
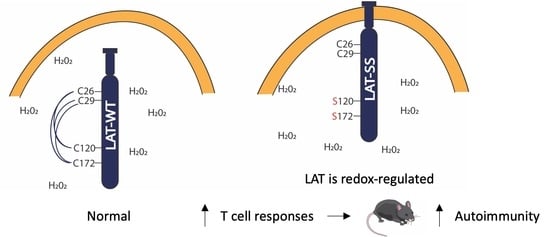
Redox Regulation of LAT Enhances T Cell-Mediated Inflammation
 , ,
, , 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
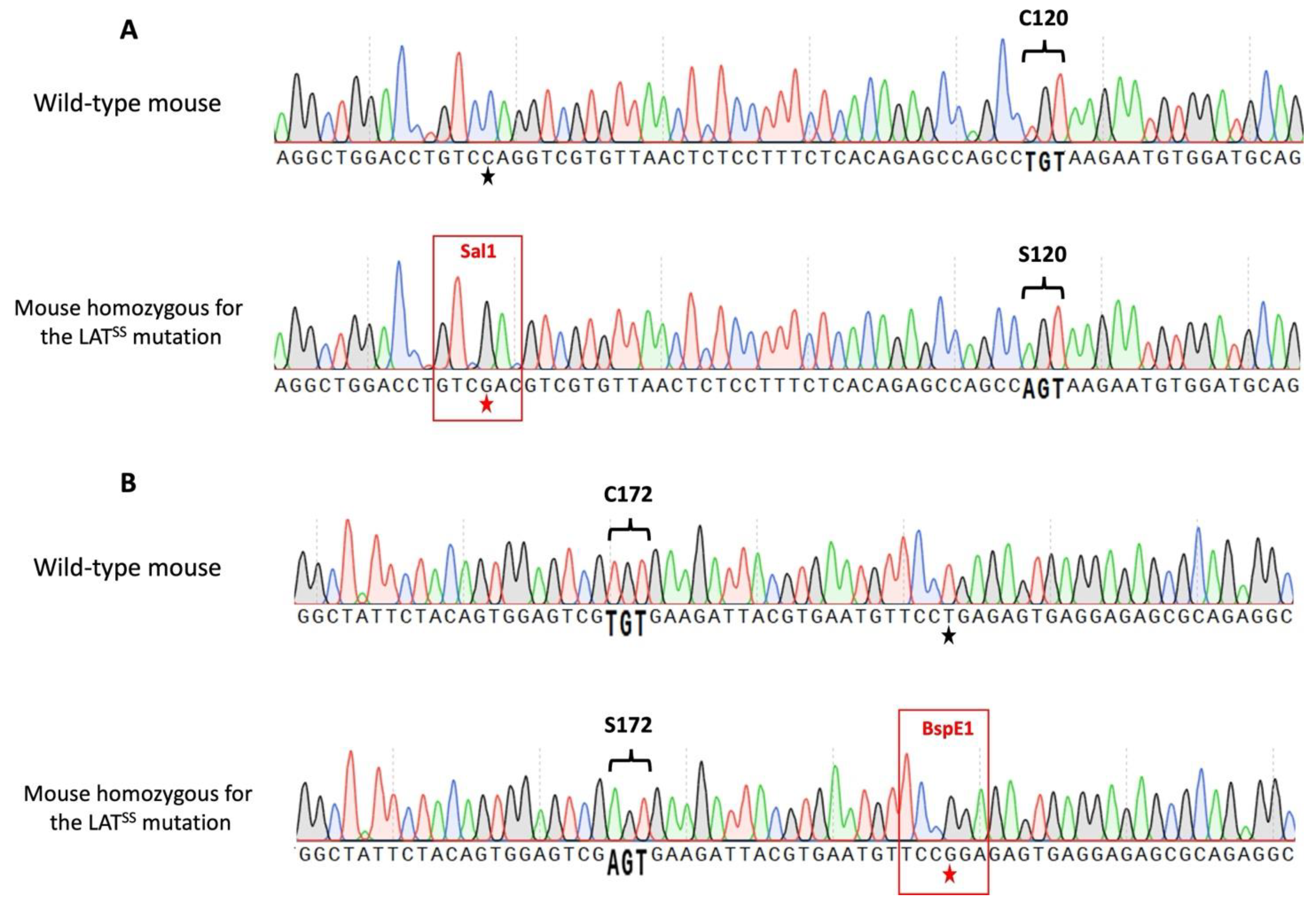
2. Materials and Methods
3. Results
3.1. LAT Phosphorylation Is NOX2-Dependent
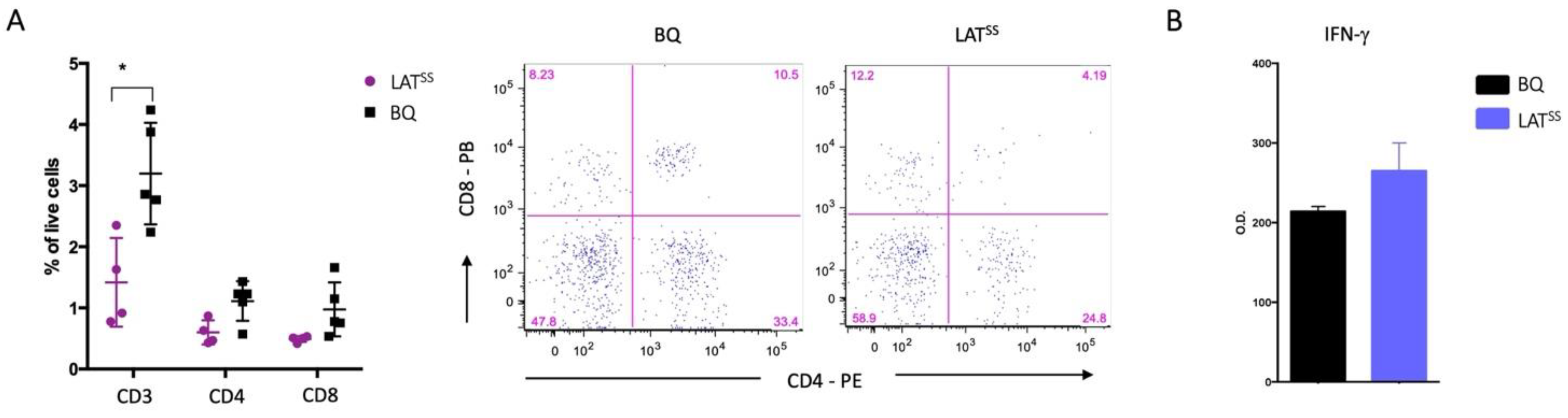
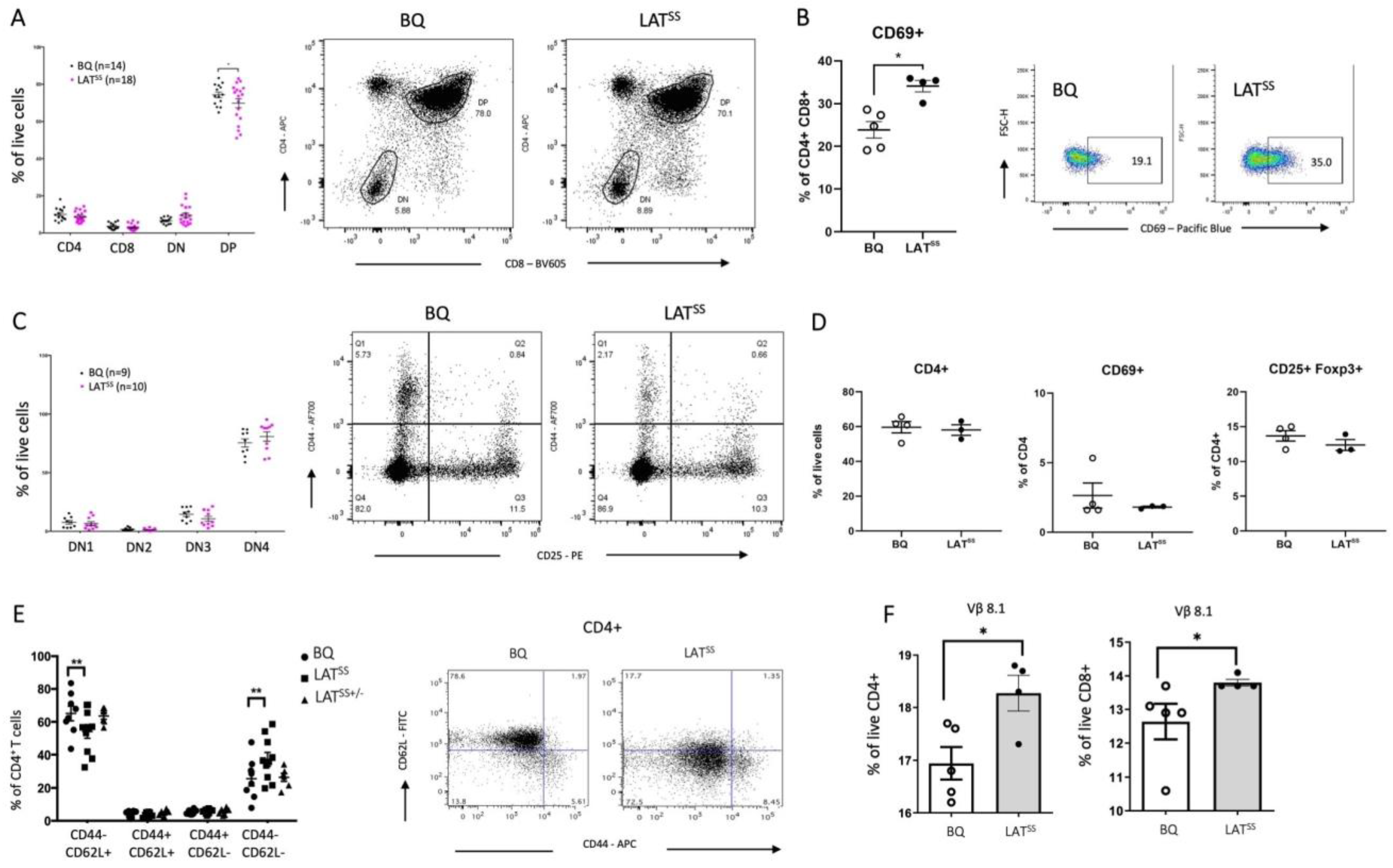
3.2. LATSS Impacts Thymic Selection and Peripheral T Cells at Steady State
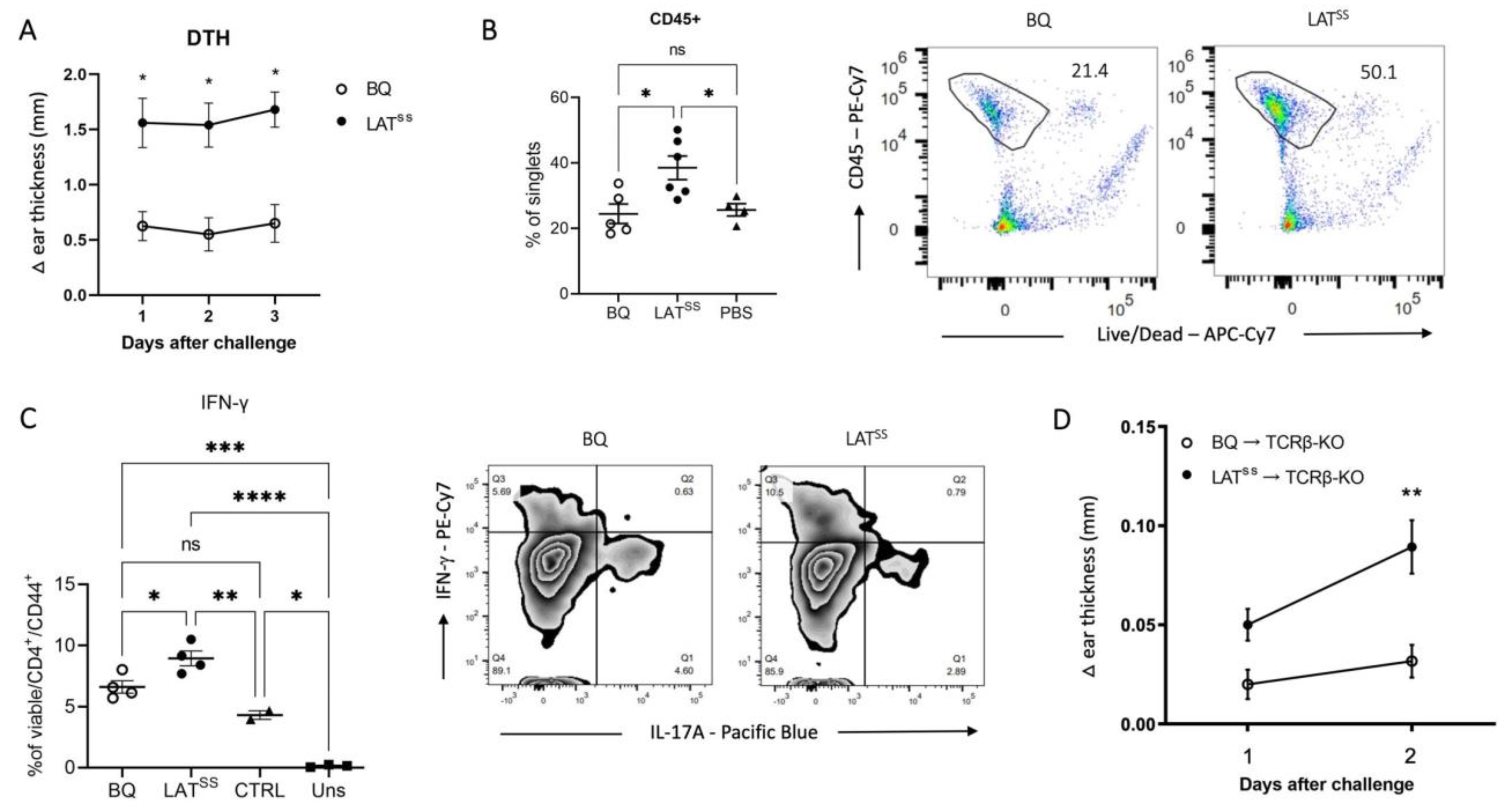
3.3. LATSS Enhances T Cell-Dependent Inflammation in DTH and CIA Models
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


References
- Olofsson, P.; Holmberg, J.; Tordsson, J.; Lu, S.; Åkerström, B.; Holmdahl, R. Positional identification of Ncf1 as a gene that regulates arthritis severity in rats. Nat. Genet. 2003, 33, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Olsson , L.M.; Johansson, Å.C.; Gullstrand, B.; Jönsen, A.; Saevarsdottir, S.; Rönnblom, L.; Leonard, D.; Wetterö, J.; Sjöwall, C.; Svenungsson, E.; et al. A single nucleotide polymorphism in the NCF1 gene leading to reduced oxidative burst is associated with systemic lupus erythematosus. Ann. Rheum. Dis. 2017, 76, 1607–1613. [Google Scholar] [CrossRef]
- Zhao, J.; Ma, J.; Deng, Y.; Kelly, J.A.; Kim, K.; Bang, S.Y.; Lee, H.-S.; Li, Q.-Z.; Wakeland, E.K.; Qiu, R.; et al. A missense variant in NCF1 is associated with susceptibility to multiple autoimmune diseases. Nat. Genet. 