Superoxide Dismutases in Pancreatic Cancer


{kind=link}
Abstract
:1. Introduction
2. Pancreatic Adenocarcinoma
3. Superoxide and Superoxide Dismutase
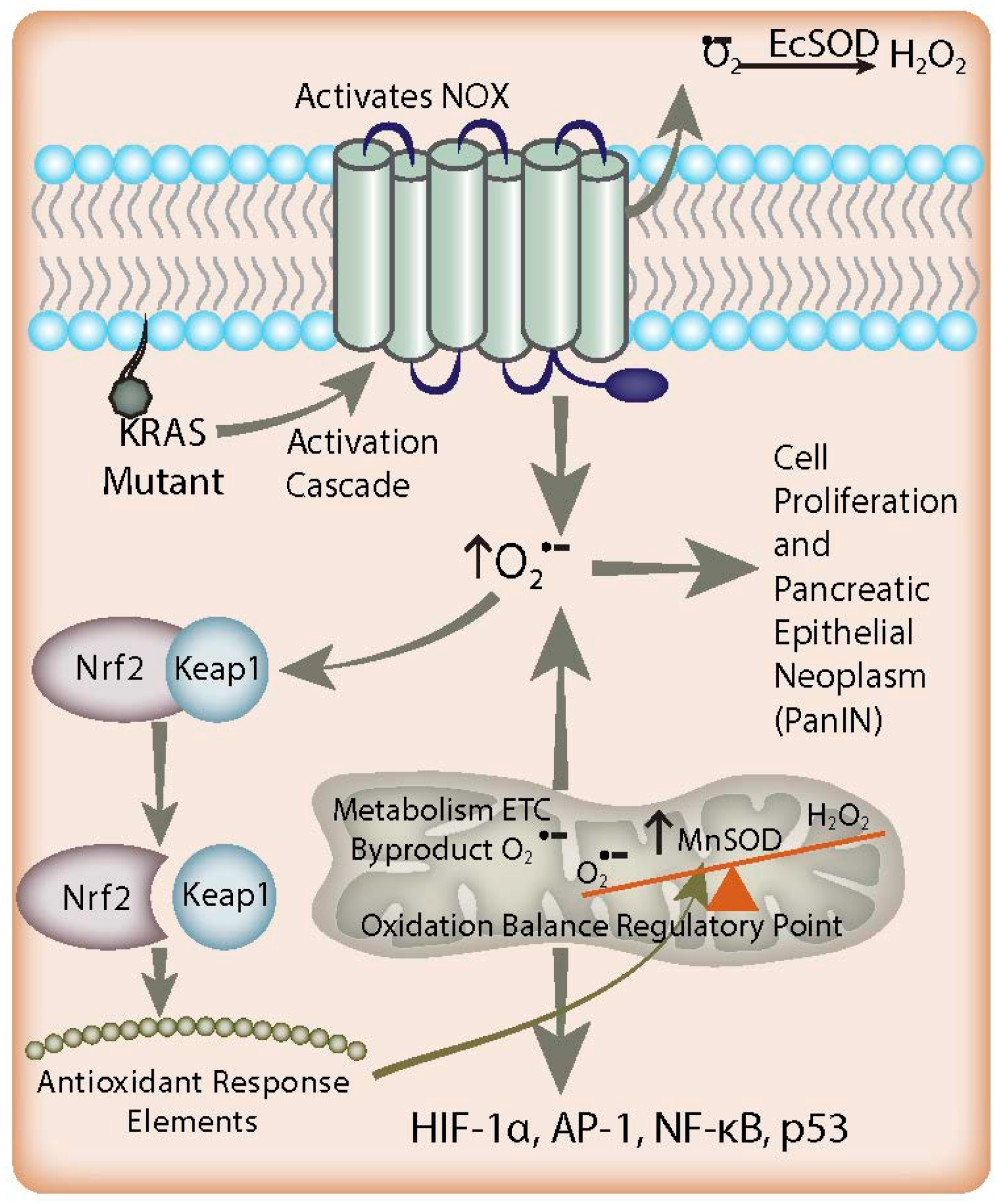
4. SOD, Superoxide and the Pancreas
5. Nrf2 Antioxidant Supports Pancreatic Cancer (PDAC) Development
6. Redox-Based Therapy in Pancreatic Cancer
6.1. Pharmacologic Ascorbate
6.2. Manganoporphyrins
6.3. Induction of ROS
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Ryerson, A.B.; Eheman, C.R.; Altekruse, S.F.; Ward, J.W.; Jemal, A.; Sherman, R.L.; Henley, S.J.; Holtzman, D.; Lake, A.; Noone, A.M.; et al. Annual report to the nation on the status of cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer 2016, 122, 1312–1337. [Google Scholar] [CrossRef] [PubMed]
- Durand, N.; Storz, P. Targeting reactive oxygen species in development and progression of pancreatic cancer. Expert Rev. Anticancer Ther. 2017, 17, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Nazee, J.; Joseph, J.C. The role of antioxidant enzymes in the growth of pancreatic carcinoma. Curr. Cancer Ther. Rev. 2007, 3, 61–65. [Google Scholar]
- Weydert, C.; Roling, B.; Liu, J.; Hinkhouse, M.M.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Suppression of the malignant phenotype in human pancreatic cancer cells by the overexpression of manganese superoxide dismutase. Mol. Cancer Ther. 2003, 2, 361–369. [Google Scholar] [PubMed]
- Teoh, M.L.; Sun, W.; Smith, B.J.; Oberley, L.W.; Cullen, J.J. Modulation of reactive oxygen species in pancreatic cancer. Clin. Cancer Res. 2007, 13, 7441–7450. [Google Scholar] [CrossRef] [PubMed]
- Cullen, J.J.; Weydert, C.; Hinkhouse, M.M.; Ritchie, J.; Domann, F.E.; Spitz, D.; Oberley, L.W. The role of manganese superoxide dismutase in the growth of pancreatic adenocarcinoma. Cancer Res. 2003, 63, 1297–1303. [Google Scholar] [PubMed]
- Sibenaller, Z.A.; Welsh, J.L.; Du, C.; Witmer, J.R.; Schrock, H.E.; Du, J.; Buettner, G.R.; Goswami, P.C.; Cieslak, J.A.; Cullen, J.J. Extracellular superoxide dismutase (EcSOD) suppresses hypoxia-inducible factor-1α (HIF-1α) in pancreatic cancer. Free Radic. Biol. Med. 2014, 69, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.E.; Armstrong, S.A.; Coussens, L.M.; Cruz-Correa, M.R.; DeBerardinis, R.J.; Doroshow, J.H.; Foti, M.; Hwu, P.; Kensler, T.W.; Morrow, M.; et al. Aacr cancer progress report 2016. Clin. Cancer Res. 2016, 22, S1–S137. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R.; Oberley, L.W.; Leuthauser, S.W. The effect of iron on the distribution of superoxide and hydroxyl radicals as seen by spin trapping and on the superoxide dismutase assay. Photochem. Photobiol. 1978, 693–695. [Google Scholar] [CrossRef]
- Buettner, G.R. Superoxide dismutase in redox biology: The roles of superoxide and hydrogen peroxide. Anticancer Agents Med. Chem. 2011, 11, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Hayyan, M.; Hashim, M.A.; AlNashef, I.M. Superoxide ion: Generation and chemical implications. Chem. Rev. 2016, 116, 3029–3085. [Google Scholar] [CrossRef] [PubMed]
- Kudin, A.P.; Bimpong-Buta, N.Y.; Vielhaber, S.; Elger, C.E.; Kunz, W.S. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004, 279, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Meitzler, J.L.; Antony, S.; Wu, Y.; Juhasz, A.; Liu, H.; Jiang, G.; Lu, J.; Roy, K.; Doroshow, J.H. NADPH oxidases: A perspective on reactive oxygen species production in tumor biology. Antioxid Redox Signal. 2014, 20, 2873–2889. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Valentine, J.S.; Pantoliano, M.W.; McDonnell, P.J.; Burger, A.R.; Lippard, S.J. pH-dependent migration of copper(II) to the vacant zinc-binding site of zinc-free bovine erythrocyte superoxide dismutase. Proc. Natl. Acad. Sci. USA 1979, 76, 4245–4249. [Google Scholar] [CrossRef] [PubMed]
