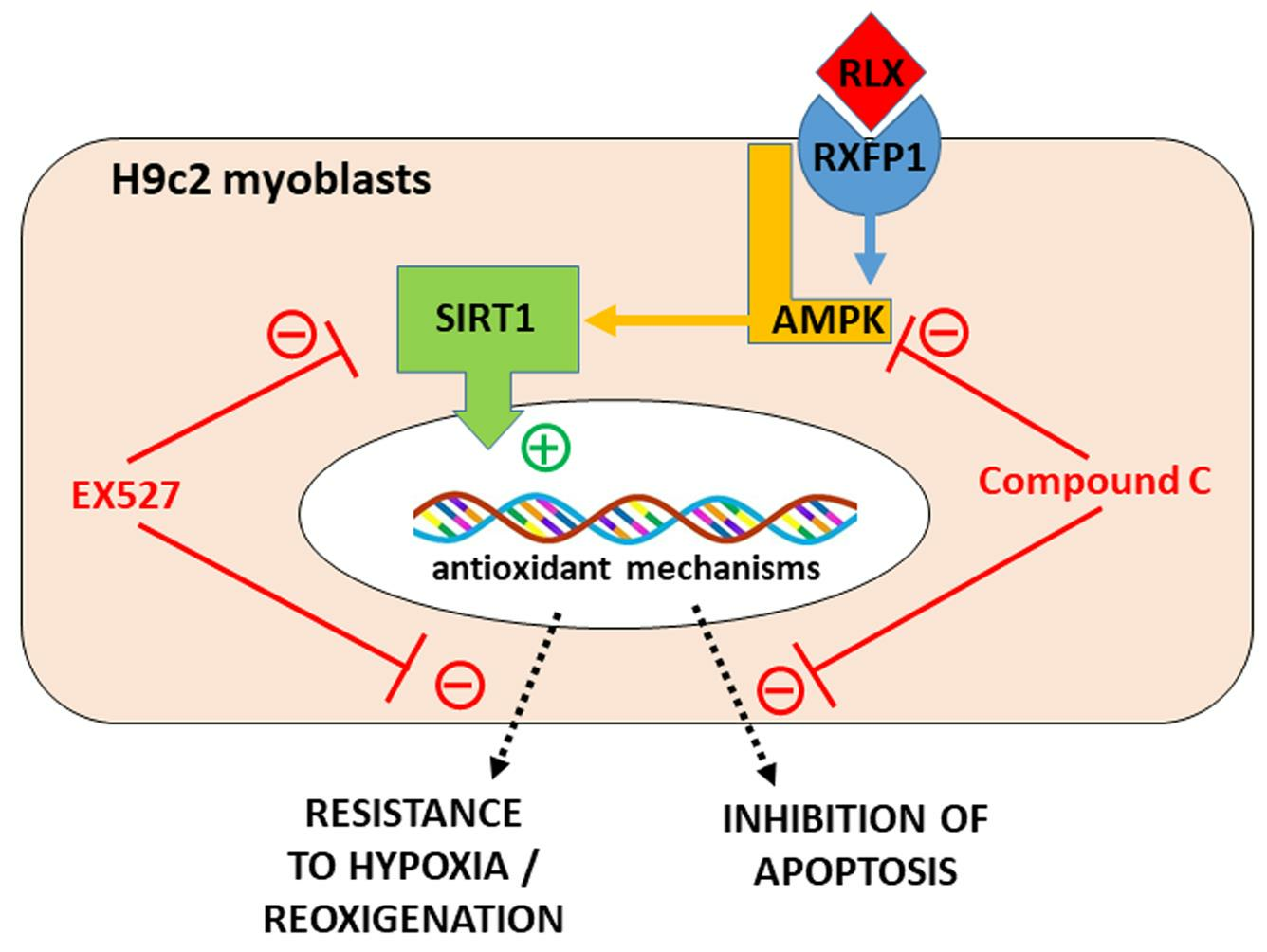
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Treatments
2.2. Western Blotting
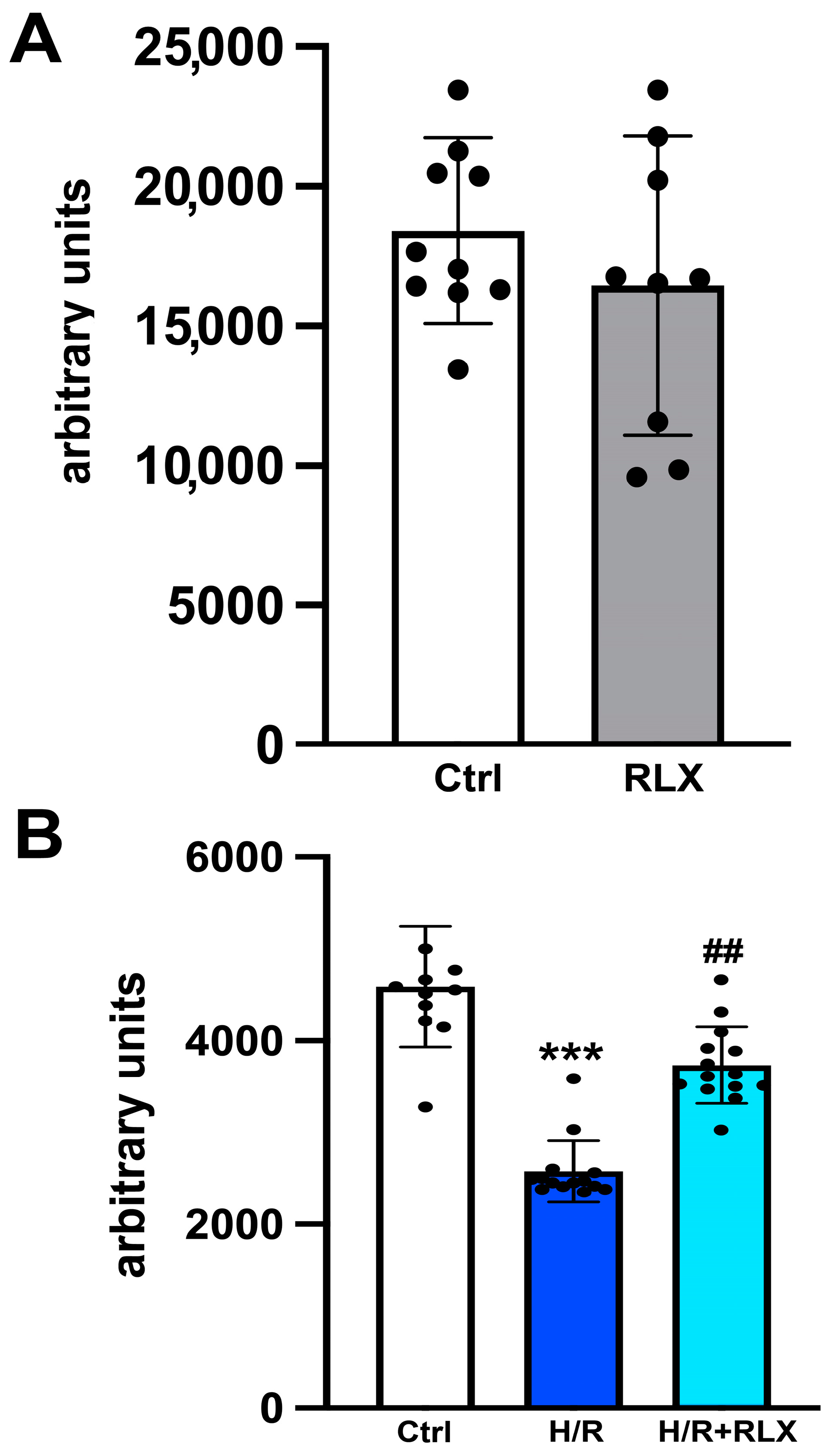
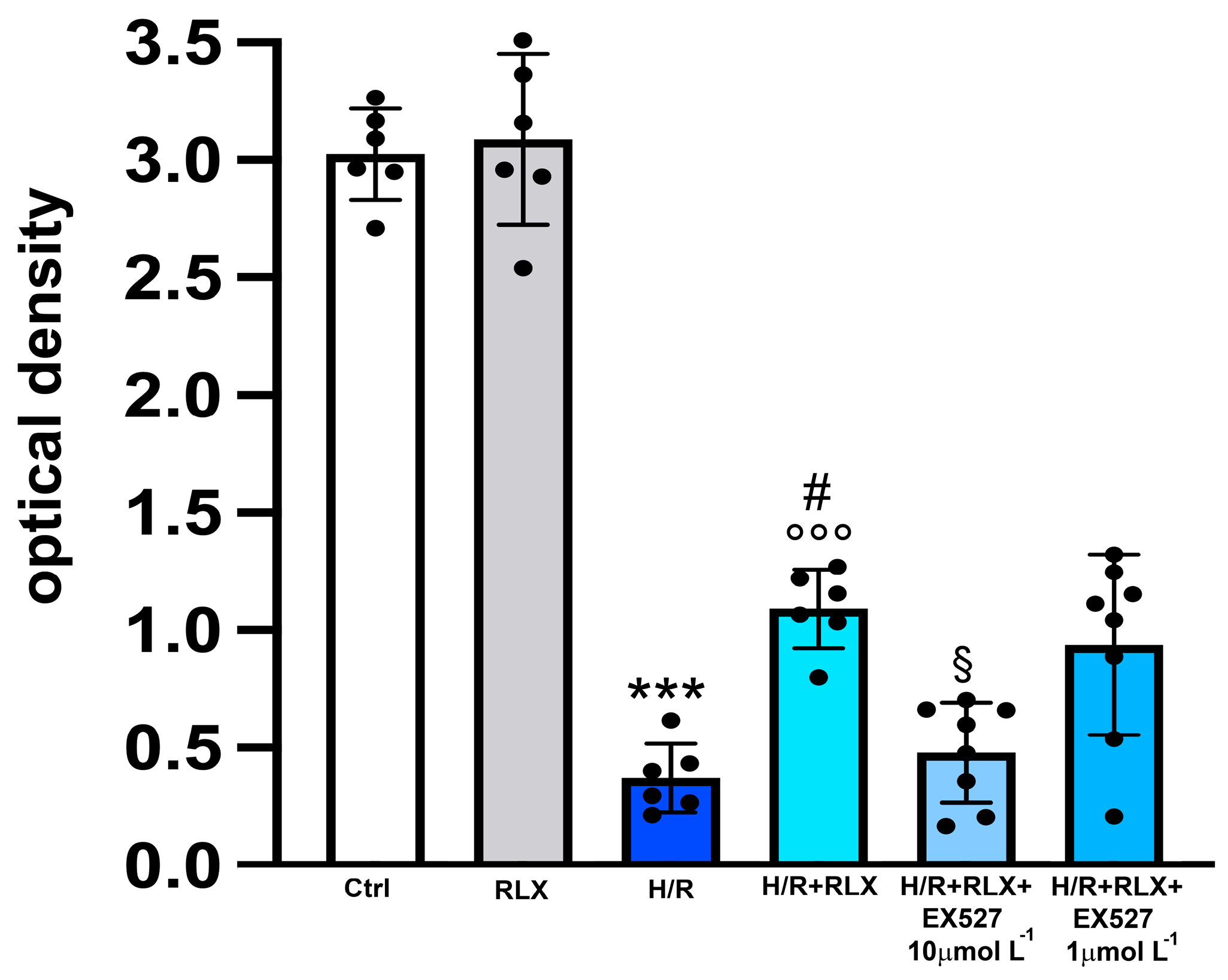
2.3. SIRT1 Activity
2.4. Mitochondrial Activity
2.5. Statistical Analysis
3. Results
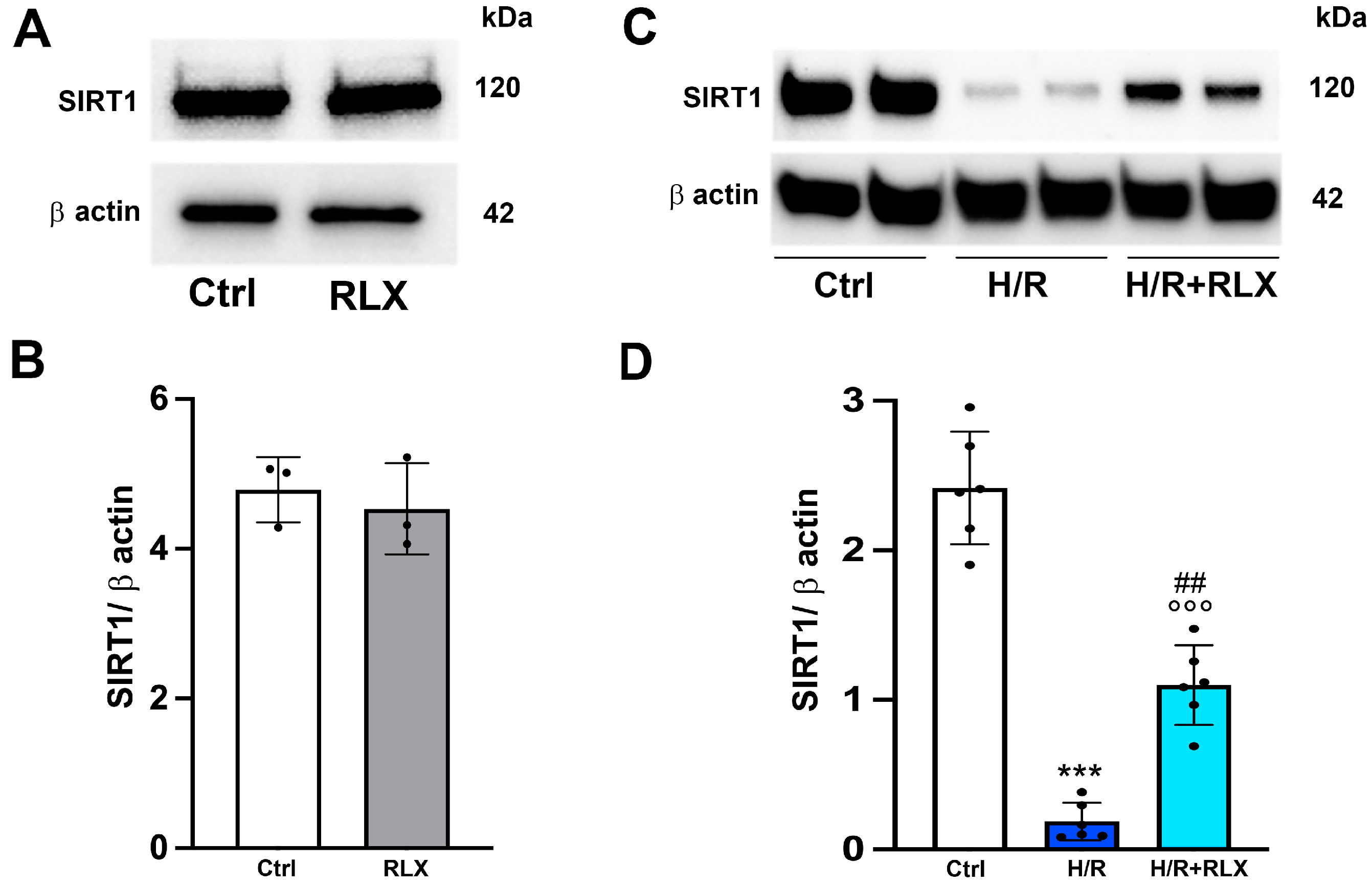
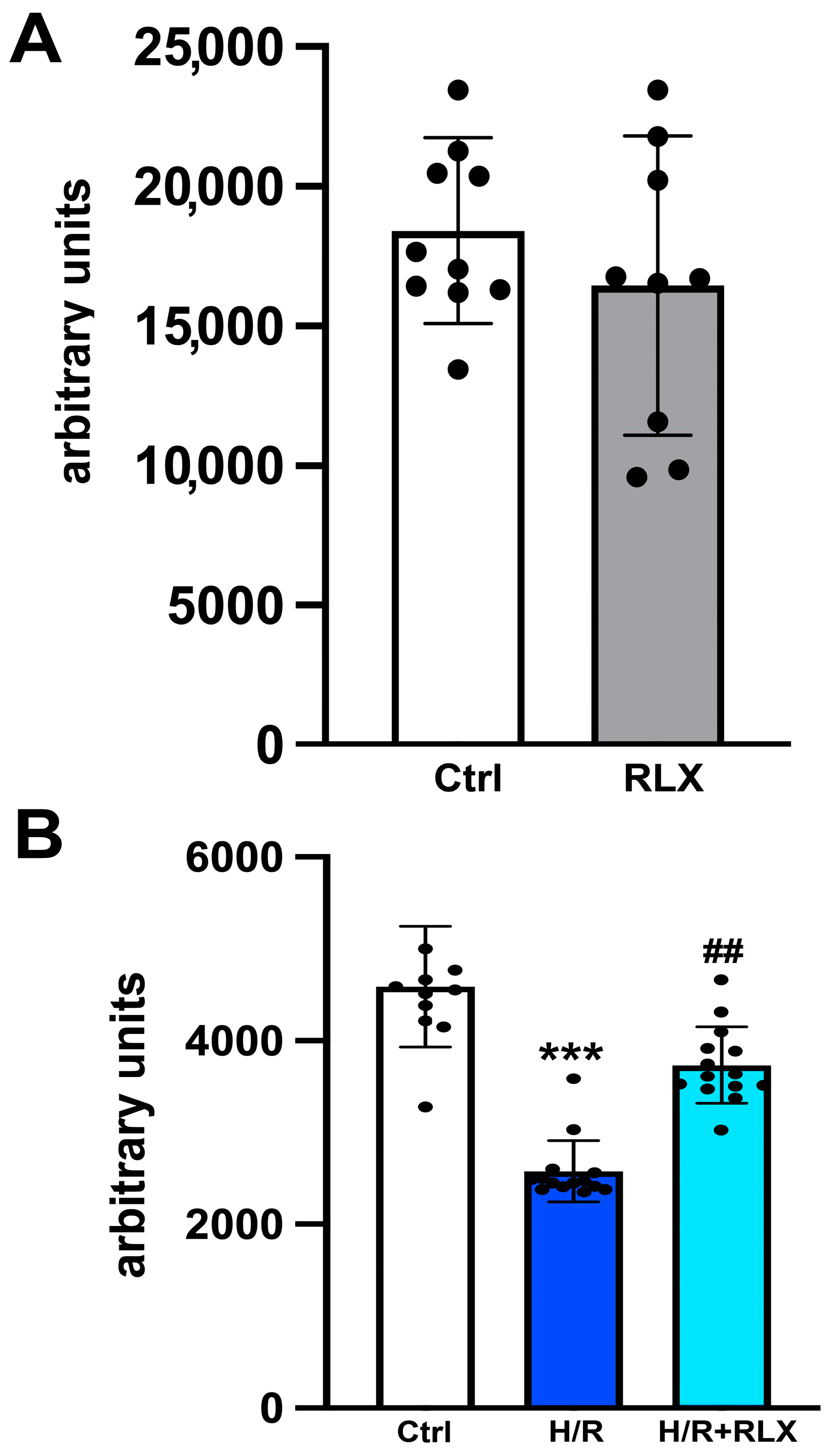
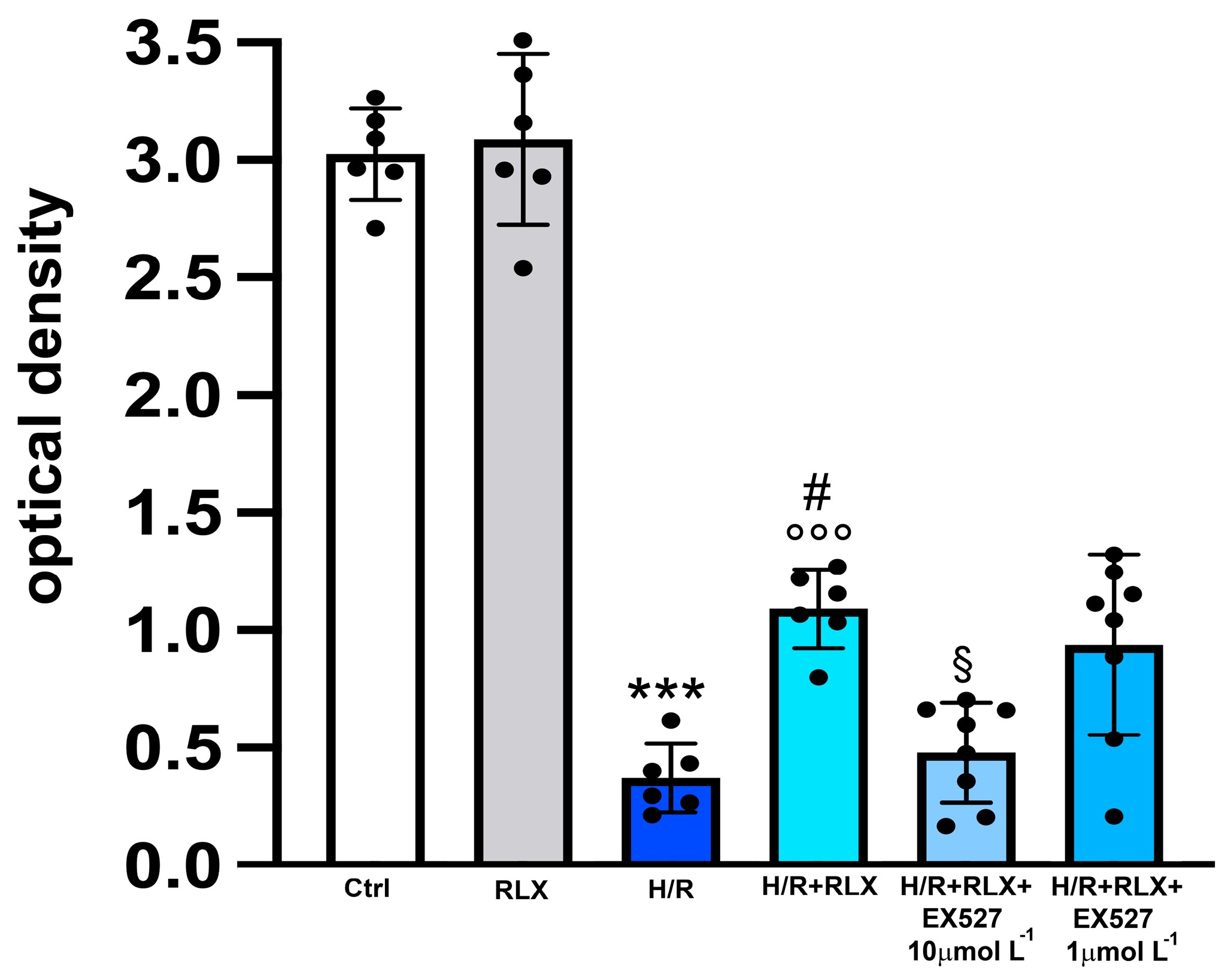
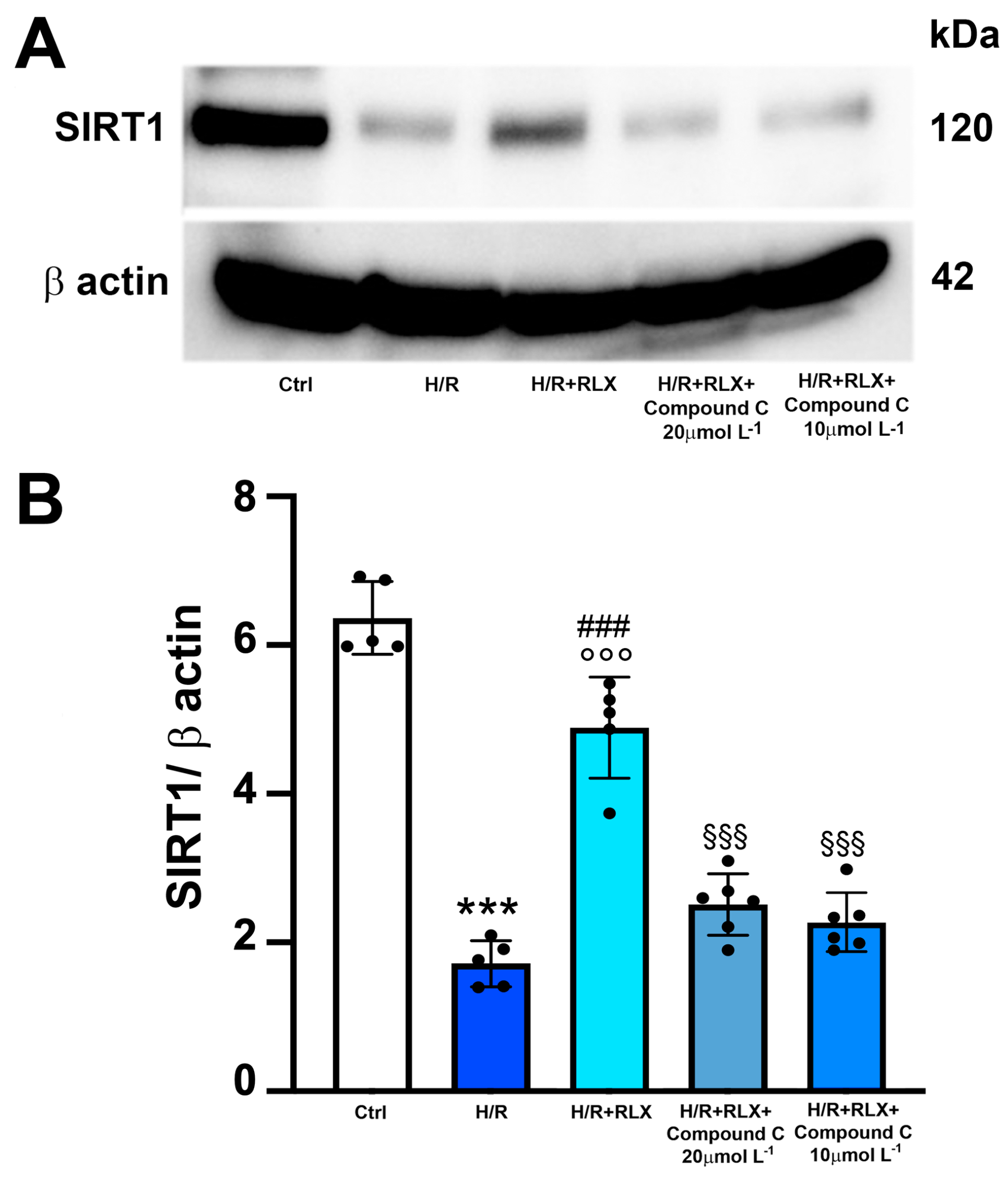
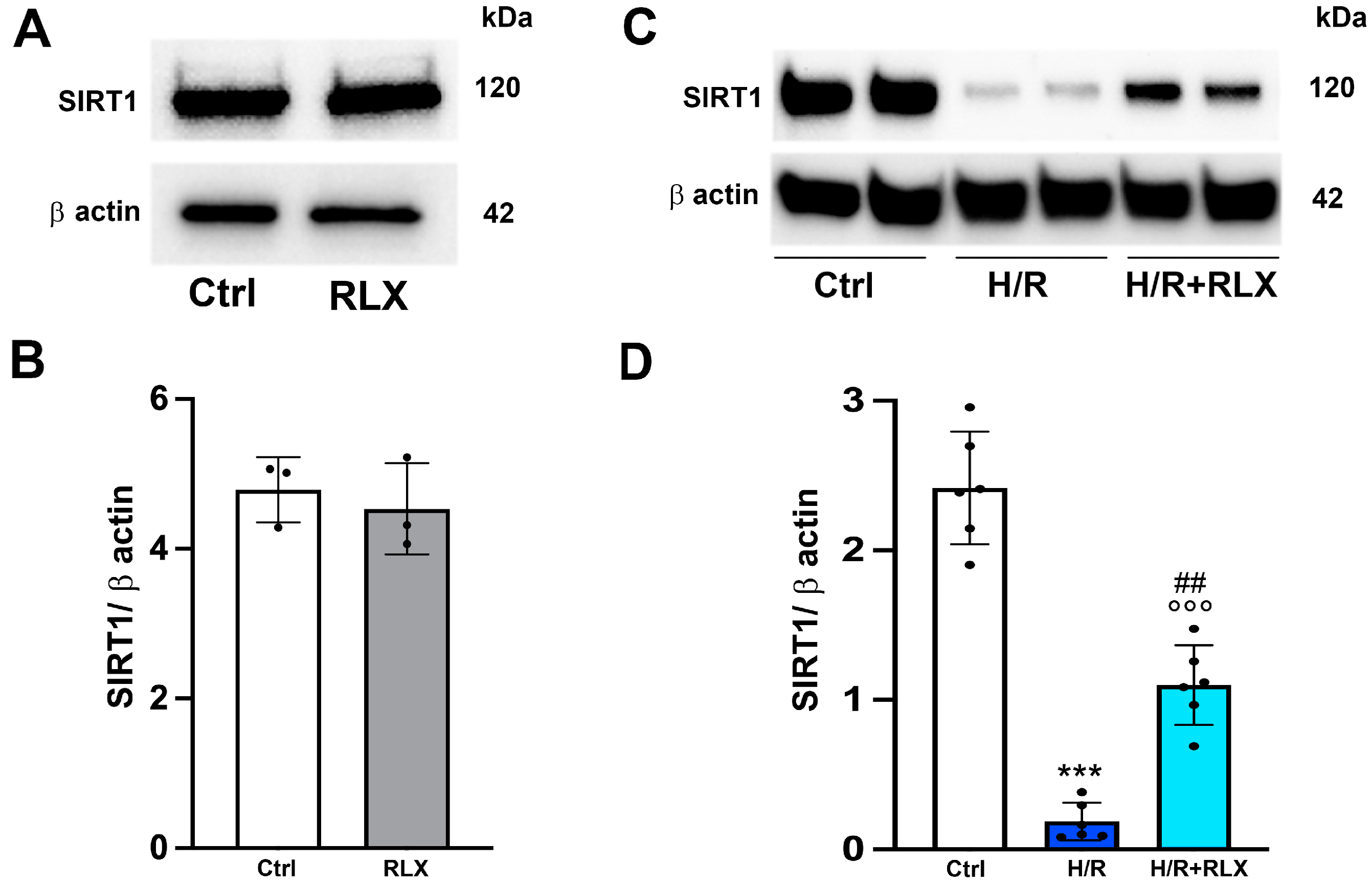
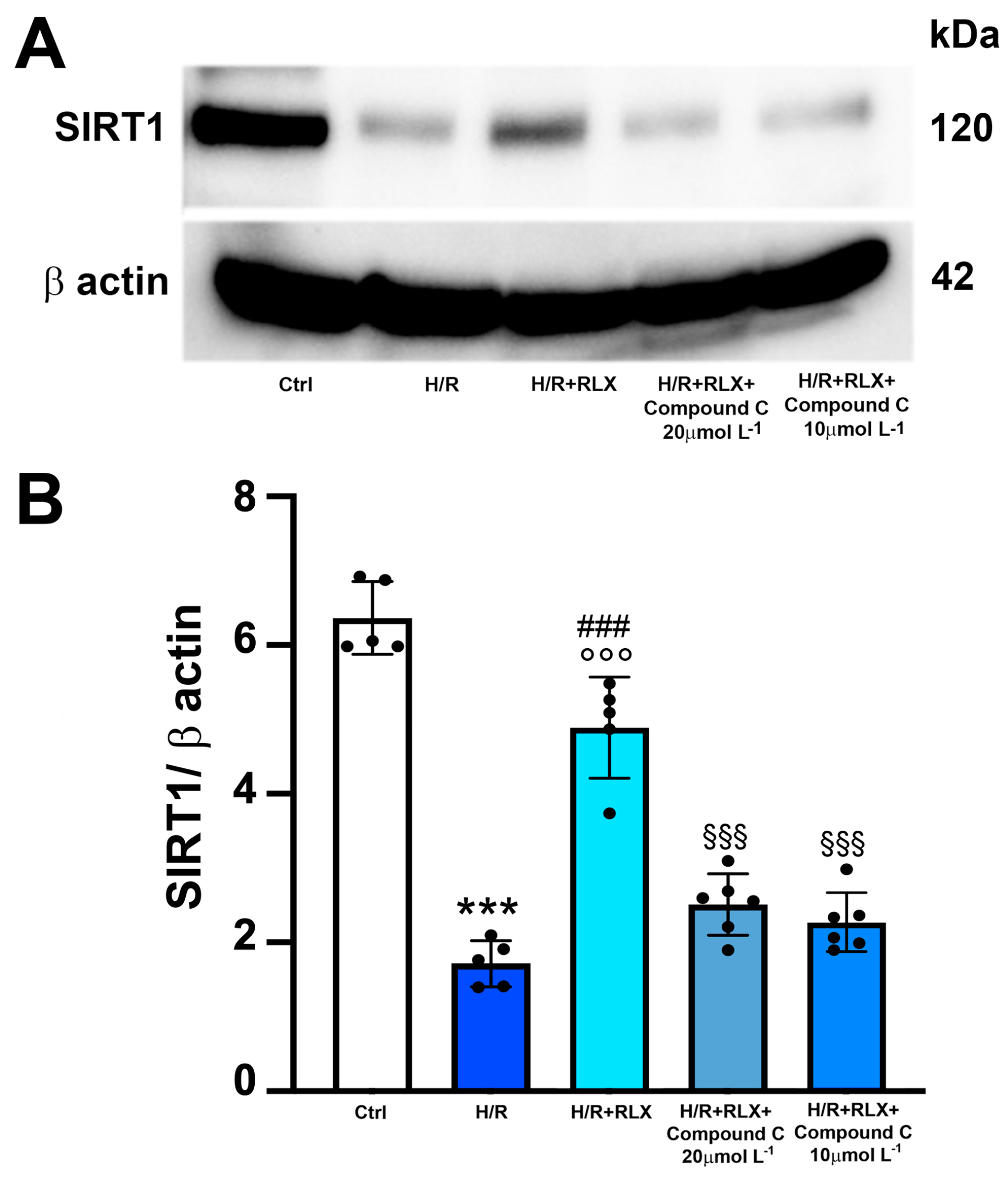
3.1. RLX Activates SIRT1 Signaling in H9c2 Cells Subjected to H/R
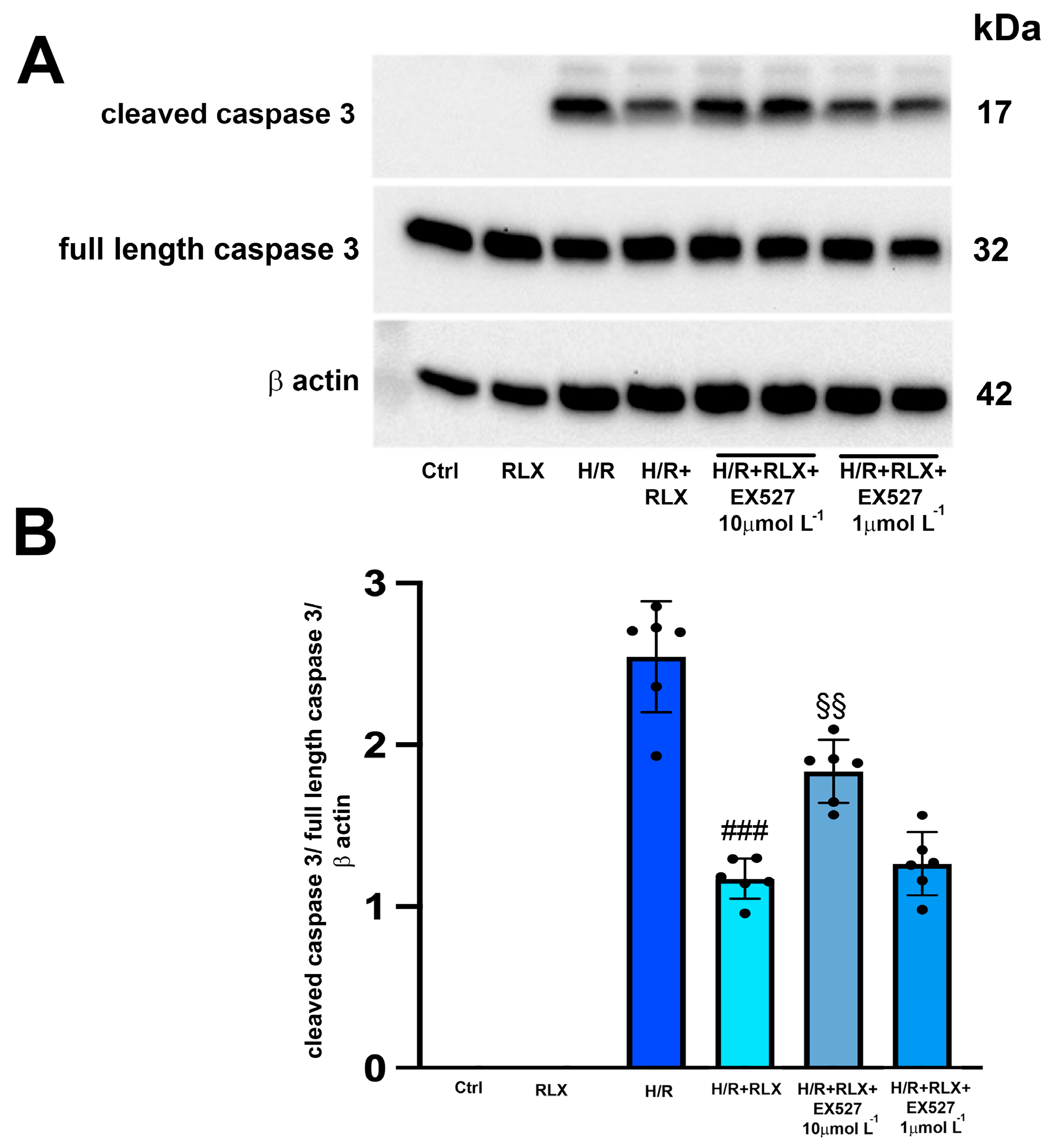
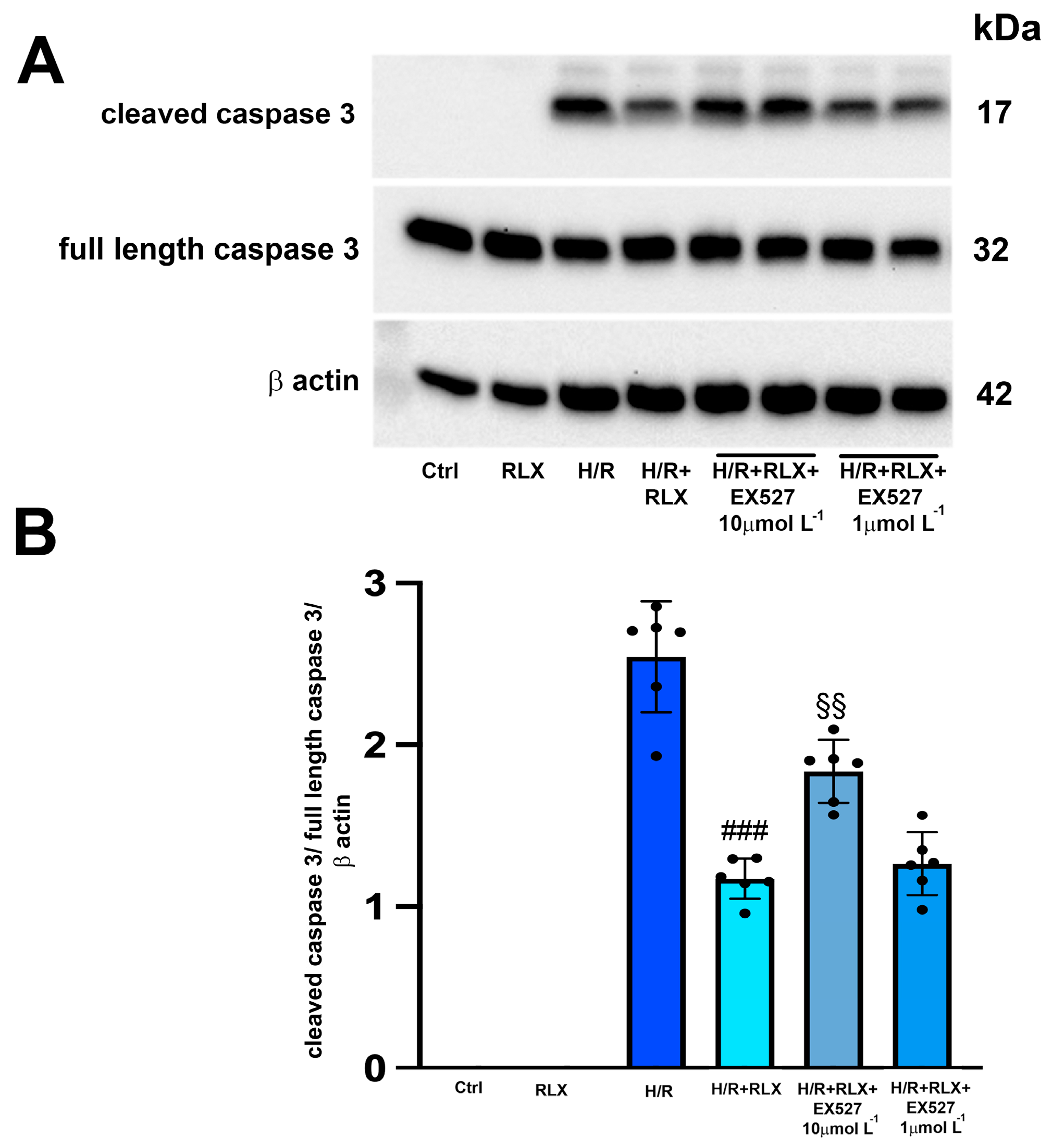
3.2. RLX Induces Cell Protection through Stimulation of SIRT1 Signaling
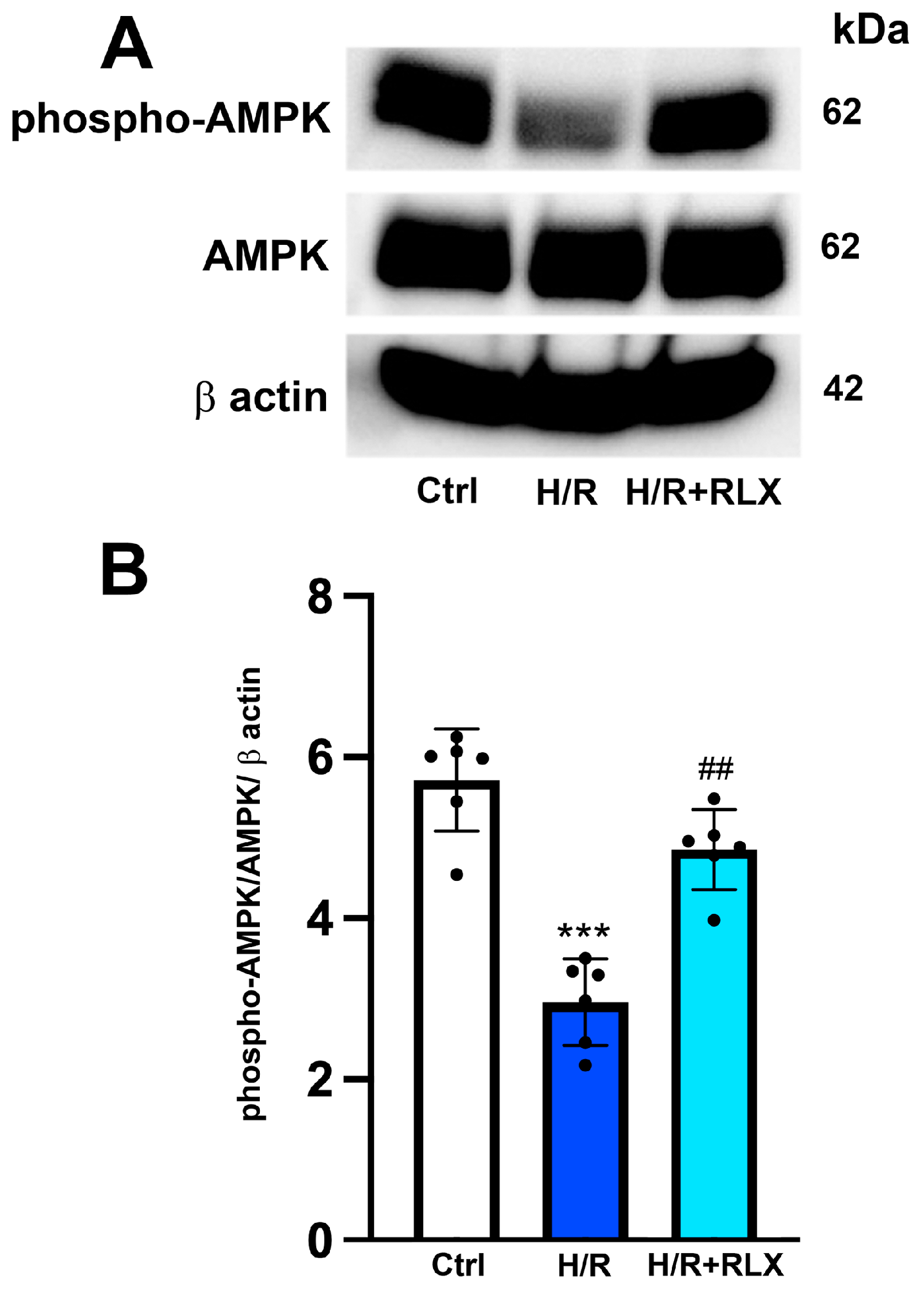
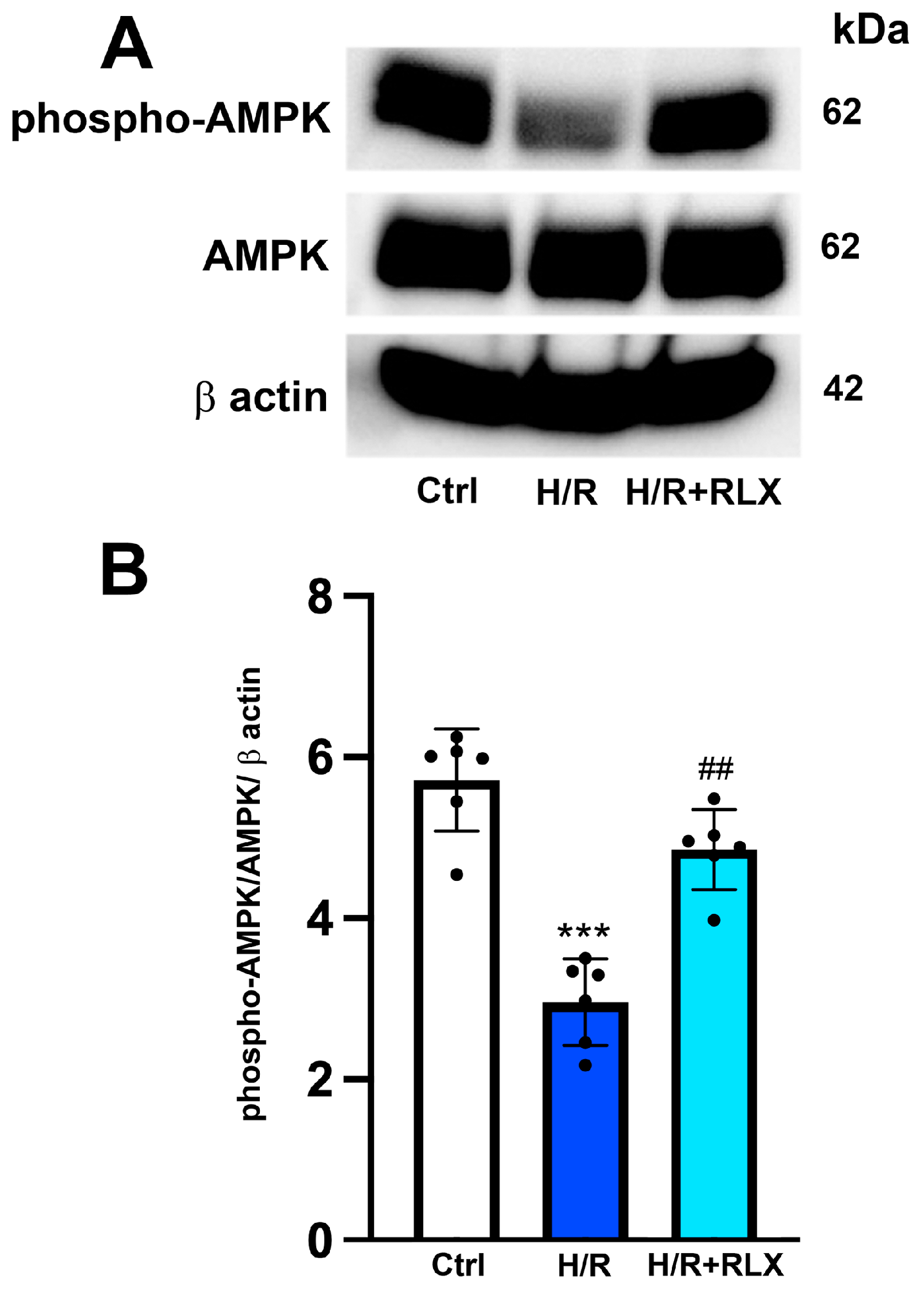
3.3. RLX Affects SIRT1 Signaling through Activation of AMPK Pathway
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sherwood, O.D. Relaxin’s physiological roles and other diverse actions. Endocrinol. Rev. 2004, 25, 205–234. [Google Scholar] [CrossRef]
- Jelinic, M.; Marshall, S.A.; Leo, C.H.; Parry, L.J.; Tare, M. From pregnancy to cardiovascular disease: Lessons from relaxin-deficient animals to understand relaxin actions in the vascular system. Microcirculation 2019, 26, e12464. [Google Scholar] [CrossRef]
- Bani, D. Recombinant human H2 relaxin (serelaxin) as a cardiovascular drug: Aiming at the right target. Drug Discov. Today 2020, 25, 1239–1244. [Google Scholar] [CrossRef]
- Taylor, M.J.; Clark, C.L. Evidence for a novel source of relaxin: Atrial cardiocytes. J. Endocrinol. 1994, 143, R5–R8. [Google Scholar] [CrossRef]