2017, 49, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, N.; Kawasaki, A.; Matsushita, T.; Furukawa, H.; Kondo, Y.; Hirano, F.; Sada, K.-E.; Matsumoto, I.; Kusaoi, M.; Amano, H.; et al. Association of NCF1 polymorphism with systemic lupus erythematosus and systemic sclerosis but not with ANCA-associated vasculitis in a Japanese population. Sci. Rep. 2019, 9, 16366. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX Family of ROS-Generating NADPH Oxidases: Physiology and Pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Hultqvist, M.; Olofsson, P.; Holmberg, J.; Bäckström, B.T.; Tordsson, J.; Holmdahl, R. Enhanced autoimmunity, arthritis, and encephalomyelitis in mice with a reduced oxidative burst due to a mutation in the Ncf1 gene. Proc. Natl. Acad. Sci. USA 2004, 101, 12646–12651. [Google Scholar] [CrossRef] [PubMed]
- Shu, P.; Liang, H.; Zhang, J.; Lin, Y.; Chen, W.; Zhang, D. Reactive oxygen species formation and its effect on CD4+ T cell-mediated inflammation. Front. Immunol. 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Gelderman, K.A.; Hultqvist, M.; Holmberg, J.; Olofsson, P.; Holmdahl, R. T cell surface redox levels determine T cell reactivity and arthritis susceptibility. Proc. Natl. Acad. Sci. USA 2006, 103, 12831–12836. [Google Scholar] [CrossRef] [PubMed]
- Chan, A.C.; Iwashima, M.; Turck, C.W.; Weiss, A. ZAP-70: A 70 kd protein-tyrosine kinase that associates with the TCR ζ chain. Cell 1992, 71, 649–662. [Google Scholar] [CrossRef]