- Hart, P.J.; Balbirnie, M.M.; Ogihara, N.L.; Nersissian, A.M.; Weiss, M.S.; Valentine, J.S.; Eisenberg, D. A structure-based mechanism for copper-zinc superoxide dismutase. Biochemistry 1999, 38, 2167–2178. [Google Scholar] [CrossRef] [PubMed]
- Fetherolf, M.M.; Boyd, S.D.; Taylor, A.B.; Kim, H.J.; Wohlschlegel, J.A.; Blackburn, N.J.; Hart, P.J.; Winge, D.R.; Winkler, D.D. Copper-zinc superoxide dismutase is activated through a sulfenic acid intermediate at a copper-ion entry site. J. Biol. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- McCord, J.M.; Fridovich, I. Superoxide dismutase: An enzymic function for erythrocuprein (hemocuprein). J. Biol. Chem. 1969, 244, 6049–6055. [Google Scholar] [PubMed]
- Miriyala, S.; Spasojevic, I.; Tovmasyan, A.; Salvemini, D.; Vujaskovic, Z.; St Clair, D.; Batinic-Haberle, I. Manganese superoxide dismutase, MnSOD and its mimics. Biochim. Biophys. Acta 2012, 1822, 794–814. [Google Scholar] [CrossRef] [PubMed]
- Lebovitz, R.M.; Zhang, H.; Vogel, H.; Cartwright, J., Jr.; Dionne, L.; Lu, N.; Huang, S.; Matzuk, M.M. Neurodegeneration, myocardial injury, and perinatal death in mitochondrial superoxide dismutase-deficient mice. Proc. Natl. Acad. Sci. USA 1996, 93, 9782–9787. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.G.; Goswami, P.C. A redox cycle within the cell cycle: Ring in the old with the new. Oncogene 2007, 26, 1101–1109. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W.; Oberley, T.D.; Buettner, G.R. Cell differentiation, aging and cancer: The possible roles of superoxide and superoxide dismutases. Med. Hypotheses 1980, 6, 249–268. [Google Scholar] [CrossRef]
- Li, S.; Yan, T.; Yang, J.Q.; Oberley, T.D.; Oberley, L.W. The role of cellular glutathione peroxidase redox regulation in the suppression of tumor cell growth by manganese superoxide dismutase. Cancer Res. 2000, 60, 3927–3939. [Google Scholar] [PubMed]
- Marklund, S.L.; Holme, E.; Hellner, L. Superoxide dismutase in extracellular fluids. Clin. Chim. Acta 1982, 126, 41–51. [Google Scholar] [CrossRef]
- Weisiger, R.A.; Fridovich, I. Superoxide dismutase. Organelle specificity. J. Biol. Chem. 1973, 248, 3582–3592. [Google Scholar] [PubMed]
- Marklund, S.L. Extracellular superoxide dismutase in human tissues and human cell lines. J. Clin. Invest. 1984, 74, 1398–1403. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, L.M.; Jonsson, J.; Edlund, T.; Marklund, S.L. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc. Natl. Acad. Sci. USA 1995, 92, 6264–6268. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, J.; Carlsson, L.; Marklund, S.L.; Edlund, T. The heparin-binding domain of extracellular superoxide dismutase C and formation of variants with reduced heparin affinity. J. Biol. Chem. 1992, 267, 18205–18209. [Google Scholar] [PubMed]
- Wheeler, M.D.; Smutney, O.M.; Samulski, R.J. Secretion of extracellular superoxide dismutase from muscle transduced with recombinant adenovirus inhibits the growth of B16 melanomas in mice. Mol. Cancer Res. 2003, 1, 871–881. [Google Scholar] [PubMed]
- Cerutti, P.A. Prooxidant states and tumor promotion. Science 1985, 227, 375–381. [Google Scholar] [CrossRef]