- Sarwar, M.; Du, X.J.; Dschietzig, T.B.; Summers, R.J. The actions of relaxin on the human cardiovascular system. Br. J. Pharmacol. 2017, 174, 933–949. [Google Scholar] [CrossRef]
- Du, X.J.; Bathgate, R.A.; Samuel, C.S.; Dart, A.M.; Summers, R.J. Cardiovascular effects of relaxin: From basic science to clinical therapy. Nat. Rev. Cardiol. 2010, 7, 48–58. [Google Scholar] [CrossRef]
- Perna, A.M.; Masini, E.; Nistri, S.; Briganti, V.; Chiappini, L.; Stefano, P.; Bigazzi, M.; Pieroni, C.; Sacchi, T.B.; Bani, D. Novel drug development opportunity for relaxin in acute myocardial infarction: Evidence from a swine model. FASEB J. 2005, 19, 1525–1527. [Google Scholar] [CrossRef]
- Boccalini, G.; Sassoli, C.; Formigli, L.; Bani, D.; Nistri, S. Relaxin protects cardiac muscle cells from hypoxia/reoxygenation injury: Involvement of the Notch-1 pathway. FASEB J. 2015, 29, 39–249. [Google Scholar] [CrossRef] [PubMed]
- Raleigh, J.V.; Mauro, A.G.; Devarakonda, T.; Marchetti, C.; He, J.; Kim, E.; Filippone, S.; Das, A.; Toldo, S.; Abbate, A.; et al. Reperfusion therapy with recombinant human relaxin-2 (Serelaxin) attenuates myocardial infarct size and NLRP3 inflammasome following ischemia/reperfusion injury via eNOS-dependent mechanism. Cardiovasc. Res. 2017, 113, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Waza, A.A.; Hamid, Z.; Bhat, S.A.; Shah, N.U.D.; Bhat, M.; Ganai, B. Relaxin protects cardiomyocytes against hypoxia-induced damage in in-vitro conditions: Involvement of Nrf2/HO-1 signaling pathway. Life Sci. 2018, 213, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.M.; Su, Y.; Moore, S.; Han, L.P.; Kiriazis, H.; Lu, Q.; Zhao, W.B.; Ruze, A.; Fang, B.B.; Duan, M.J.; et al. Relaxin mitigates microvascular damage and inflammation following cardiac ischemia-reperfusion. Basic Res. Cardiol. 2019, 114, 30. [Google Scholar] [CrossRef]