- Guy, C.S.; Vignali, D.A.A. Organization of proximal signal initiation at the TCR:CD3 complex. Immunol. Rev. 2009, 232, 7–21. [Google Scholar] [CrossRef]
- Lo, W.L.; Shah, N.H.; Ahsan, N.; Horkova, V.; Stepanek, O.; Salomon, A.R.; Kuriyan, J.; Weiss, A. Lck promotes Zap70-dependent LAT phosphorylation by bridging Zap70 to LAT. Nat. Immunol. 2018, 19, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Sommers, C.L.; Menon, R.K.; Grinberg, A.; Zhang, W.; Samelson, L.E.; Love, P.E. Knock-in Mutation of the Distal Four Tyrosines of Linker for Activation of T Cells Blocks Murine T Cell Development. J. Exp. Med. 2001, 194, 135–142. [Google Scholar] [CrossRef]
- Finco, T.S.; Kadlecek, T.; Zhang, W.; Samelson, L.E.; Weiss, A. LAT Is Required for TCR-Mediated Activation of PLCγ1 and the Ras Pathway. Immunity 1998, 9, 617–626. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sommers, C.L.; Burshtyn, D.N.; Stebbins, C.C.; DeJarnette, J.B.; Trible, R.P.; Grinberg, A.; Tsay, H.C.; Jacobs, H.M.; Kessler, C.M.; et al. Essential role of LAT in T cell development. Immunity 1999, 10, 323–332. [Google Scholar] [CrossRef]
- Shen, S.; Zhu, M.; Lau, J.; Chuck, M.; Zhang, W. The Essential Role of LAT in Thymocyte Development during Transition from the Double-Positive to Single-Positive Stage. J. Immunol. 2009, 182, 5596–5604. [Google Scholar] [CrossRef] [PubMed]
- Sommers, C.L.; Park, C.S.; Lee, J.; Feng, C.; Fuller, C.L.; Grinberg, A.; Hildebrand, J.A.; Lacaná, E.; Menon, R.K.; Shores, E.W.; et al. A LAT Mutation That Inhibits T Cell Development Yet Induces Lymphoproliferation. Science 2002, 296, 2040–2043. [Google Scholar] [CrossRef] [PubMed]