- Du, J.; Nelson, E.S.; Simons, A.L.; Olney, K.E.; Moser, J.C.; Schrock, H.E.; Wagner, B.A.; Buettner, G.R.; Smith, B.J.; Teoh, M.L.; et al. Regulation of pancreatic cancer growth by superoxide. Mol. Carcinog. 2013, 52, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.G.; Balin, A.K. Oxidative influence on development and differentiation: An overview of a free radical theory of development. Free Radic. Biol. Med. 1989, 6, 631–661. [Google Scholar] [CrossRef]
- Hockenbery, D.M.; Oltvai, Z.N.; Yin, X.M.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993, 75, 241–251. [Google Scholar] [CrossRef]
- Tang, H.; Dong, X.; Day, R.S.; Hassan, M.M.; Li, D. Antioxidant genes, diabetes and dietary antioxidants in association with risk of pancreatic cancer. Carcinogenesis 2010, 31, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Wheatley-Price, P.; Asomaning, K.; Reid, A.; Zhai, R.; Su, L.; Zhou, W.; Zhu, A.; Ryan, D.P.; Christiani, D.C.; Liu, G. Myeloperoxidase and superoxide dismutase polymorphisms are associated with an increased risk of developing pancreatic adenocarcinoma. Cancer 2008, 112, 1037–1042. [Google Scholar] [CrossRef] [PubMed]
- Mohelnikova-Duchonova, B.; Marsakova, L.; Vrana, D.; Holcatova, I.; Ryska, M.; Smerhovsky, Z.; Slamova, A.; Schejbalova, M.; Soucek, P. Superoxide dismutase and nicotinamide adenine dinucleotide phosphate: Quinone oxidoreductase polymorphisms and pancreatic cancer risk. Pancreas 2011, 40, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Kikuchi, S.; Yagyu, K.; Ishibashi, T.; Kurosawa, M.; Ito, Y.; Watanabe, Y.; Inaba, Y.; Tajima, K.; Nakachi, K.; et al. Serum soluble fas levels and superoxide dismutase activity and the risk of death from pancreatic cancer: A nested case-control study within the Japanese collaborative cohort study. Asian Pac. J. Cancer Prev. 2009, 10 Suppl, 81–85. [Google Scholar] [PubMed]
- Vaquero, E.C.; Edderkaoui, M.; Pandol, S.J.; Gukovsky, I.; Gukovskaya, A.S. Reactive oxygen species produced by NAD(P)H oxidase inhibit apoptosis in pancreatic cancer cells. J. Biol. Chem. 2004, 279, 34643–34654. [Google Scholar] [CrossRef] [PubMed]
- MacMillan-Crow, L.A.; Greendorfer, J.S.; Vickers, S.M.; Thompson, J.A. Tyrosine nitration of c-SRC tyrosine kinase in human pancreatic ductal adenocarcinoma. Arch. Biochem. Biophys. 2000, 377, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-Q.; Li, S.; Domann, F.E.; Buettner, G.R.; Oberley, L.W. Superoxide generation in v-Ha-ras–transduced human keratinocyte hacat cells. Mol. Carcinog. 1999, 26, 180–188. [Google Scholar] [CrossRef]
- Qian, J.; Niu, J.; Li, M.; Chiao, P.J.; Tsao, M.S. In vitro modeling of human pancreatic duct epithelial cell transformation defines gene expression changes induced by K-ras oncogenic activation in pancreatic carcinogenesis. Cancer Res. 2005, 65, 5045–5053. [Google Scholar] [CrossRef] [PubMed]
- Ju, H.Q.; Ying, H.; Tian, T.; Ling, J.; Fu, J.; Lu, Y.; Wu, M.; Yang, L.; Achreja, A.; Chen, G.; et al. Mutant Kras- and p16-regulated NOX4 activation overcomes metabolic checkpoints in development of pancreatic ductal adenocarcinoma. Nat. Commun. 2017, 8, 14437. [Google Scholar] [CrossRef] [PubMed]
- Liou, G.-Y.; Döppler, H.; DelGiorno, K.E.; Zhang, L.; Leitges, M.; Crawford, H.C.; Murphy, M.P.; Storz, P. Mutant KRas-Induced mitochondrial oxidative stress in acinar cells upregulates EGFR signaling to drive formation of pancreatic precancerous lesions. Cell Rep. 2016, 14, 2325–2336. [Google Scholar] [CrossRef] [PubMed]
