
Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia



Abstract
:1. Introduction
2. Materials and Methods
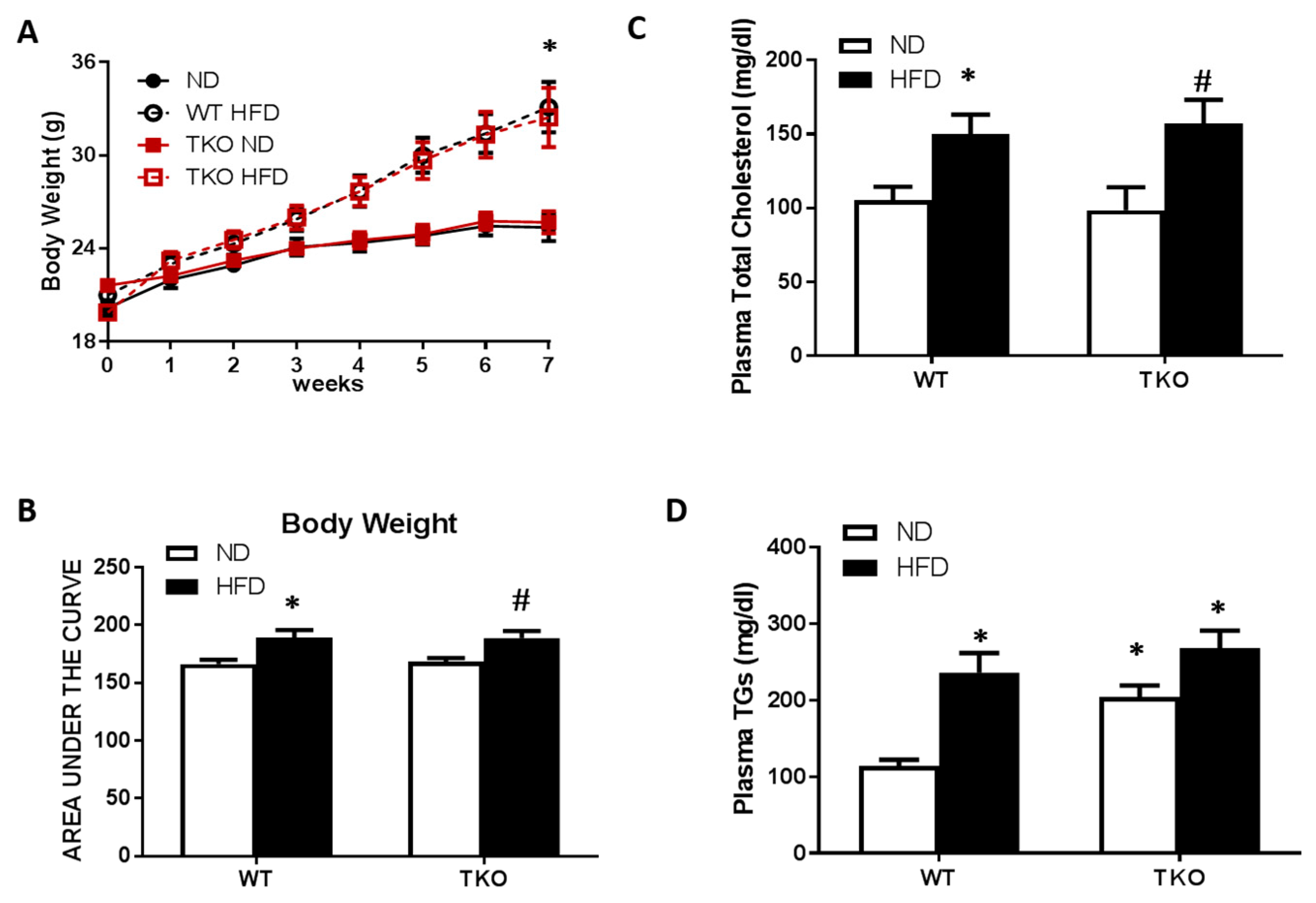
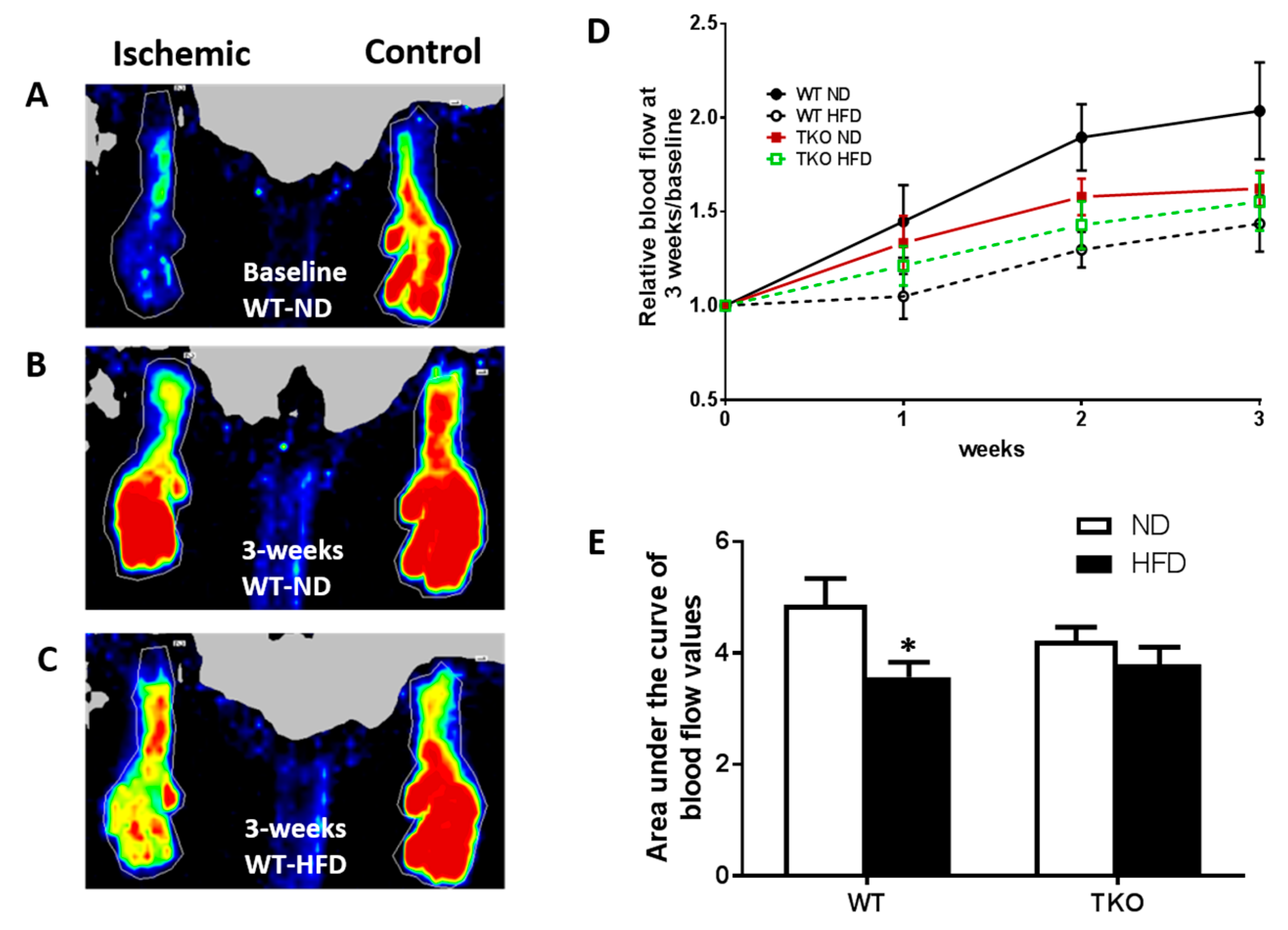
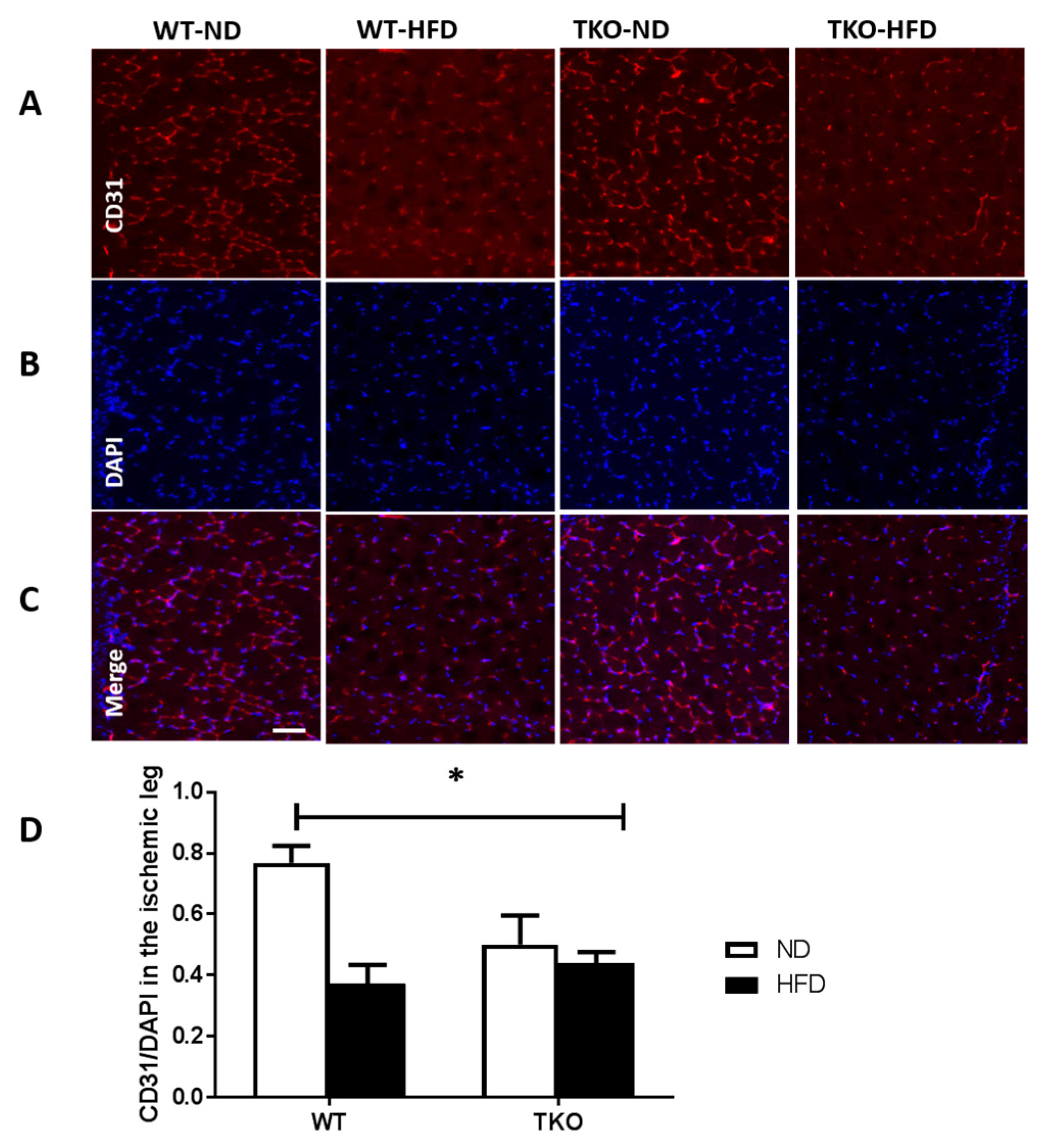
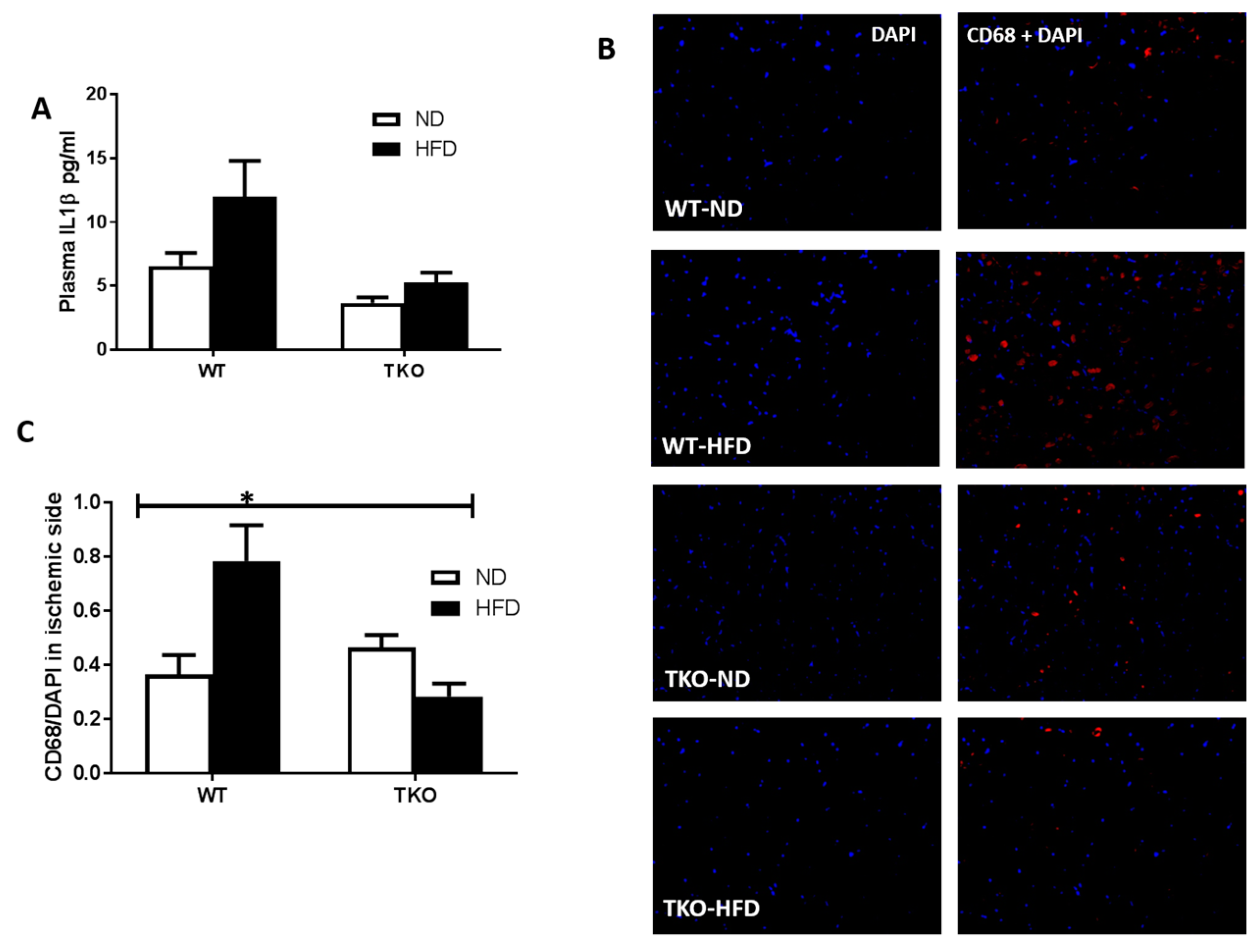
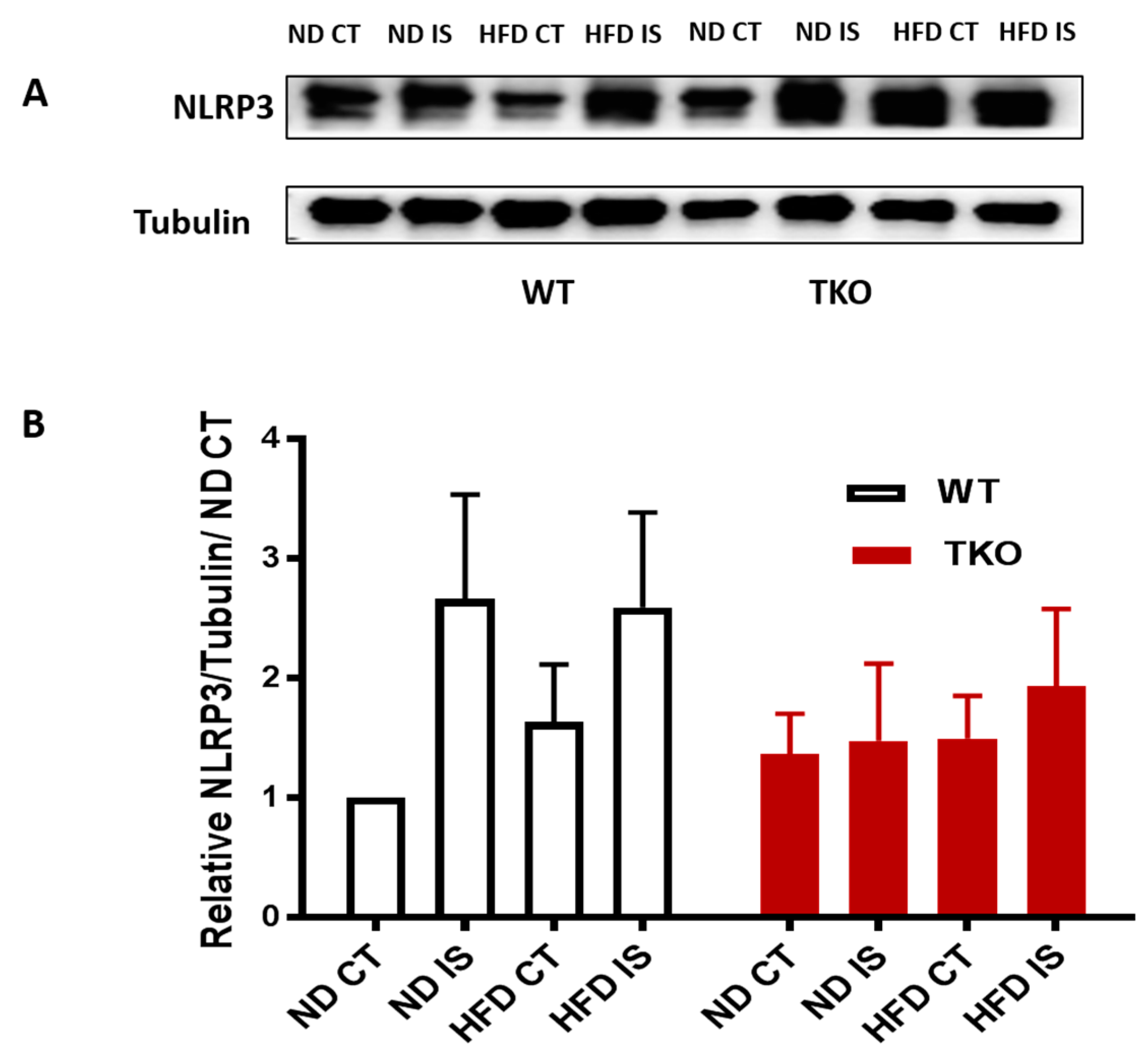
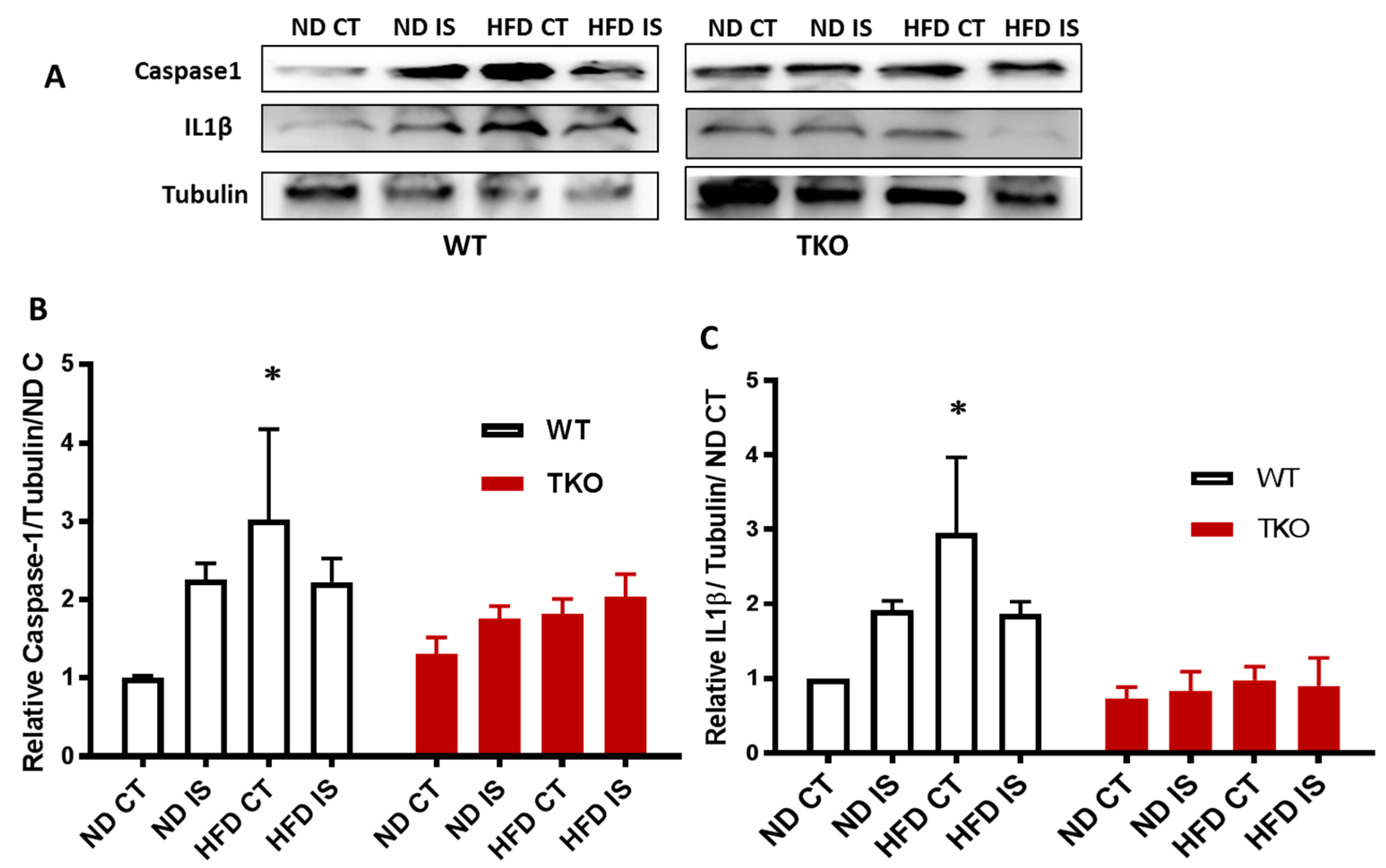
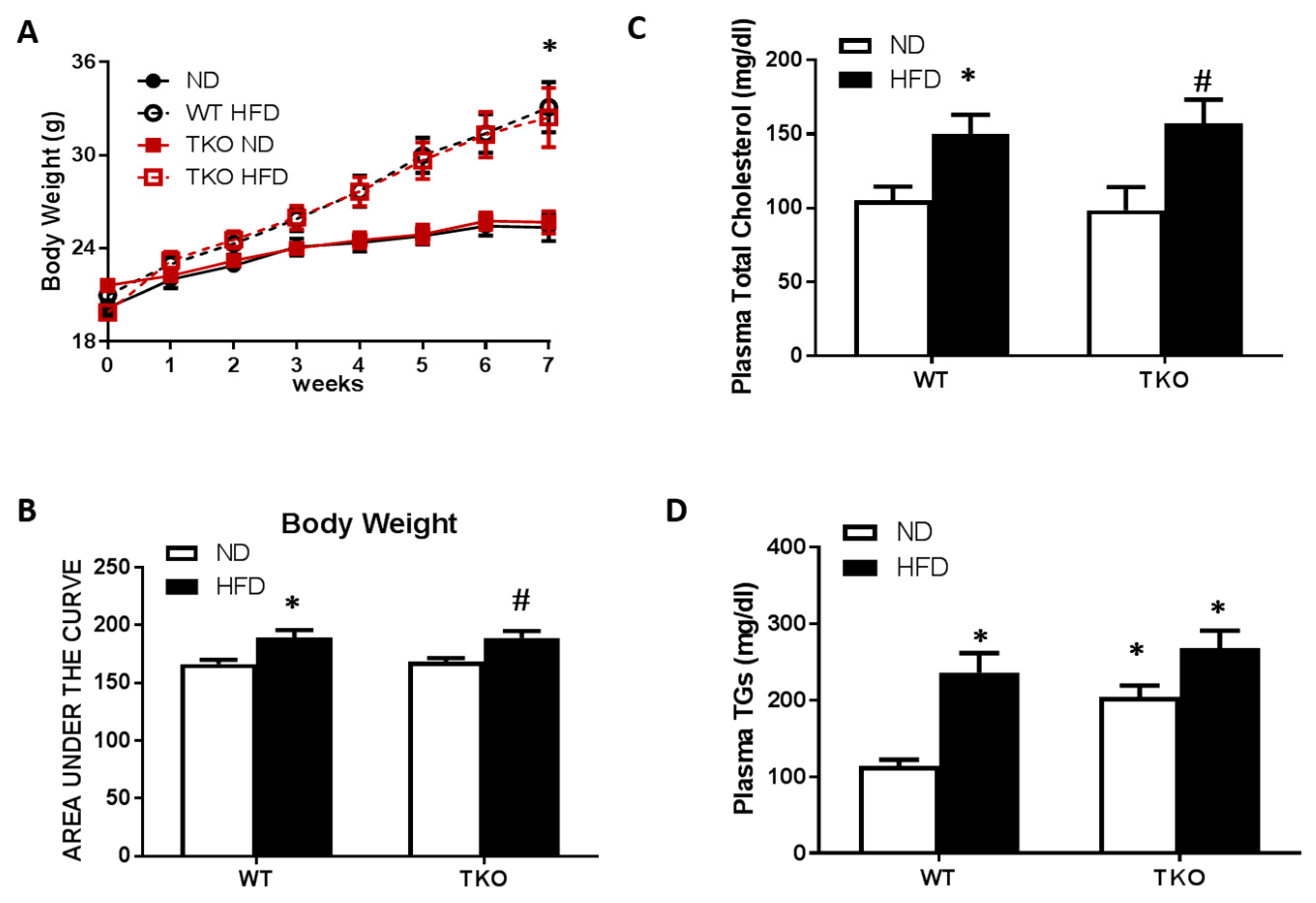
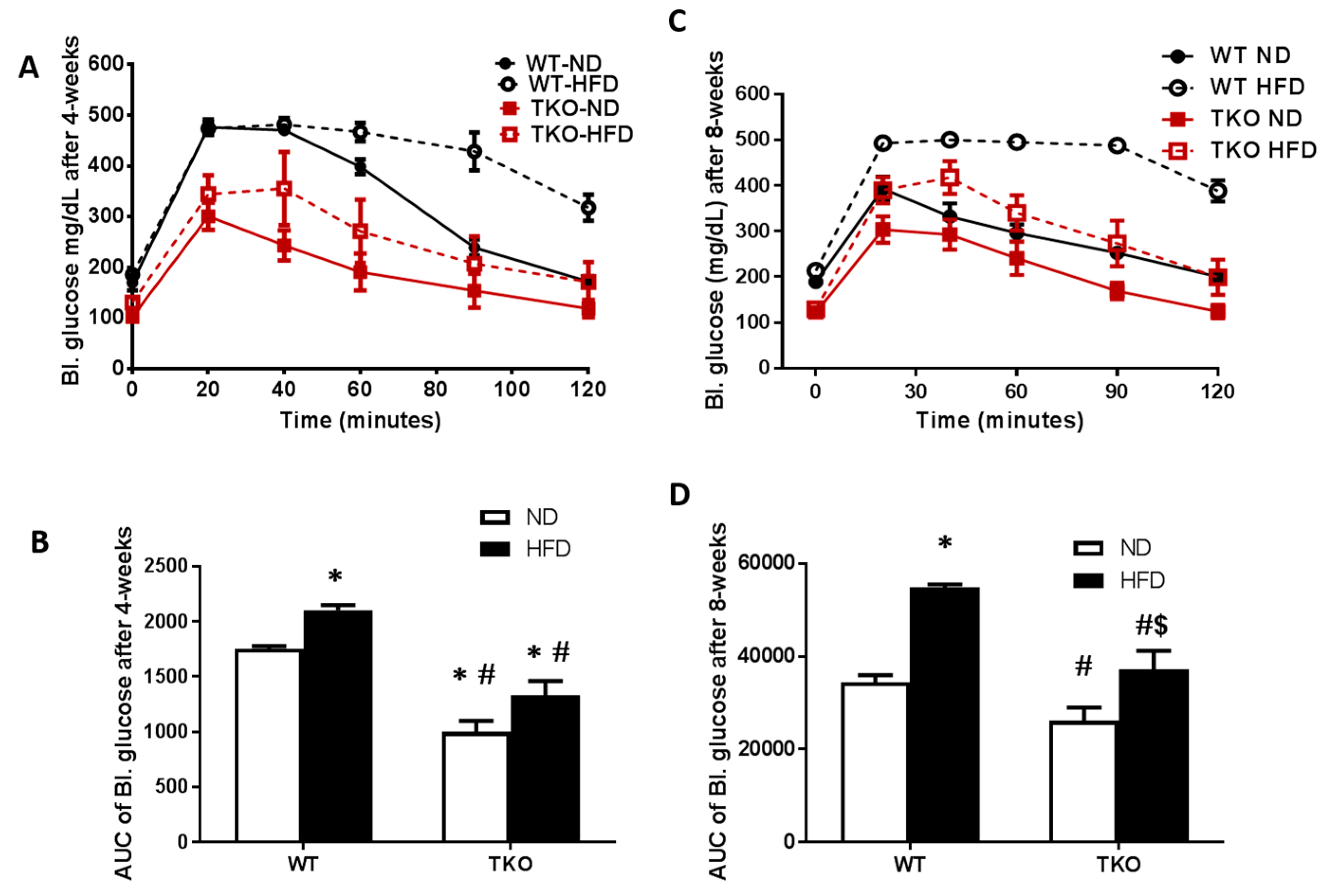
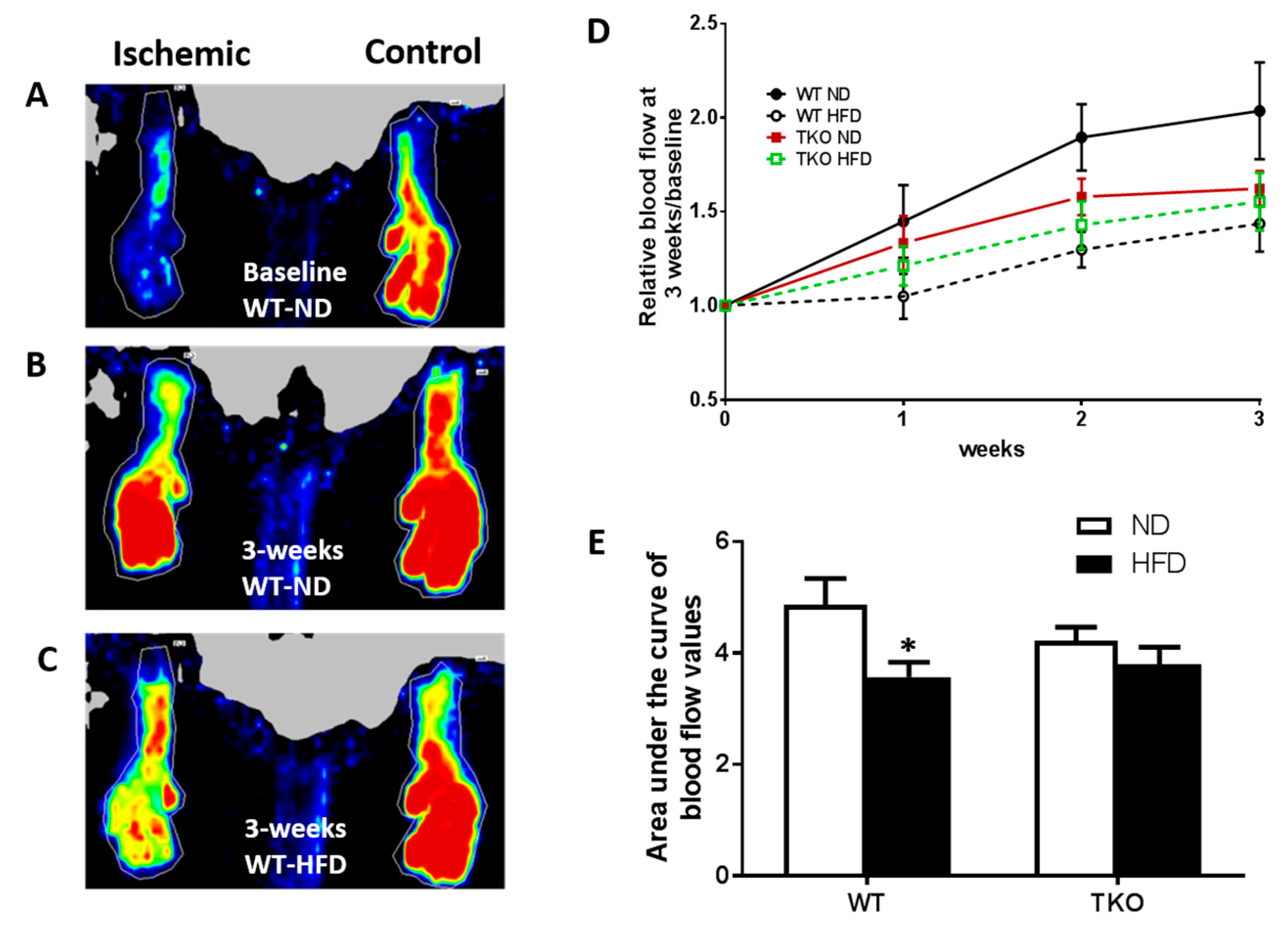
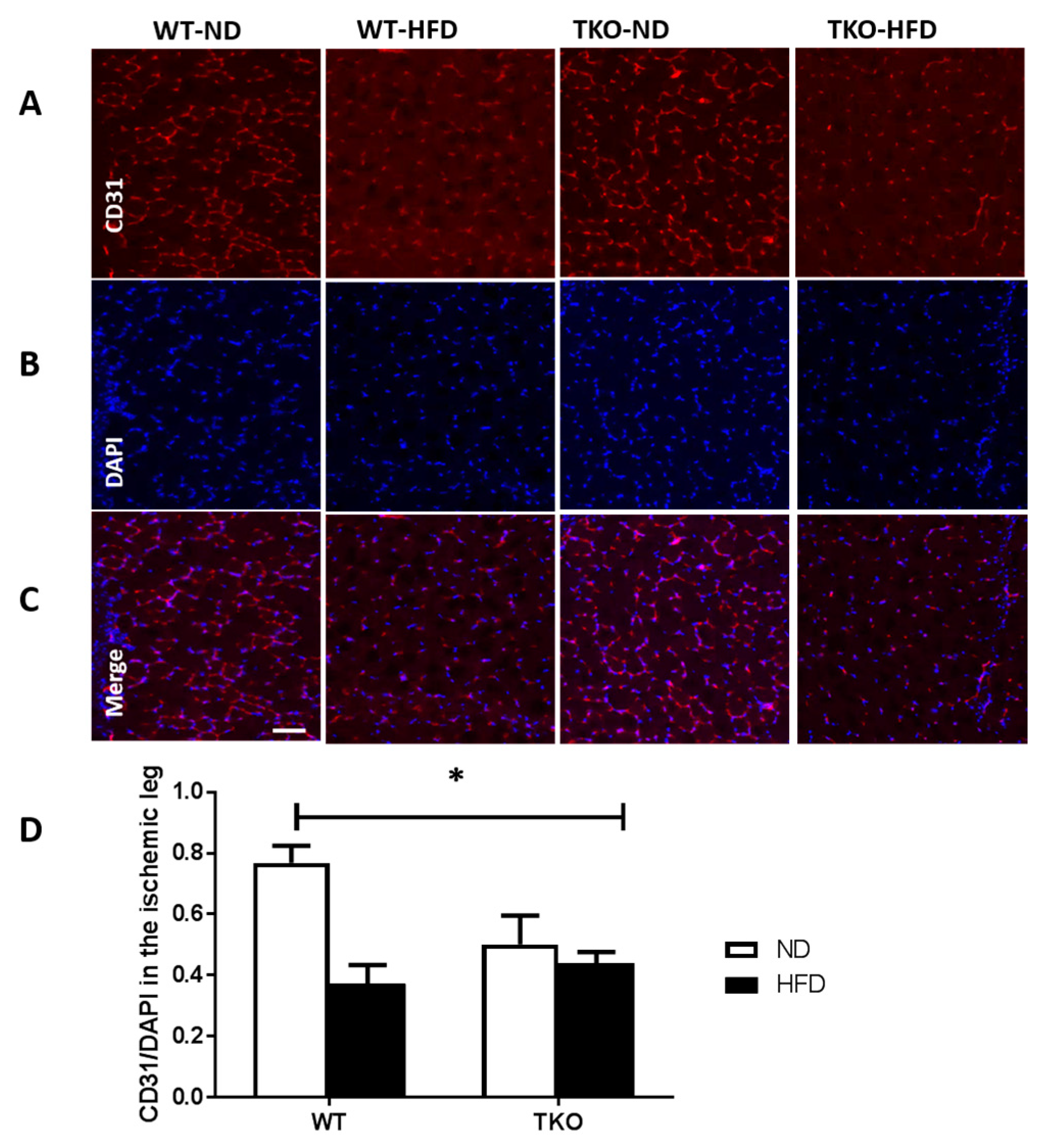
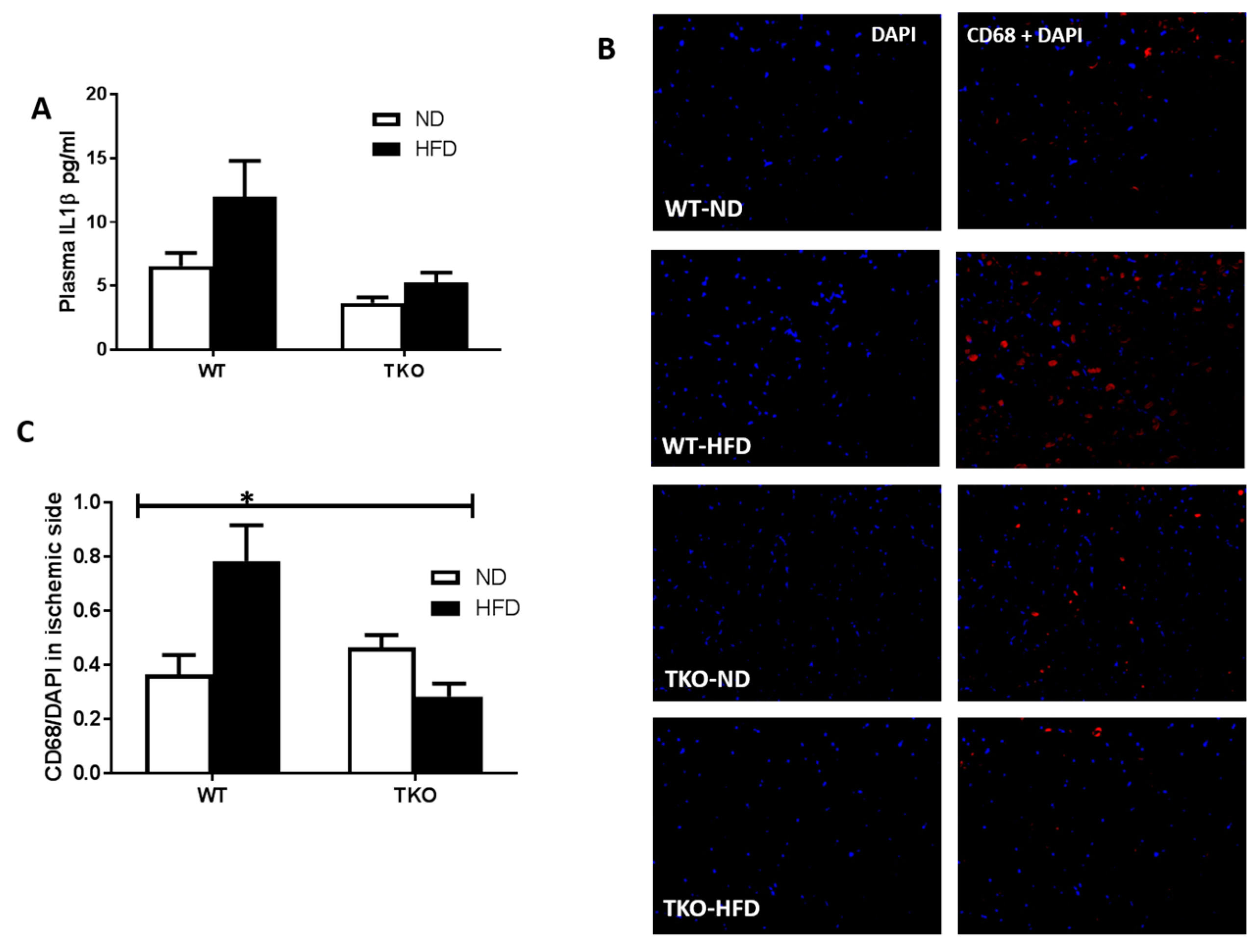
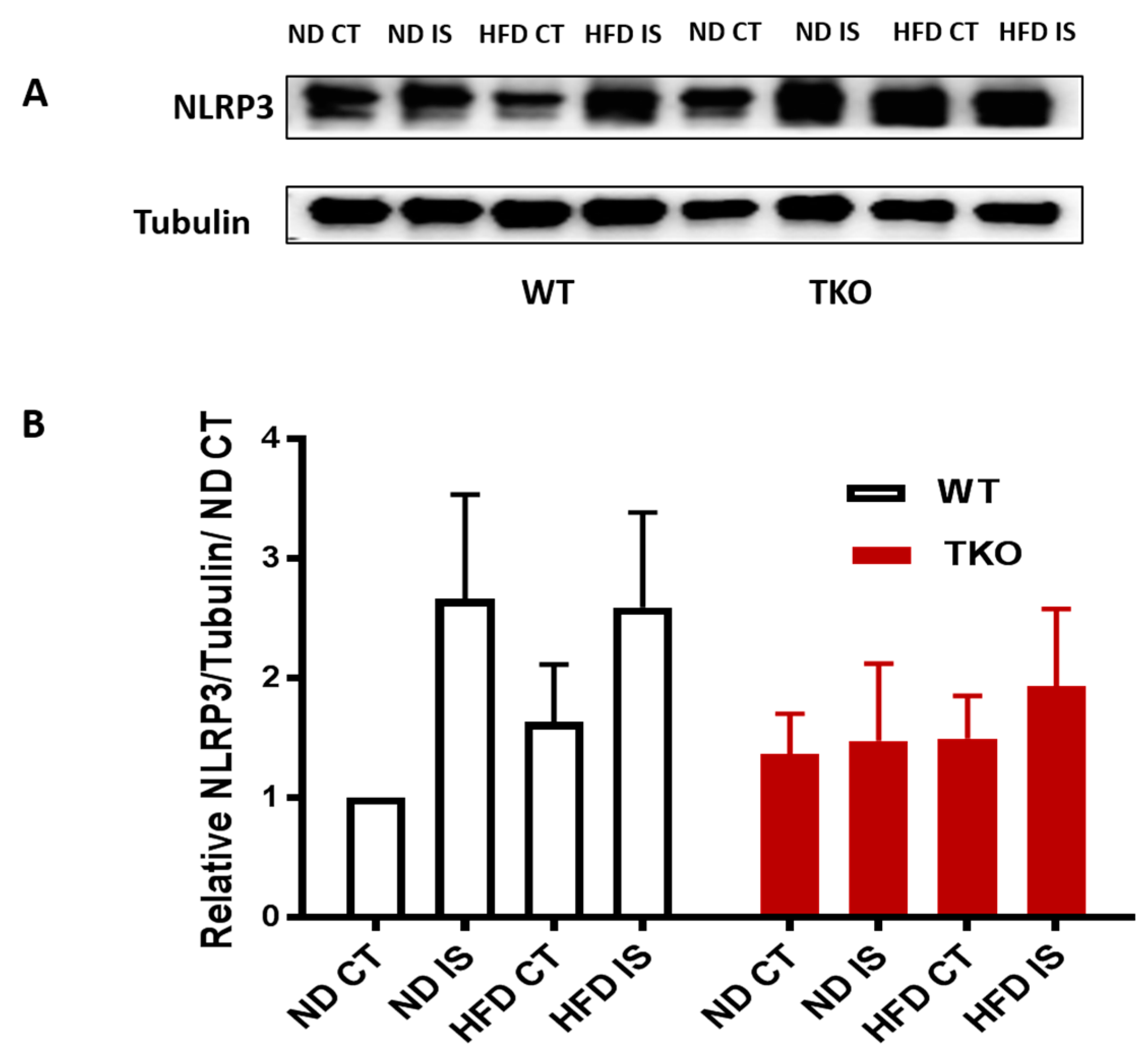
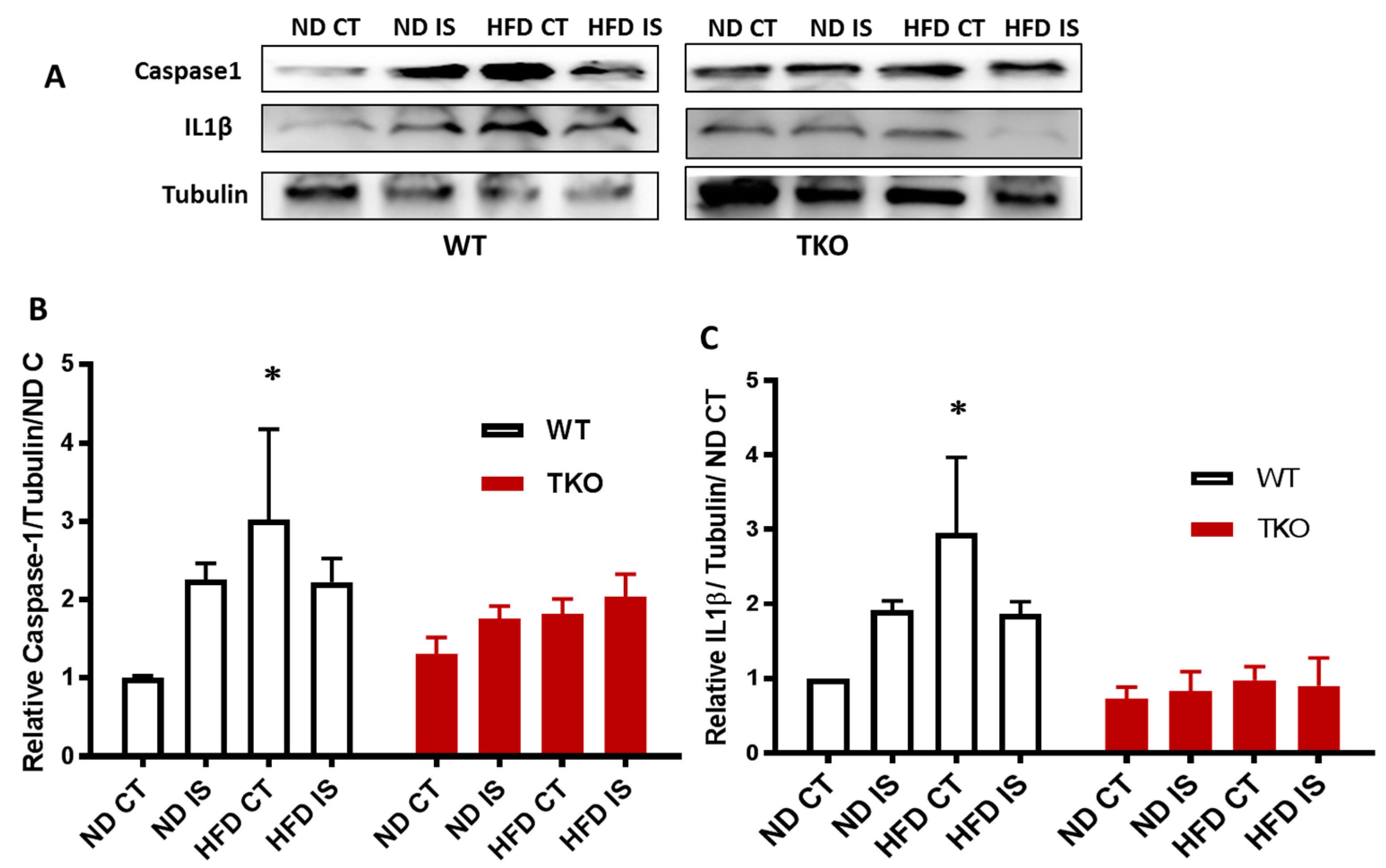
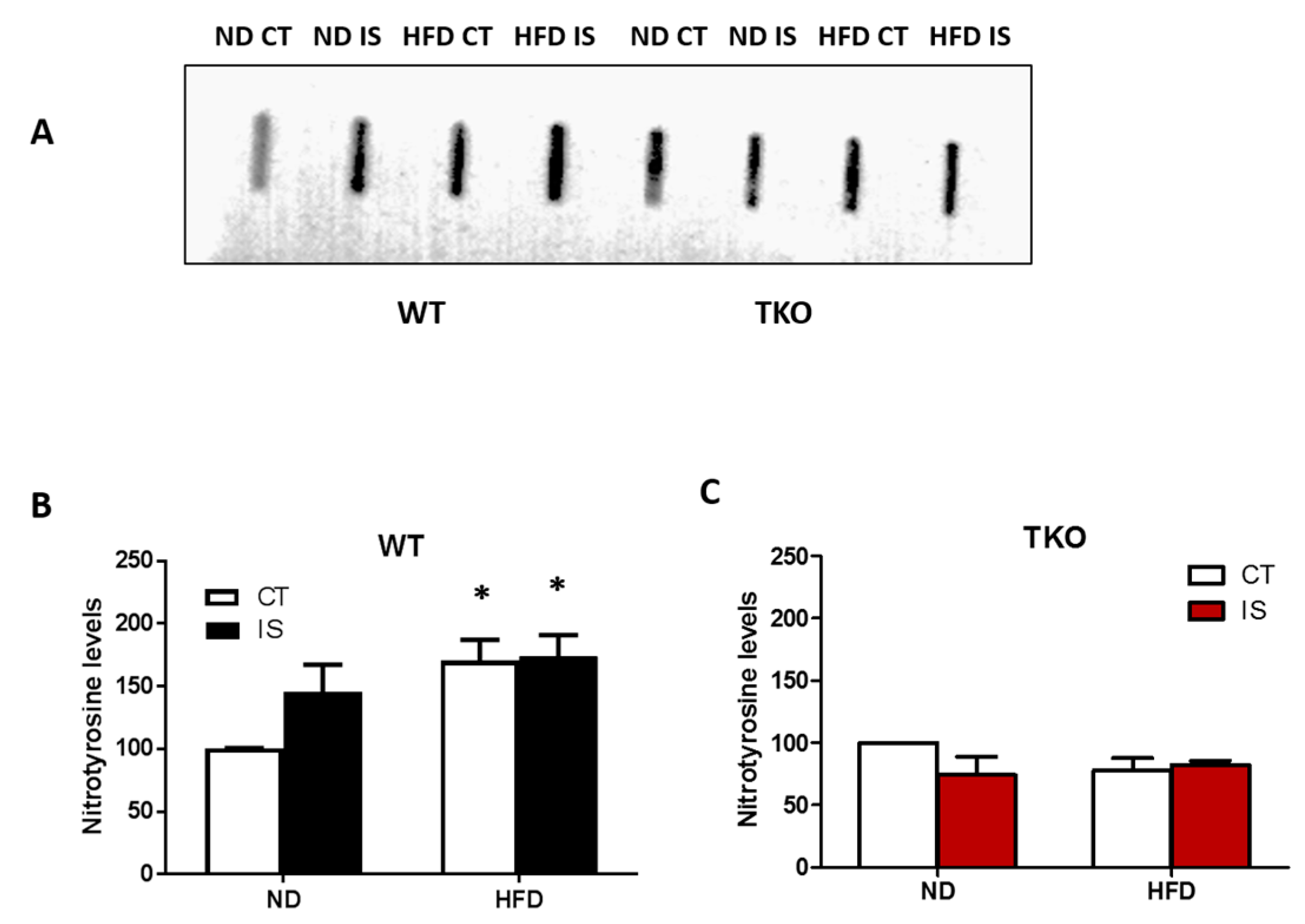
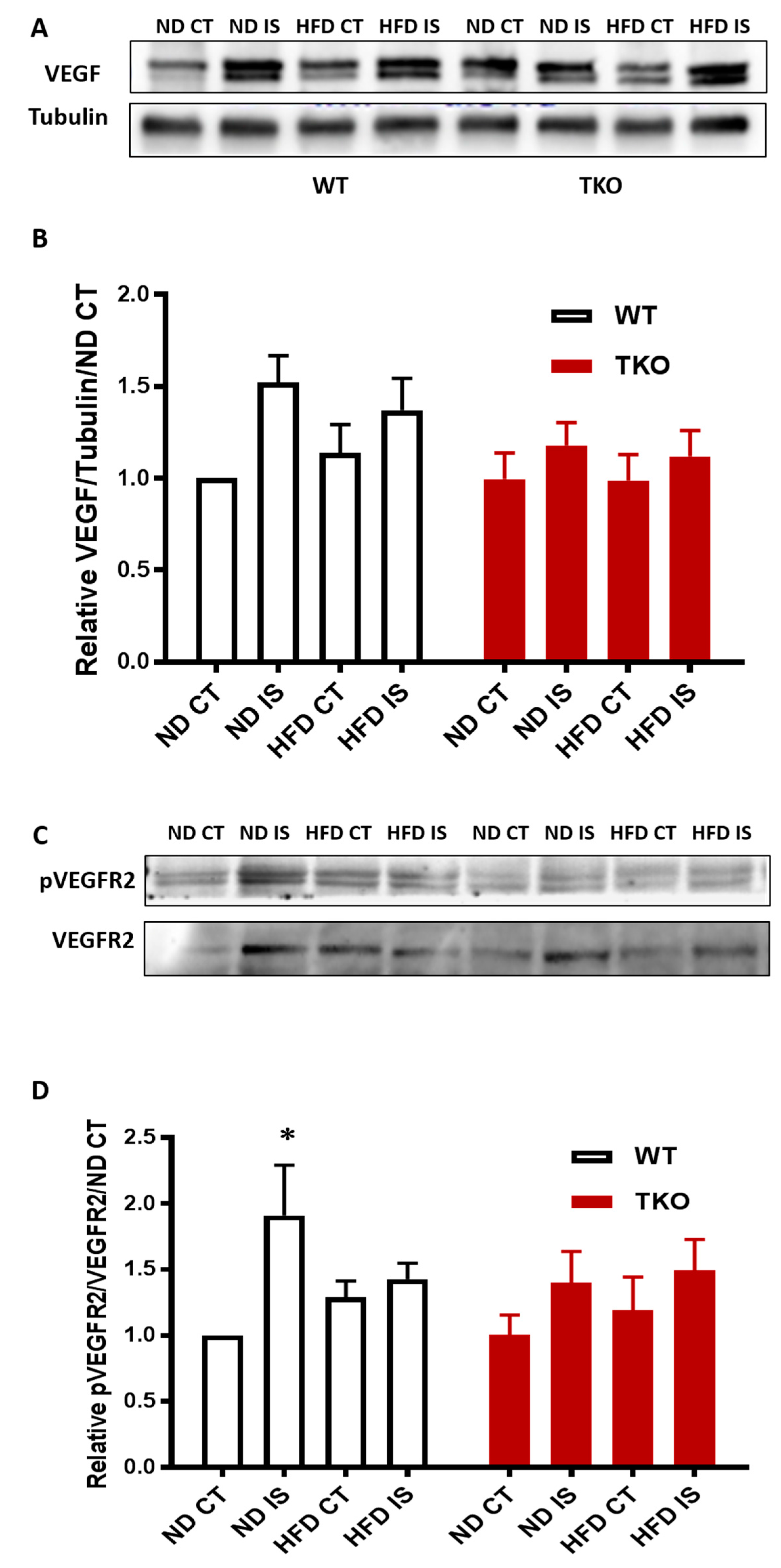
3. Results
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Roger, V.L.; Go, A.S.; Lloyd-Jones, D.M.; Adams, R.J.; Berry, J.D.; Brown, T.M.; Carnethon, M.R.; Dai, S.; de Simone, G.; Ford, E.S.; et al. Heart disease and stroke statistics—2011 update: A report from the American heart association. Circulation 2011, 123, e18–e209. [Google Scholar] [CrossRef] [PubMed]
- Annex, B.H. Therapeutic angiogenesis for critical limb ischaemia. Nat. Rev. Cardiol. 2013, 10, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Norgren, L.; Hiatt, W.R.; Dormandy, J.A.; Nehler, M.R.; Harris, K.A.; Fowkes, F.G. Inter-society consensus for the management of peripheral arterial disease (TASC II). J. Vasc. Surg. 2007, 45, S5–S67. [Google Scholar] [CrossRef] [PubMed]
- Elshaer, S.L. Cell therapy and critical limb ischemia: Evidence and window of opportunity in obesity. Obes. Control Ther. 2016, 3, 1–8. [Google Scholar]
- Albadawi, H.; Oklu, R.; Cormier, N.R.; O’Keefe, R.M.; Heaton, J.T.; Kobler, J.B.; Austen, W.G.; Watkins, M.T. Hind limb ischemia-reperfusion injury in diet-induced obese mice. J. Surg. Res. 2014, 190, 683–691. [Google Scholar] [CrossRef] [PubMed]