- Joachim, A.; Aussel, R.; Gélard, L.; Zhang, F.; Mori, D.; Grégoire, C.; Merino, S.V.; Gaya, M.; Liang, Y.; Malissen, M.; et al. Defective LAT signalosome pathology in mice mimics human IgG4-related disease at single-cell level. J. Exp. Med. 2023, 220, e20231028. [Google Scholar] [CrossRef] [PubMed]
- Balagopalan, L.; Ashwell, B.A.; Bernot, K.M.; Akpan, I.O.; Quasba, N.; Barr, V.A.; Samelson, L.E. Enhanced T-cell signaling in cells bearing linker for activation of T-cell (LAT) molecules resistant to ubiquitylation. Proc. Natl. Acad. Sci. USA 2011, 108, 2885–2890. [Google Scholar] [CrossRef] [PubMed]
- Balagopalan, L.; Malik, H.; McIntire, K.M.; Garvey, J.A.; Nguyen, T.; Rodriguez-Peña, A.B.; Samelson, L.E. Bypassing ubiquitination enables LAT recycling to the cell surface and enhanced signaling in T cells. PLoS ONE 2020, 15, e0229036. [Google Scholar] [CrossRef] [PubMed]
- Hundt, M.; Harada, Y.; De Giorgio, L.; Tanimura, N.; Zhang, W.; Altman, A. Palmitoylation-Dependent Plasma Membrane Transport but Lipid Raft-Independent Signaling by Linker for Activation of T Cells. J. Immunol. 2009, 183, 1685–1694. [Google Scholar] [CrossRef]
- Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. [Google Scholar] [CrossRef] [PubMed]
- Reth, M. Hydrogen peroxide as second messenger in lymphocyte activation. Nat. Immunol. 2002, 3, 1129–1134. [Google Scholar] [CrossRef] [PubMed]