- Löhr, M.; Klöppel, G.; Maisonneuve, P.; Lowenfels, A.B.; Lüttges, J. Frequency of K-ras mutations in pancreatic intraductal neoplasias associated with pancreatic ductal adenocarcinoma and chronic pancreatitis: A meta-analysis. Neoplasia 2005, 7, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Irani, K.; Xia, Y.; Zweier, J.L.; Sollott, S.J.; Der, C.J.; Fearon, E.R.; Sundaresan, M.; Finkel, T.; Goldschmidt-Clermont, P.J. Mitogenic signaling mediated by oxidants in ras-transformed fibroblasts. Science (New York, N.Y.) 1997, 275, 1649–1652. [Google Scholar] [CrossRef]
- Wang, W.; Abbruzzese, J.L.; Evans, D.B.; Larry, L.; Cleary, K.R.; Chiao, P.J. The nuclear factor-κB RelAl transcription factor is constitutively activated in human pancreatic adenocarcinoma cells. Clin. Cancer Res. 1999, 5, 119–127. [Google Scholar] [PubMed]
- Wu, Y.; Antony, S.; Meitzler, J.L.; Doroshow, J.H. Molecular mechanisms underlying chronic inflammation-associated cancers. Cancer Lett. 2014, 345, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Song, M.; Zeng, Z.-L.; Zhu, C.-F.; Lu, W.-H.; Yang, J.; Ma, M.-Z.; Huang, A.M.; Hu, Y.; Huang, P. Identification of NDUFAF1 in mediating K-Ras induced mitochondrial dysfunction by a proteomic screening approach. Oncotarget 2015, 6, 3947–3962. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Hinkhouse, M.M.; Sun, W.; Weydert, C.J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Redox regulation of pancreatic cancer cell growth: Role of glutathione peroxidase in the suppression of the malignant phenotype. Hum. Gene Ther. 2004, 15, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.G.; Sarsour, E.H.; Kalen, A.L.; Venkataraman, S.; Hitchler, M.J.; Domann, F.E.; Oberley, L.W.; Goswami, P.C. Superoxide signaling mediates N-acetyl-L-cysteine-induced G1 arrest: Regulatory role of cyclin D1 and manganese superoxide dismutase. Cancer Res. 2007, 67, 6392–6399. [Google Scholar] [CrossRef] [PubMed]
- Menon, S.G.; Sarsour, E.H.; Spitz, D.R.; Higashikubo, R.; Sturm, M.; Zhang, H.; Goswami, P.C. Redox regulation of the G1 to S phase transition in the mouse embryo fibroblast cell cycle. Cancer Res. 2003, 63, 2109–2117. [Google Scholar] [PubMed]
- Sarsour, E.H.; Kalen, A.L.; Xiao, Z.; Veenstra, T.D.; Chaudhuri, L.; Venkataraman, S.; Reigan, P.; Buettner, G.R.; Goswami, P.C. Manganese superoxide dismutase regulates a metabolic switch during the mammalian cell cycle. Cancer Res. 2012, 72, 3807–3816. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; Posener, K.; Negelein, E. Ueber den stoffwechsel der tumoren. Biochem. Zeitschrift 1924, 319–344. [Google Scholar]
- Hu, Y.; Rosen, D.G.; Zhou, Y.; Feng, L.; Yang, G.; Liu, J.; Huang, P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: Role in cell proliferation and response to oxidative stress. J. Biol. Chem. 2005, 280, 39485–39492. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W. Anticancer therapy by overexpression of superoxide dismutase. Antioxid. Redox Signal. 2001, 3, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, M.; Tamae, D.; Hou, D.X.; Wang, T.; Lebon, T.; Spitz, D.R.; Li, J.J. Response of cyclin B1 to ionizing radiation: Regulation by NF-κB and mitochondrial antioxidant enzyme MnSOD. Anticancer Res. 2004, 24, 2657–2663. [Google Scholar] [PubMed]