- Nistri, S.; Fiorillo, C.; Becatti, M.; Bani, D. Human Relaxin-2 (Serelaxin) attenuates oxidative stress in cardiac muscle cells exposed in vitro to hypoxia-reoxygenation. Evidence for the involvement of reduced glutathione up-regulation. Antioxidants 2020, 21, 9–774. [Google Scholar] [CrossRef]
- Bathgate, R.A.; Halls, M.L.; van der Westhuizen, E.T.; Callander, G.E.; Kocan, M.; Summers, R.J. Relaxin family peptides and their receptors. Physiol. Rev. 2013, 93, 405–480. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.Y.; Li, X.; Hung, C.H.; Bahudhanapati, H.; Tan, J.; Kass, D.J.; Zhang, Y. The relaxin family peptide receptor 1 (RXFP1): An emerging player in human health and disease. Mol. Genet. Genomic Med. 2020, 8, e1194. [Google Scholar] [CrossRef]
- Boccalini, G.; Sassoli, C.; Bani, D.; Nistri, S. Relaxin induces up-regulation of ADAM10 metalloprotease in RXFP1-expressing cells by PI3K/AKT signaling. Mol. Cell. Endocrinol. 2018, 472, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Valkovic, A.L.; Bathgate, R.A.; Samuel, C.S.; Kocan, M. Understanding relaxin signalling at the cellular level. Mol. Cell. Endocrinol. 2019, 487, 24–33. [Google Scholar] [CrossRef]
- Aragón-Herrera, A.; Feijóo-Bandín, S.; Rodríguez-Penas, D.; Roselló-Lletí, E.; Portolés, M.; Rivera, M.; Bigazzi, M.; Bani, D.; Gualillo, O.; González-Juanatey, J.R.; et al. Relaxin activates AMPK-AKT signaling and increases glucose uptake by cultured cardiomyocytes. Endocrine 2018, 60, 103–111. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 signaling pathways in cardiovascular disease protection. Antiox. Redox Signal. 2018, 28, 711–732. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Ubaid, S. Role of Silent Information Regulator 1 (SIRT1) in regulating oxidative stress and inflammation. Inflammation 2020, 43, 1589–1598. [Google Scholar] [CrossRef]
- Hsu, C.P.; Zhai, P.; Yamamoto, T.; Maejima, Y.; Matsushima, S.; Hariharan, N.; Shao, D.; Takagi, H.; Oka, S.; Sadoshima, J. Silent information regulator 1 protects the heart from ischemia/reperfusion. Circulation 2010, 122, 2170–2182. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Li, Q.; Yu, B.; Yang, Y.; Jin, Z.; Duan, W.; Zhao, G.; Zhai, M.; Liu, L.; Yi, D.; et al. Berberine attenuates myocardial ischemia/reperfusion injury by reducing oxidative stress and inflammation response: Role of silent information regulator 1. Oxid. Med. Cell. Longev. 2016, 2016, 1689602. [Google Scholar] [CrossRef]
- Tian, L.; Cao, W.; Yue, R.; Yuan, Y.; Guo, X.; Qin, D.; Xing, J.; Wang, X. Pretreatment with Tilianin improves mitochondrial energy metabolism and oxidative stress in rats with myocardial ischemia/reperfusion injury via AMPK/SIRT1/PGC-1 alpha signaling pathway. J. Pharmacol. Sci. 2019, 139, 352–360. [Google Scholar] [CrossRef]
- Cai, J.; Chen, X.; Liu, X.; Li, Z.; Shi, A.; Tang, X.; Xia, P.; Zhang, J.; Yu, P. AMPK: The key to ischemia-reperfusion injury. J. Cell Physiol. 2022, 237, 4079–4096. [Google Scholar] [CrossRef]