- Ke, K.; Sul, O.J.; Choi, E.K.; Safdar, A.M.; Kim, E.S.; Choi, H.S. Reactive oxygen species induce the association of SHP-1 with c-Src and the oxidation of both to enhance osteoclast survival. Am. J. Physiol.-Endocrinol. Metab. 2014, 307, E61–E70. [Google Scholar] [CrossRef] [PubMed]
- Kuban-Jankowska A, Knap N, Gorska M, Popowska U, Wozniak M. Protein tyrosine phosphatase CD45 as a molecular biosensor of hydrogen peroxide generation in cell culture media. Biochem. Biophys. Res. Commun. 2011, 415, 270–273.
- James, J.; Chen, Y.; Hernandez, C.M.; Forster, F.; Dagnell, M.; Cheng, Q.; Saei, A.A.; Gharibi, H.; Lahore, G.F.; Åstrand, A.; et al. Redox regulation of PTPN22 affects the severity of T-cell-dependent autoimmune inflammation. eLife 2022, 11, e74549. [Google Scholar] [CrossRef] [PubMed]
- Simeoni, L.; Bogeski, I. Redox regulation of T-cell receptor signaling. Biol. Chem. 2015, 396, 555–568. [Google Scholar] [CrossRef]
- Belikov, A.V.; Schraven, B.; Simeoni, L. TCR-triggered extracellular superoxide production is not required for T-cell activation. Cell Commun. Signal. 2014, 12, 50. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; Leow, A.; Papendrecht-van der Voort, E.A.M.; Remans, P.H.J.; Breedveld, F.C.; Verweij, C.L. Displacement of Linker for Activation of T Cells from the Plasma Membrane Due to Redox Balance Alterations Results in Hyporesponsiveness of Synovial Fluid T Lymphocytes in Rheumatoid Arthritis. J. Immunol. 2000, 164, 2170–2179. [Google Scholar] [CrossRef]