- Tsai, T.H.; Chai, H.T.; Sun, C.K.; Yen, C.H.; Leu, S.; Chen, Y.L.; Chung, S.Y.; Ko, S.F.; Chang, H.W.; Wu, C.J.; et al. Obesity suppresses circulating level and function of endothelial progenitor cells and heart function. J. Transl. Med. 2012, 10, 137. [Google Scholar] [CrossRef] [PubMed]
- Kolluru, G.K.; Bir, S.C.; Kevil, C.G. Endothelial dysfunction and diabetes: Effects on angiogenesis, vascular remodeling, and wound healing. Int. J. Vasc. Med. 2012, 2012, 918267. [Google Scholar] [CrossRef] [PubMed]
- Spindel, O.N.; World, C.; Berk, B.C. Thioredoxin interacting protein: Redox dependent and independent regulatory mechanisms. Antioxid. Redox Signal. 2012, 16, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.N.; Ishrat, T.; Fagan, S.C.; El-Remessy, A.B. Role of inflammasome activation in the pathophysiology of vascular diseases of the neurovascular unit. Antioxid. Redox Signal. 2015, 22, 1188–1206. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Mohamed, I.N.; Hafez, S.S.; Fairaq, A.; Ergul, A.; Imig, J.D.; El-Remessy, A.B. Thioredoxin-interacting protein is required for endothelial nlrp3 inflammasome activation and cell death in a rat model of high-fat diet. Diabetologia 2014, 57, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hui, S.T.; Couto, F.M.; Mungrue, I.N.; Davis, D.B.; Attie, A.D.; Lusis, A.J.; Davis, R.A.; Shalev, A. Thioredoxin-interacting protein deficiency induces AKT/BCL-XL signaling and pancreatic beta-cell mass and protects against diabetes. FASEB J. 2008, 22, 3581–3594. [Google Scholar] [CrossRef] [PubMed]
- Coucha, M.; Mohamed, I.N.; Elshaer, S.L.; Mbata, O.; Bartasis, M.L. High fat diet dysregulates microRNA-17–5p and triggers retinal inflammation: Role of er-stress. World J. Diabetes 2011, 8, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Falcon, B.L.; Chintharlapalli, S.; Uhlik, M.T.; Pytowski, B. Antagonist antibodies to vascular endothelial growth factor receptor 2 (VEGFR-2) as anti-angiogenic agents. Pharmacol. Ther. 2016, 164, 204–225. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.L.; Simpson, P.J.; Prosser, H.C.; Lecce, L.; Yuen, G.S.; Buckle, A.; Sieveking, D.P.; Vanags, L.Z.; Lim, P.R.; Chow, R.W.; et al. A critical role for thioredoxin-interacting protein in diabetes-related impairment of angiogenesis. Diabetes 2014, 63, 675–687. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.L.; Buckle, A.M.; Cooke, J.P.; Ng, M.K. The emerging role of the thioredoxin system in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2089–2098. [Google Scholar] [CrossRef] [PubMed]
- Perrone, L.; Devi, T.S.; Hosoya, K.; Terasaki, T.; Singh, L.P. Thioredoxin interacting protein (TXNIP) induces inflammation through chromatin modification in retinal capillary endothelial cells under diabetic conditions. J. Cell. Physiol. 2009, 221, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Farrell, M.R.; Rogers, L.K.; Liu, Y.; Welty, S.E.; Tipple, T.E. Thioredoxin-interacting protein inhibits hypoxia-inducible factor transcriptional activity. Free Radic. Biol. Med. 2010, 49, 1361–1367. [Google Scholar] [CrossRef] [PubMed]
- Adluri, R.S.; Thirunavukkarasu, M.; Zhan, L.; Akita, Y.; Samuel, S.M.; Otani, H.; Ho, Y.S.; Maulik, G.; Maulik, N. Thioredoxin 1 enhances neovascularization and reduces ventricular remodeling during chronic myocardial infarction: A study using thioredoxin 1 transgenic mice. J. Mol. Cell Cardiol. 2011, 50, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.N.; Soliman, S.A.; Alhusban, A.; Matragoon, S.; Pillai, B.A.; Elmarkaby, A.A.; El-Remessy, A.B. Diabetes exacerbates retinal oxidative stress, inflammation, and microvascular degeneration in spontaneously hypertensive rats. Mol. Vis. 2012, 18, 1457–1466. [Google Scholar] [PubMed]
- Hui, S.T.; Andres, A.M.; Miller, A.K.; Spann, N.J.; Potter, D.W.; Post, N.M.; Chen, A.Z.; Sachithanantham, S.; Jung, D.Y.; Kim, J.K.; et al. Txnip balances metabolic and growth signaling via PTEN disulfide reduction. Proc. Natl. Acad. Sci. USA 2008, 105, 3921–3926. [Google Scholar] [CrossRef] [PubMed]