- Canto, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliot, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Hescheler, J.; Meyer, R.; Plant, S.; Krautwurst, D.; Rosenthal, W.; Schultz, G. Morphological, biochemical, and electrophysiological characterization of a clonal cell (H9c2) line from rat heart. Circ. Res. 1991, 69, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, C.; Zhang, L.; Lv, J.; Ni, H. Rutin alleviates hypoxia/reoxygenation-induced injury in myocardial cells by up-regulating SIRT1 expression. Chem. Biol. Interact. 2019, 297, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Feng, J.; Zhang, J.; Kang, X.; Qian, D. Quercetin modulates AMPK/SIRT1/NF-κB signaling to inhibit inflammatory/oxidative stress responses in diabetic high fat diet-induced atherosclerosis in the rat carotid artery. Exp. Ther. Med. 2020, 20, 280. [Google Scholar] [CrossRef]
- Cen, Y.; Liao, W.; Wang, T.; Zhang, D. APPL1 ameliorates myocardial ischemia-reperfusion injury by regulating the AMPK signaling pathway. Exp. Ther. Med. 2022, 23, 157. [Google Scholar] [CrossRef]
- Pini, A.; Boccalini, G.; Baccari, M.C.; Becatti, M.; Garella, R.; Fiorillo, C.; Calosi, L.; Bani, D.; Nistri, S. Protection from cigarette smoke-induced vascular injury by recombinant human relaxin-2 (serelaxin). J. Cell. Mol. Med. 2016, 20, 891–902. [Google Scholar] [CrossRef]
- Chen, L.; Li, S.; Zhu, J.; You, A.; Huang, X.; Yi, X.; Xue, M. Mangiferin prevents myocardial infarction-induced apoptosis and heart failure in mice by activating the Sirt1/FoxO3a pathway. J. Cell. Mol. Med. 2021, 25, 2944–2955. [Google Scholar] [CrossRef]
- He, R.; Cui, M.; Lin, H.; Zhao, L.; Wang, J.; Chen, S.; Shao, Z. Melatonin resists oxidative stress-induced apoptosis in nucleus pulposus cells. Life Sci. 2018, 199, 122–130. [Google Scholar] [CrossRef]