- Zhang, Y.; Muyrers, J.P.P.; Testa, G.; Stewart, A.F. DNA cloning by homologous recombination in Escherichia coli. Nat. Biotechnol. 2000, 18, 1314–1317. [Google Scholar] [CrossRef]
- Bunting, M.; Bernstein, K.E.; Greer, J.M.; Capecchi, M.R.; Thomas, K.R. Targeting genes for self-excision in the germ line. Genes Dev. 1999, 13, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Soriano, P. The PDGFα receptor is required for neural crest cell development and for normal patterning of the somites. Development 1997, 124, 2691–2700. [Google Scholar] [CrossRef] [PubMed]
- Pettitt, S.J.; Liang, Q.; Rairdan, X.Y.; Moran, J.L.; Prosser, H.M.; Beier, D.R.; Lloyd, K.C.; Bradley, A.; Skarnes, W.C. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat. Methods 2009, 6, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Bäcklund, J.; Li, C.; Jansson, E.; Carlsen, S.; Merky, P.; Nandakumar, K.S.; Haag, S.; Ytterberg, J.; A Zubarev, R.; Holmdahl, R. C57BL/6 mice need MHC class II Aq to develop collagen-induced arthritis dependent on autoreactive T cells. Ann. Rheum. Dis. 2013, 72, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Mac-Daniel, L.; Buckwalter, M.R.; Gueirard, P.; Ménard, R. Myeloid cell isolation from mouse skin and draining lymph node following intradermal immunization with live attenuated plasmodium sporozoites. J. Vis. Exp. 2016, 2016, 53796. Available online: http://pmc/articles/PMC4927700/?report=abstract (accessed on 16 October 2020).