- Abdelsaid, M.A.; Matragoon, S.; El-Remessy, A.B. Thioredoxin-interacting protein expression is required for VEGF-mediated angiogenic signal in endothelial cells. Antioxid. Redox Signal. 2013, 19, 2199–2212. [Google Scholar] [CrossRef] [PubMed]
- Teraa, M.; Conte, M.S.; Moll, F.L.; Verhaar, M.C. Critical limb ischemia: Current trends and future directions. J Am Heart Assoc 2016, 5, e002938. [Google Scholar] [CrossRef] [PubMed]
- Ogden, C.L.; Carroll, M.D.; Fryar, C.D.; Flegal, K.M. Prevalence of obesity among adults and youth: United States, 2011–2014. NCHS Data Brief 2015, 1, 1–8. [Google Scholar]
- Chong, C.R.; Chan, W.P.; Nguyen, T.H.; Liu, S.; Procter, N.E.; Ngo, D.T.; Sverdlov, A.L.; Chirkov, Y.Y.; Horowitz, J.D. Thioredoxin-interacting protein: Pathophysiology and emerging pharmacotherapeutics in cardiovascular disease and diabetes. Cardiovasc. Drugs Ther. 2014, 28, 347–360. [Google Scholar] [CrossRef] [PubMed]
- Minn, A.H.; Hafele, C.; Shalev, A. Thioredoxin-interacting protein is stimulated by glucose through a carbohydrate response element and induces beta-cell apoptosis. Endocrinology 2005, 146, 2397–2405. [Google Scholar] [CrossRef] [PubMed]
- Stoltzman, C.A.; Peterson, C.W.; Breen, K.T.; Muoio, D.M.; Billin, A.N.; Ayer, D.E. Glucose sensing by MondoA:Mlx complexes: A role for hexokinases and direct regulation of thioredoxin-interacting protein expression. Proc. Natl. Acad. Sci. USA 2008, 105, 6912–6917. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Chai, T.F.; He, H.; Hagen, T.; Luo, Y. Thioredoxin-interacting protein (TXNIP) gene expression: Sensing oxidative phosphorylation status and glycolytic rate. J. Biol. Chem. 2010, 285, 25822–25830. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, J.S.; Chatterjee, A.; Castellani, L.W.; Ross, D.A.; Ohmen, J.; Cavalcoli, J.; Wu, C.; Dains, K.M.; Catanese, J.; Chu, M.; et al. Positional cloning of the combined hyperlipidemia gene Hyplip1. Nat. Genet. 