- Hausenloy, D.J.; Chilian, W.; Crea, F.; Davidson, S.M.; Ferdinandy, P.; Garcia-Dorado, D.; van Royen, N.; Schulz, R.; Heusch, G. The coronary circulation in acute myocardial ischaemia/reperfusion injury: A target for cardioprotection. Cardiovasc. Res. 2019, 115, 1143–1155. [Google Scholar] [CrossRef]
- Caito, S.; Rajendrasozhan, S.; Cook, S.; Chung, S.; Yao, H.; Friedman, A.E.; Brookes, P.S.; Rahman, I. SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 2010, 24, 3145–3159. [Google Scholar] [CrossRef]
- Wang, L.; Quan, N.; Sun, W.; Chen, X.; Cates, C.; Rousselle, T.; Zhou, X.; Zhao, X.; Li, J. Cardiomyocyte-specific deletion of Sirt1 gene sensitizes myocardium to ischaemia and reperfusion injury. Cardiovasc. Res. 2018, 114, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Guan, Y.; Mu, F.; Guo, C.; Zhang, E.; Yin, Y.; Wei, G.; Zhu, Y.; Cui, J.; Cao, J.; et al. Protective effect of butin against ischemia/reperfusion-induced myocardial injury in diabetic mice: Involvement of the AMPK/GSK-3beta/Nrf2 signaling pathway. Sci. Rep. 2017, 7, 41491. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, X.; Zhang, W.; He, J.; Xu, B.; Lei, B.; Wang, Z.; Cates, C.; Rousselle, T.; Li, J. Activation of AMPK inhibits inflammatory response during hypoxia and reoxygenation through modulating JNK-mediated NF-κB pathway. Metabolism 2018, 83, 56–270. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Hong, J.; Hu, C.; Huang, C.; Gao, J.; Huang, J.; Wang, D.; Geng, Q.; Dong, Y. Palmatine protects against cerebral ischemia/reperfusion injury by activation of the AMPK/Nrf2 pathway. Oxid. Med. Cell. Longev. 2021, 2021, 6660193. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, A.; Suuronen, T.; Ojala, J.; Kaarniranta, K.; Salminen, A. Antagonistic crosstalk between NF-κB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013, 25, 1939–1948. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.; Chung, J.W.; Bae, H.R.; Choi, J.S.; Kim, C.M.; Kim, N.D. Humulus japonicus extract exhibits antioxidative and anti-aging effects via modulation of the AMPK-SIRT1 pathway. Exp. Ther. Med. 2015, 9, 1819–1826. [Google Scholar] [CrossRef]
- Han, X.; Tai, H.; Wang, X.; Wang, Z.; Zhou, J.; Wei, X.; Ding, Y.; Gong, H.; Mo, C.; Zhang, J.; et al. AMPK activation protects cells from oxidative stress-induced senescence via autophagic flux restoration and intracellular NAD(+) elevation. Aging Cell. 2016, 15, 416–427. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Yang, M.; Lv, H.; Liu, Q.; Zhang, L.; Zhang, R.; Huang, X.; Wang, X.; Han, B.; Hou, S.; Liu, D.; et al. Colchicine alleviates cholesterol crystal-induced endothelial cell pyroptosis through activating AMPK/SIRT1 pathway. Oxid. Med. Cell. Longev. 2020, 2020, 9173530. [Google Scholar] [CrossRef]
- Lu, C.; Jiang, B.; Xu, J.; Zhang, X.; Jiang, N. Neferine protected cardiomyocytes against hypoxia/oxygenation injury through SIRT1/Nrf2/HO-1 signaling. J. Biochem. Mol. Toxicol. 2023, 37, e23398. [Google Scholar] [CrossRef] [PubMed]
- Harvey, C.J.; Thimmulappa, R.K.; Singh, A.; Blake, D.J.; Ling, G.; Wakabayashi, N.; Fujii, J.; Myers, A.; Biswal, S. Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 2009, 46, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Samuel, C.S.; Du, X.J.; Bathgate, R.A.; Summers, R.J. ‘Relaxin’ the stiffened heart and arteries: The therapeutic potential for relaxin in the treatment of cardiovascular disease. Pharmacol. Ther. 2006, 112, 529–552. [Google Scholar] [CrossRef] [PubMed]