- Gringhuis, S.I.; Papendrecht-van der Voort, E.A.M.; Leow, A.; Levarht, E.W.N.; Breedveld, F.C.; Verweij, C.L. Effect of Redox Balance Alterations on Cellular Localization of LAT and Downstream T-Cell Receptor Signaling Pathways. Mol. Cell. Biol. 2002, 22, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.T.; Anderson, M.T.; Staal, G.E.J.; Herzenberg, L.A.; Gitler, C.; Herzenberg, L.A. Redox regulation of signal transduction: Tyrosine phosphorylation and calcium influx (inflammatory cytoklnes/human immunodeficency virus infection). Proc. Natl. Acad. Sci. USA 1994, 91, 3619–3622. [Google Scholar] [CrossRef] [PubMed]
- Heng, T.S.P.; Painter, M.W.; The Immunological Genome Project Consortium; Elpek, K.; Lukacs-Kornek, V.; Mauermann, N.; Turley, S.J.; Koller, D.; Kim, F.S.; Wagers, A.J.; et al. The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef]
- Chiocchia, G.; Boissier, M.; Fournier, C. Therapy against murine collagen-induced arthritis with T cell receptor Vβ-specific antibodies. Eur. J. Immunol. 1991, 21, 2899–2905. [Google Scholar] [CrossRef]
- Allen, I.C. Delayed-type hypersensitivity models in mice. Methods Mol. Biol. 2013, 1031, 101–107. Available online: https://pubmed.ncbi.nlm.nih.gov/23824893/ (accessed on 27 August 2020). [PubMed]
- Corthay, A.; Bäcklund, J.; Broddefalk, J.; Michaëlsson, E.; Goldschmidt, T.J.; Kihlberg, J.; Holmdahl, R. Epitope glycosylation plays a critical role for T cell recognition of type II collagen in collagen-induced arthritis. Eur. J. Immunol. 1998, 28, 2580–2590. [Google Scholar] [CrossRef]
- Xu, Z.; Xu, B.; Lundström, S.L.; Moreno-Giró, À.; Zhao, D.; Martin, M.; Lönnblom, E.; Li, Q.; Krämer, A.; Ge, C.; et al. A subset of type-II collagen-binding antibodies prevents experimental arthritis by inhibiting FCGR3 signaling in neutrophils. Nat. Commun. 2023, 14, 5949. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Shadel, G.S.; Ghosh, S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011, 11, 389–402. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, M.M.; Röth, D.; Krammer, P.H.; Gülow, K. Mitochondria as Oxidative Signaling Organelles in T-cell Activation: Physiological Role and Pathological Implications. Arch. Immunol. Ther. Exp. 2013, 61, 367–384. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, M.L.; DeFranco, A.L. Prolonged Production of Reactive Oxygen Species in Response to B Cell Receptor Stimulation Promotes B Cell Activation and Proliferation. J. Immunol. 2012, 189, 4405–4416. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.K.; Kumar, D.; Siddiqui, Z.; Basu, S.K.; Kumar, V.; Rao, K.V.S. The Strength of Receptor Signaling Is Centrally Controlled through a Cooperative Loop between Ca2+ and an Oxidant Signal. Cell 2005, 121, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Chaudhri, G.; Clark, I.A.; Hunt, N.H.; Cowden, W.B.; Ceredig, R. Effect of antioxidants on primary alloantigen-induced T cell activation and proliferation. J. Immunol. 1986, 137, 2646–2652. [Google Scholar] [CrossRef] [PubMed]
- Laniewski, N.G.; Grayson, J.M. Antioxidant Treatment Reduces Expansion and Contraction of Antigen-Specific CD8 + T Cells during Primary but Not Secondary Viral Infection. J. Virol. 2004, 78, 11246–11257. [Google Scholar] [CrossRef] [PubMed]