2002, 30, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Chutkow, W.A.; Birkenfeld, A.L.; Brown, J.D.; Lee, H.Y.; Frederick, D.W.; Yoshioka, J.; Patwari, P.; Kursawe, R.; Cushman, S.W.; Plutzky, J.; et al. Deletion of the alpha-arrestin protein Txnip in mice promotes adiposity and adipogenesis while preserving insulin sensitivity. Diabetes 2010, 59, 1424–1434. [Google Scholar] [CrossRef] [PubMed]
- Yoshihara, E.; Fujimoto, S.; Inagaki, N.; Okawa, K.; Masaki, S.; Yodoi, J.; Masutani, H. Disruption of TBP-2 ameliorates insulin sensitivity and secretion without affecting obesity. Nat. Commun. 2010, 1, 127. [Google Scholar] [CrossRef] [PubMed]
- Pande, R.L.; Perlstein, T.S.; Beckman, J.A.; Creager, M.A. Association of insulin resistance and inflammation with peripheral arterial disease: The national health and nutrition examination survey, 1999 to 2004. Circulation 2008, 118, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kumagai, H.; Yamaguchi, S.; Kozono, H.; Takahashi, T.; Inoue, M.; Itoh, S.; Takamoto, I.; Sasako, T.; et al. Impaired insulin signaling in endothelial cells reduces insulin-induced glucose uptake by skeletal muscle. Cell Metab. 2011, 13, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Kubota, T.; Kubota, N.; Kadowaki, T. The role of endothelial insulin signaling in the regulation of glucose metabolism. Rev. Endocr. Metab. Disord. 2013, 14, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Tie, G.; Park, B.; Yan, Y.; Nowicki, P.T.; Messina, L.M. Recovery from hind limb ischemia is less effective in type 2 than in type 1 diabetic mice: Roles of endothelial nitric oxide synthase and endothelial progenitor cells. J. Vasc. Surg. 2009, 50, 1412–1422. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lloyd, S.G. High-fat, low-carbohydrate diet alters myocardial oxidative stress and impairs recovery of cardiac function after ischemia and reperfusion in obese rats. Nutr. Res. 2013, 33, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Xiong, L.; Zuo, Z. Trans-sodium crocetinate provides neuroprotection