- Venkataraman, S.; Jiang, X.; Weydert, C.; Zhang, Y.; Zhang, H.J.; Goswami, P.C.; Ritchie, J.M.; Oberley, L.W.; Buettner, G.R. Manganese superoxide dismutase overexpression inhibits the growth of androgen-independent prostate cancer cells. Oncogene 2005, 24, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Weydert, C.J.; Waugh, T.A.; Ritchie, J.M.; Iyer, K.S.; Smith, J.L.; Li, L.; Spitz, D.R.; Oberley, L.W. Overexpression of manganese or copper-zinc superoxide dismutase inhibits breast cancer growth. Free Radic. Biol. Med. 2006, 41, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Oberley, L.W.; Buettner, G.R. Role of superoxide dismutase in cancer: A review. Cancer Res. 1979, 39, 1141–1149. [Google Scholar] [PubMed]
- Loven, D.P.; Oberley, L.W.; Rousseau, F.M.; Stevens, R.H. Superoxide dismutase activity in 1,2-dimethylhydrazine-induced rat colon adenocarcinoma. J. Natl. Cancer Inst. 1980, 65, 377–381. [Google Scholar] [PubMed]
- Kodydkova, J.; Vavrova, L.; Stankova, B.; Macasek, J.; Krechler, T.; Zak, A. Antioxidant status and oxidative stress markers in pancreatic cancer and chronic pancreatitis. Pancreas 2013, 42, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Belinsky, G.; Sagheer, U.; Zhang, X.; Grippo, P.J.; Chung, C. Pigment epithelium-derived factor (PEDF) blocks Wnt3a protein-induced autophagy in pancreatic intraepithelial neoplasms. J. Biol. Chem. 2016, 291, 22074–22085. [Google Scholar] [CrossRef] [PubMed]
- Hurt, E.M.; Thomas, S.B.; Peng, B.; Farrar, W.L. Molecular consequences of SOD2 expression in epigenetically silenced pancreatic carcinoma cell lines. Br. J. Cancer 2007, 97, 1116–1123. [Google Scholar] [CrossRef] [PubMed]
- Pandit, H.; Zhang, W.; Li, Y.; Agle, S.; Li, X.; Li, S.P.; Cui, G.; Li, Y.; Martin, R.C. Manganese superoxide dismutase (MnSOD) expression is negatively associated with microRNA-301a in human Pancreatic Ductal Adenocarcinoma (PDAC). Cancer Gene Ther. 2015, 22, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Kilian, M.; Gregor, J.I.; Heukamp, I.; Helmecke, K.; Hanel, M.; Wassersleben, B.; Walz, M.K.; Schimke, I.; Kristiansen, G.; Wenger, F.A. Impact of Octreotide and SOM-230 on liver metastasis and hepatic lipidperoxidation in ductal pancreatic adenocarcinoma in Syrian Hamster. Clin. Exp. Metastasis 2009, 26, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Smith, B.J.; Oberley, L.W. Enzymatic activity is necessary for the tumor-suppressive effects of MnSOD. Antioxid. Redox Signal. 2006, 8, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Ough, M.; Lewis, A.; Zhang, Y.; Hinkhouse, M.M.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Inhibition of cell growth by overexpression of manganese superoxide dismutase (MnSOD) in human pancreatic carcinoma. Free Radic. Res. 2004, 38, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Du, J.; Liu, J.; Ritchie, J.M.; Oberley, L.W.; Cullen, J.J. Metastatic progression of pancreatic cancer: Changes in antioxidant enzymes and cell growth. Clin. Exp. Metastasis 2005, 22, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Heukamp, I.; Gregor, J.I.; Kilian, M.; Kiewert, C.; Jacobi, C.A.; Schimke, I.; Walz, M.K.; Guski, H.; Wenger, F.A. Influence of different dietary fat intake on liver metastasis and hepatic lipid peroxidation in BOP-induced pancreatic cancer in syrian hamsters. Pancreatology 2006, 6, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Ma, Z.; Ma, J.; Li, X.; Xu, Q.; Duan, W.; Chen, X.; Lv, Y.; Zhou, S.; Wu, E.; et al. Hydrogen peroxide mediates hyperglycemia-induced invasive activity via ERK and p38 MAPK in human pancreatic cancer. Oncotarget 2015, 6, 31119–31133. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Li, J.; Chen, X.; Chen, K.; Li, W.; Li, X.; Zhang, L.; Duan, W.; Lei, J.; Xu, Q.; et al. Lipoxin A4 attenuates cell invasion by inhibiting ROS/ERK/MMP pathway in pancreatic cancer. Oxid. Med. Cell. Longev. 2016, 2016, 6815727. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Ewton, D.Z.; Friedman, E. Mirk/Dyrk1B maintains the viability of quiescent pancreatic cancer cells by reducing levels of reactive oxygen species. Cancer Res. 2009, 69, 3317–3324. [Google Scholar] [CrossRef] [PubMed]
- Ewton, D.Z.; Hu, J.; Vilenchik, M.; Deng, X.; Luk, K.C.; Polonskaia, A.; Hoffman, A.F.; Zipf, K.; Boylan, J.F.; Friedman, E.A. Inactivation of Mirk/Dyrk1B kinase targets quiescent pancreatic cancer cells. Mol. Cancer Ther. 2011, 10, 2104–2114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.X.; Qin, Y.M.; Guo, L.K. Correlations between polymorphisms of extracellular superoxide dismutase, aldehyde dehydrogenase-2 genes, as well as drinking behavior and pancreatic cancer. Chin. Med. Sci. J. 2014, 29, 162–166. [Google Scholar] [CrossRef]
- O’Leary, B.R.; Fath, M.A.; Bellizzi, A.M.; Hrabe, J.E.; Button, A.M.; Allen, B.G.; Case, A.J.; Altekruse, S.; Wagner, B.A.; Buettner, G.R.; et al. Loss of SOD3 (EcSOD) expression promotes an aggressive phenotype in human pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2015, 21, 1741–1751. [Google Scholar] [CrossRef] [PubMed]
- Maitra, A.; Hruban, R.H. Pancreatic cancer. Annu. Rev. Pathol. 2008, 3, 157–188. [Google Scholar] [CrossRef] [PubMed]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Meier, B.; Radeke, H.H.; Selle, S.; Younes, M.; Sies, H.; Resch, K.; Habermehl, G.G. Human fibroblasts release reactive oxygen species in response to interleukin-1 or tumour necrosis factor-alpha. Biochem. J. 1989, 263, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Shiga, K.; Hara, M.; Nagasaki, T.; Sato, T.; Takahashi, H.; Takeyama, H. Cancer-associated fibroblasts: Their characteristics and their roles in tumor growth. Cancers 2015, 7, 2443–2458. [Google Scholar] [CrossRef] [PubMed]
- Sarsour, E.H.; Goswami, M.; Kalen, A.L.; Goswami, P.C. Mnsod activity protects mitochondrial morphology of quiescent fibroblasts from age associated abnormalities. Mitochondrion 2010, 10, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N. Nrf2: Friend or foe for chemoprevention? Carcinogenesis 2010, 31, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Pickett, C.B. Regulatory mechanisms controlling gene expression mediated by the antioxidant response element. Annu. Rev. Pharmacol Toxicol 2003, 43, 233–260. [Google Scholar] [CrossRef] [PubMed]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Hybertson, B.M.; Gao, B.; Bose, S.K.; McCord, J.M. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol. Aspects Med. 2011, 32, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, H.M.; Kansanen, E.; Polonen, P.; Heinaniemi, M.; Levonen, A.L. Role of the keap1-Nrf2 pathway in cancer. Adv. Cancer Res. 2014, 122, 281–320. [Google Scholar] [PubMed]
- Chio, I.I.C.; Jafarnejad, S.M.; Ponz-Sarvise, M.; Park, Y.; Rivera, K.; Palm, W.; Wilson, J.; Sangar, V.; Hao, Y.; Ohlund, D.; et al. Nrf2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell 2016, 166, 963–976. [Google Scholar]