- Yarosz, E.L.; Chang, C.H. Role of reactive oxygen species in regulating T cell-mediated immunity and disease. Immune Netw. 2018, 18, e14. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria Are Required for Antigen-Specific T Cell Activation through Reactive Oxygen Species Signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Carpier, J.M.; Zucchetti, A.E.; Bataille, L.; Dogniaux, S.; Shafaq-Zadah, M.; Bardin, S.; Lucchino, M.; Maurin, M.; Joannas, L.D.; Magalhaes, J.G.; et al. Rab6-dependent retrograde traffic of LAT controls immune synapse formation and T cell activation. J. Exp. Med. 2018, 215, 1245–1265. [Google Scholar] [CrossRef] [PubMed]
- Balagopalan, L.; Barr, V.A.; Sommers, C.L.; Barda-Saad, M.; Goyal, A.; Isakowitz, M.S.; Samelson, L.E. c-Cbl-Mediated Regulation of LAT-Nucleated Signaling Complexes. Mol. Cell. Biol. 2007, 27, 8622–8636. [Google Scholar] [CrossRef] [PubMed]
- Necci, M.; Piovesan, D.; Tosatto, S.C.E. Critical assessment of protein intrinsic disorder prediction. Nat. Methods 2021, 18, 472–481. Available online: https://www.nature.com/articles/s41592-021-01117-3 (accessed on 16 July 2021). [CrossRef] [PubMed]
- Balagopalan, L.; Kortum, R.L.; Coussens, N.P.; Barr, V.A.; Samelson, L.E. The linker for activation of T cells (LAT) signaling hub: From signaling complexes to microclusters. J. Biol. Chem. 2015, 290, 26422–26429. Available online: http://www.jbc.org/article/S0021925820495171/fulltext (accessed on 17 June 2021). [CrossRef]
- Paster, W.; Brockmeyer, C.; Fu, G.; Simister, P.C.; de Wet, B.; Martinez-Riaño, A.; Hoerter, J.A.H.; Feller, S.M.; Wülfing, C.; Gascoigne, N.R.J.; et al. GRB2-Mediated Recruitment of THEMIS to LAT Is Essential for Thymocyte Development. J. Immunol. 2013, 190, 3749–3756. [Google Scholar] [CrossRef] [PubMed]
- Clements, J.L.; Yang, B.; Ross-Barta, S.E.; Eliason, S.L.; Hrstka, R.F.; Williamson, R.A.; Koretzky, G.A. Requirement for the leukocyte-specific adapter protein SLP-76 for normal T cell development. Science 1998, 281, 416–419. Available online: https://pubmed.ncbi.nlm.nih.gov/9665885/ (accessed on 16 July 2021). [CrossRef] [PubMed]
- van Oers, N.S.C.; Lowin-Kropf, B.; Finlay, D.; Connolly, K.; Weiss, A. αβ T Cell Development Is Abolished in Mice Lacking Both Lck and Fyn Protein Tyrosine Kinases. Immunity 1996, 5, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. Available online: https://www.nature.com/articles/1208074 (accessed on 16 July 2021). [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
James, J.; Coelho, A.; Lahore, G.F.; Hernandez, C.M.; Forster, F.; Malissen, B.; Holmdahl, R. Redox Regulation of LAT Enhances T Cell-Mediated Inflammation. Antioxidants 2024, 13, 499. https://doi.org/10.3390/antiox13040499
James J, Coelho A, Lahore GF, Hernandez CM, Forster F, Malissen B, Holmdahl R. Redox Regulation of LAT Enhances T Cell-Mediated Inflammation. Antioxidants. 2024; 13(4):499. https://doi.org/10.3390/antiox13040499
Chicago/Turabian StyleJames, Jaime, Ana Coelho, Gonzalo Fernandez Lahore, Clara M. Hernandez, Florian Forster, Bernard Malissen, and Rikard Holmdahl. 2024. "Redox Regulation of LAT Enhances T Cell-Mediated Inflammation" Antioxidants 13, no. 4: 499. https://doi.org/10.3390/antiox13040499
APA StyleJames, J., Coelho, A., Lahore, G. F., Hernandez, C. M., Forster, F., Malissen, B., & Holmdahl, R. (2024). Redox Regulation of LAT Enhances T Cell-Mediated Inflammation. Antioxidants, 13(4), 499. https://doi.org/10.3390/antiox13040499