against cerebral ischemia and reperfusion in obese mice. J. Neurosci. Res. 2015, 93, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; He, F.F.; Tang, H.; Lei, C.T.; Chen, S.; Meng, X.F.; Su, H.; Zhang, C. NADPH oxidase-induced NALP3 inflammasome activation is driven by thioredoxin-interacting protein which contributes to podocyte injury in hyperglycemia. J. Diabetes Res. 2015, 2015, 504761. [Google Scholar] [CrossRef] [PubMed]
- 40. Zhang, X.; Zhang, J.H.; Chen, X.Y.; Hu, Q.H.; Wang, M.X.; Jin, R.; Zhang, Q.Y.; Wang, W.; Wang, R.; Kang, L.L.; et al. Reactive oxygen species-induced TXNIP drives fructose-mediated hepatic inflammation and lipid accumulation through NLRP3 inflammasome activation. Antioxid. Redox Signal. 2015, 22, 848–870. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Bao, S.; Yang, W.; Zhang, J.; Li, L.; Shan, Z.; Teng, W. Epigallocatechin gallate prevents inflammation by reducing macrophage infiltration and inhibiting tumor necrosis factor-alpha signaling in the pancreas of rats on a high-fat diet. Nutr. Res. 2014, 34, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.G.; Ko, H.J.; Cho, Y.R.; Kim, H.J.; Ma, Z.; Yu, T.Y.; Friedline, R.H.; Kurt-Jones, E.; Finberg, R.; Fischer, M.A.; et al. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 2009, 58, 2525–2535. [Google Scholar] [CrossRef] [PubMed]
- Abderrazak, A.; El Hadri, K.; Bosc, E.; Blondeau, B.; Slimane, M.N.; Buchele, B.; Simmet, T.; Couchie, D.; Rouis, M. Inhibition of the inflammasome NLRP3 by arglabin attenuates inflammation, protects pancreatic beta-cells from apoptosis, and prevents type 2 diabetes mellitus development in apoe2ki mice on a chronic high-fat diet. J. Pharmacol. Exp. Ther. 2016, 357, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Finucane, O.M.; Lyons, C.L.; Murphy, A.M.; Reynolds, C.M.; Klinger, R.; Healy, N.P.; Cooke, A.A.; Coll, R.C.; McAllan, L.; Nilaweera, K.N.; et al. Monounsaturated fatty acid-enriched high-fat diets impede adipose NLRP3 inflammasome-mediated IL-1beta secretion and insulin resistance despite obesity. Diabetes 2015, 64, 2116–2128. [Google Scholar] [CrossRef] [PubMed]