- Zhang, P.; Singh, A.; Yegnasubramanian, S.; Esopi, D.; Kombairaju, P.; Bodas, M.; Wu, H.; Bova, S.G.; Biswal, S. Loss of Kelch-like ECH-associated protein 1 function in prostate cancer cells causes chemoresistance and radioresistance and promotes tumor growth. Mol. Cancer Ther. 2010, 9, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.B.; Kang, H.J.; Kwon, S.Y.; Kim, H.J.; Kwon, K.Y.; Cho, C.H.; Lee, J.-M.; Kallakury, B.V.S.; Bae, I. Nrf2 regulates drug resistance in pancreatic cancer cells. Pancreas 2010, 39, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, J.; Zong, L.; Chen, X.; Chen, K.; Jiang, Z.; Nan, L.; Li, X.; Li, W.; Shan, T.; et al. Reactive oxygen species and targeted therapy for pancreatic cancer. Oxid. Med. Cell. Longev. 2016, 2016, 1616781. [Google Scholar] [CrossRef] [PubMed]
- Che, M.; Wang, R.; Li, X.; Wang, H.Y.; Zheng, X.F. Expanding roles of superoxide dismutases in cell regulation and cancer. Drug Discov. Today 2016, 21, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Deeb, D.; Gao, X.; Liu, Y.; Varma, N.R.S.; Arbab, A.S.; Gautam, S.C. Inhibition of telomerase activity by oleanane triterpenoid CDDO-Me in pancreatic cancer cells is ROS-dependent. Molecules 2013, 18, 3250–3265. [Google Scholar] [CrossRef] [PubMed]
- Deeb, D.; Gao, X.; Liu, Y.B.; Gautam, S.C. Inhibition of cell proliferation and induction of apoptosis by CDDO-Me in pancreatic cancer cells is ROS-dependent. J. Exp. Ther. Oncol. 2012, 10, 51–64. [Google Scholar] [PubMed]
- Pramanik, K.C.; Boreddy, S.R.; Srivastava, S.K. Role of mitochondrial electron transport chain complexes in capsaicin mediated oxidative stress leading to apoptosis in pancreatic cancer cells. PloS ONE 2011, 6, e20151. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Daniels, D.H.; Asbury, C.; Venkataraman, S.; Liu, J.; Spitz, D.R.; Oberley, L.W.; Cullen, J.J. Mitochondrial production of reactive oxygen species mediate dicumarol-induced cytotoxicity in cancer cells. J. Biol. Chem. 2006, 281, 37416–37426. [Google Scholar] [CrossRef] [PubMed]
- Heukamp, I.; Kilian, M.; Gregor, J.I.; Neumann, A.; Jacobi, C.A.; Guski, H.; Schimke, I.; Walz, M.K.; Wenger, F.A. Effects of the antioxidative vitamins A, C and E on liver metastasis and intrametastatic lipid peroxidation in BOP-induced pancreatic cancer in syrian hamsters. Pancreatology 2005, 5, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Espey, M.G.; Krishna, M.C.; Mitchell, J.B.; Corpe, C.P.; Buettner, G.R.; Shacter, E.; Levine, M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: Action as a pro-drug to deliver hydrogen peroxide to tissues. Proc. Natl. Acad. Sci. USA 2005, 102, 13604–13609. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, J.D.; Sibenaller, Z.A.; Mapuskar, K.A.; Wagner, B.A.; Cramer-Morales, K.L.; Furqan, M.; Sandhu, S.; Carlisle, T.L.; Smith, M.C.; Abu Hejleh, T.; et al. O●2− and H2O2-mediated disruption of Fe metabolism causes the differential susceptibility of NSCLC and GBM cancer cells to pharmacological ascorbate. Cancer Cell 2017, 31, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Batinic-Haberle, I.; Reboucas, J.S.; Spasojevic, I. Superoxide dismutase mimics: Chemistry, pharmacology, and therapeutic potential. Antioxid. Redox Signal. 2010, 13, 877–918. [Google Scholar] [CrossRef] [PubMed]
- Archibald, F.S.; Fridovich, I. The scavenging of superoxide radical by manganous complexes: In vitro. Arch. Biochem. Biophys. 1982, 214, 452–463. [Google Scholar] [CrossRef]