- Nowaczyk, A.; Kowalska, M.; Nowaczyk, J.; Grześk, G. Carbon monoxide and nitric oxide as examples of the youngest class of transmitters. Int. J. Mol. Sci. 2021, 22, 6029. [Google Scholar] [CrossRef] [PubMed]
- Bice, J.S.; Jones, B.R.; Chamberlain, G.R.; Baxter, G.F. Nitric oxide treatments as adjuncts to reperfusion in acute myocardial infarction: A systematic review of experimental and clinical studies. Basic Res. Cardiol. 2016, 111, 23. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zizi, V.; Becatti, M.; Bani, D.; Nistri, S. Serelaxin Protects H9c2 Cardiac Myoblasts against Hypoxia and Reoxygenation-Induced Damage through Activation of AMP Kinase/Sirtuin1: Further Insight into the Molecular Mechanisms of the Cardioprotection of This Hormone. Antioxidants 2024, 13, 163. https://doi.org/10.3390/antiox13020163
Zizi V, Becatti M, Bani D, Nistri S. Serelaxin Protects H9c2 Cardiac Myoblasts against Hypoxia and Reoxygenation-Induced Damage through Activation of AMP Kinase/Sirtuin1: Further Insight into the Molecular Mechanisms of the Cardioprotection of This Hormone. Antioxidants. 2024; 13(2):163. https://doi.org/10.3390/antiox13020163
Chicago/Turabian StyleZizi, Virginia, Matteo Becatti, Daniele Bani, and Silvia Nistri. 2024. "Serelaxin Protects H9c2 Cardiac Myoblasts against Hypoxia and Reoxygenation-Induced Damage through Activation of AMP Kinase/Sirtuin1: Further Insight into the Molecular Mechanisms of the Cardioprotection of This Hormone" Antioxidants 13, no. 2: 163. https://doi.org/10.3390/antiox13020163