- Tseng, H.H.; Vong, C.T.; Kwan, Y.W.; Lee, S.M.; Hoi, M.P. TRPM2 regulates TXNIP-mediated NLRP3 inflammasome activation via interaction with p47 phox under high glucose in human monocytic cells. Sci. Rep. 2016, 6, 35016. [Google Scholar] [CrossRef] [PubMed]
- Abderrazak, A.; Syrovets, T.; Couchie, D.; El Hadri, K.; Friguet, B.; Simmet, T.; Rouis, M. NLRP3 inflammasome: From a danger signal sensor to a regulatory node of oxidative stress and inflammatory diseases. Redox Biol. 2015, 4, 296–307. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Jiang, N.; Hu, Y.; Zhang, Y.; Wang, G.; Yin, M.; Ma, X.; Zhou, K.; Qi, J.; Yu, B.; et al. Ruscogenin attenuates cerebral ischemia-induced blood-brain barrier dysfunction by suppressing TXNIP/NLRP3 inflammasome activation and the mapk pathway. Int. J. Mol. Sci. 2016, 17, 1418. [Google Scholar] [CrossRef] [PubMed]
- Bauters, C.; Asahara, T.; Zheng, L.P.; Takeshita, S.; Bunting, S.; Ferrara, N.; Symes, J.F.; Isner, J.M. Site-specific therapeutic angiogenesis after systemic administration of vascular endothelial growth factor. J. Vasc. Surg. 1995, 21, 314–324. [Google Scholar] [CrossRef]
- Walder, C.E.; Errett, C.J.; Bunting, S.; Lindquist, P.; Ogez, J.R.; Heinsohn, H.G.; Ferrara, N.; Thomas, G.R. Vascular endothelial growth factor augments muscle blood flow and function in a rabbit model of chronic hindlimb ischemia. J. Cardiovasc. Pharmacol. 1996, 27, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Shi, X.; Pang, J.; Yan, C.; Berk, B.C. Thioredoxin-interacting protein mediates sustained VEGFR2 signaling in endothelial cells required for angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Spindel, O.N.; Yan, C.; Berk, B.C. Thioredoxin-interacting protein mediates nuclear-to-plasma membrane communication: Role in vascular endothelial growth factor 2 signaling. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1264–1270. [Google Scholar] [CrossRef] [PubMed]