- Du, J.; Cullen, J.J.; Buettner, G.R. Ascorbic acid: Chemistry, biology and the treatment of cancer. Biochim. Biophys. Acta 2012, 1826, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Rawal, M.; Schroeder, S.R.; Wagner, B.A.; Cushing, C.M.; Welsh, J.L.; Button, A.M.; Du, J.; Sibenaller, Z.A.; Buettner, G.R.; Cullen, J.J. Manganoporphyrins increase ascorbate-induced cytotoxicity by enhancing H2O2 generation. Cancer Res. 2013, 73, 5232–5241. [Google Scholar] [CrossRef] [PubMed]
- Cieslak, J.A.; Strother, R.K.; Rawal, M.; Du, J.; Doskey, C.M.; Schroeder, S.R.; Button, A.; Wagner, B.A.; Buettner, G.R.; Cullen, J.J. Manganoporphyrins and ascorbate enhance gemcitabine cytotoxicity in pancreatic cancer. Free radic. Biol. Med. 2015, 83, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Welsh, J.L.; Wagner, B.A.; van’t Erve, T.J.; Zehr, P.S.; Berg, D.J.; Halfdanarson, T.R.; Yee, N.S.; Bodeker, K.L.; Du, J.; Roberts, L.J.; et al. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): Results from a phase I clinical trial. Cancer Chemother. Pharmacol. 2013, 71, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Du, Y. Acquisition of resistance of pancreatic cancer cells to 2-methoxyestradiol is associated with the upregulation of manganese superoxide dismutase. Mol. Cancer Res. R 2012, 10, 768–777. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.J.; Goswami, P.C. Mitochondria-targeted antioxidant enzyme activity regulates radioresistance in human pancreatic cancer cells. Cancer Biol. Ther. 2008, 7, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Fan, M.; Candas, D.; Qin, L.; Zhang, X.; Eldridge, A.; Zou, J.X.; Zhang, T.; Juma, S.; Jin, C.; et al. CDK1-mediated SIRT3 activation enhances mitochondrial function and tumor radioresistance. Mol. Cancer Ther. 2015, 14, 2090–2102. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Nakayama, H.; Yoshida, R.; Hirosue, A.; Nagata, M.; Tanaka, T.; Kawahara, K.; Sakata, J.; Arita, H.; Nakashima, H.; et al. IL-6 controls resistance to radiation by suppressing oxidative stress via the Nrf2-antioxidant pathway in oral squamous cell carcinoma. Br. J. Cancer 2016, 115, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Patel, G.K.; Khan, M.A.; Bhardwaj, A.; Srivastava, S.K.; Zubair, H.; Patton, M.C.; Singh, S.; Khushman, M.; Singh, A.P. Exosomes confer chemoresistance to pancreatic cancer cells by promoting ROS detoxification and miR-155-mediated suppression of key gemcitabine-metabolising enzyme, DCK. Br. J. Cancer 2017, 116, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Shang, B.-Y.; Sheng, W.-J.; Zhang, S.-H.; Li, Y.; Miao, Q.-F.; Zhen, Y.-S. A recombinantly tailored β-defensin that displays intensive macropinocytosis-mediated uptake exerting potent efficacy against K-ras mutant pancreatic cancer. Oncotarget 2016, 7, 58418–58434. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wilkes, J.G.; Alexander, M.S.; Cullen, J.J. Superoxide Dismutases in Pancreatic Cancer. Antioxidants 2017, 6, 66. https://doi.org/10.3390/antiox6030066
Wilkes JG, Alexander MS, Cullen JJ. Superoxide Dismutases in Pancreatic Cancer. Antioxidants. 2017; 6(3):66. https://doi.org/10.3390/antiox6030066
Chicago/Turabian StyleWilkes, Justin G., Matthew S. Alexander, and Joseph J. Cullen. 2017. "Superoxide Dismutases in Pancreatic Cancer" Antioxidants 6, no. 3: 66. https://doi.org/10.3390/antiox6030066
APA StyleWilkes, J. G., Alexander, M. S., & Cullen, J. J. (2017). Superoxide Dismutases in Pancreatic Cancer. Antioxidants, 6(3), 66. https://doi.org/10.3390/antiox6030066