- Abdelsaid, M.A.; Matragoon, S.; Ergul, A.; El-Remessy, A.B. Deletion of thioredoxin interacting protein (TXNIP) augments hyperoxia-induced vaso-obliteration in a mouse model of oxygen induced-retinopathy. PLoS ONE 2014, 9, e110388. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibodies | Catalog Number | Company |
|---|---|---|
| anti-VEGF | Cat.# 676472 Cat.# ABS82 | Abcam Millipore |
| anti-pVEGF receptor-2 (Tyr996) | Cat.#2474 | Cell Signaling |
| anti-VEGF receptor 2 | Cat.#2472 | |
| anti-GAPDH | Cat.# 5174 | |
| anti-IL-1β | Cat.# ab9722 | Abcam |
| anti-tubulin | Cat.# ab4074 | |
| anti-NLRP-3 | Cat.# LS-B4321 | LifeSpan Biosciences |
| anti-nitrotyrosine | Cat.# 05-2333 | Millipore |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elshaer, S.L.; Mohamed, I.N.; Coucha, M.; Altantawi, S.; Eldahshan, W.; Bartasi, M.L.; Shanab, A.Y.; Lorys, R.; El-Remessy, A.B. Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia. Antioxidants 2017, 6, 47. https://doi.org/10.3390/antiox6030047
Elshaer SL, Mohamed IN, Coucha M, Altantawi S, Eldahshan W, Bartasi ML, Shanab AY, Lorys R, El-Remessy AB. Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia. Antioxidants. 2017; 6(3):47. https://doi.org/10.3390/antiox6030047
Chicago/Turabian StyleElshaer, Sally L., Islam N. Mohamed, Maha Coucha, Sara Altantawi, Wael Eldahshan, Megan L. Bartasi, Ahmed Y. Shanab, Renee Lorys, and Azza B. El-Remessy. 2017. "Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia" Antioxidants 6, no. 3: 47. https://doi.org/10.3390/antiox6030047
APA StyleElshaer, S. L., Mohamed, I. N., Coucha, M., Altantawi, S., Eldahshan, W., Bartasi, M. L., Shanab, A. Y., Lorys, R., & El-Remessy, A. B. (2017). Deletion of TXNIP Mitigates High-Fat Diet-Impaired Angiogenesis and Prevents Inflammation in a Mouse Model of Critical Limb Ischemia. Antioxidants, 6(3), 47. https://doi.org/10.3390/antiox6030047