Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses

Abstract
1. Introduction
2. Mechanisms of Chemotherapy-Induced Cardiomyopathy
3. Oxidized Protein Removal Pathways
3.1. Protein Unfolding System
3.2. Ubiquitin-Proteasome System (UPS)
3.3. Autophagy
3.4. Mitophagy
4. Extracellular Vesicles
4.1. Exosome Biogenesis
4.2. Microvesicles (MVs) Biogenesis
4.3. Apoptotic Bodies Biogenesis
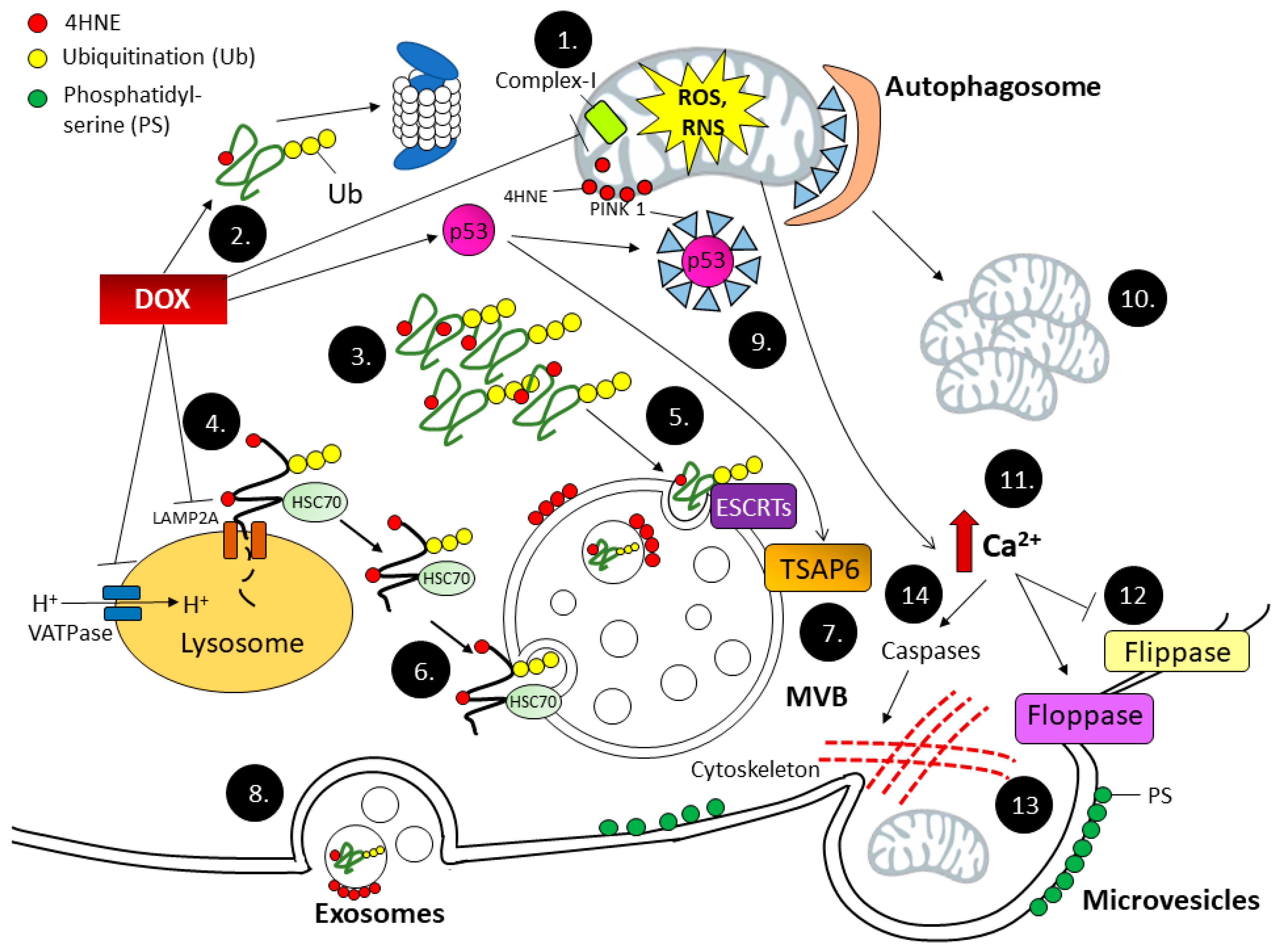
4.4. EVs Serve as a Bypass Highway for Oxidized Proteins Removal during Dox-Induced Cardiotoxicity
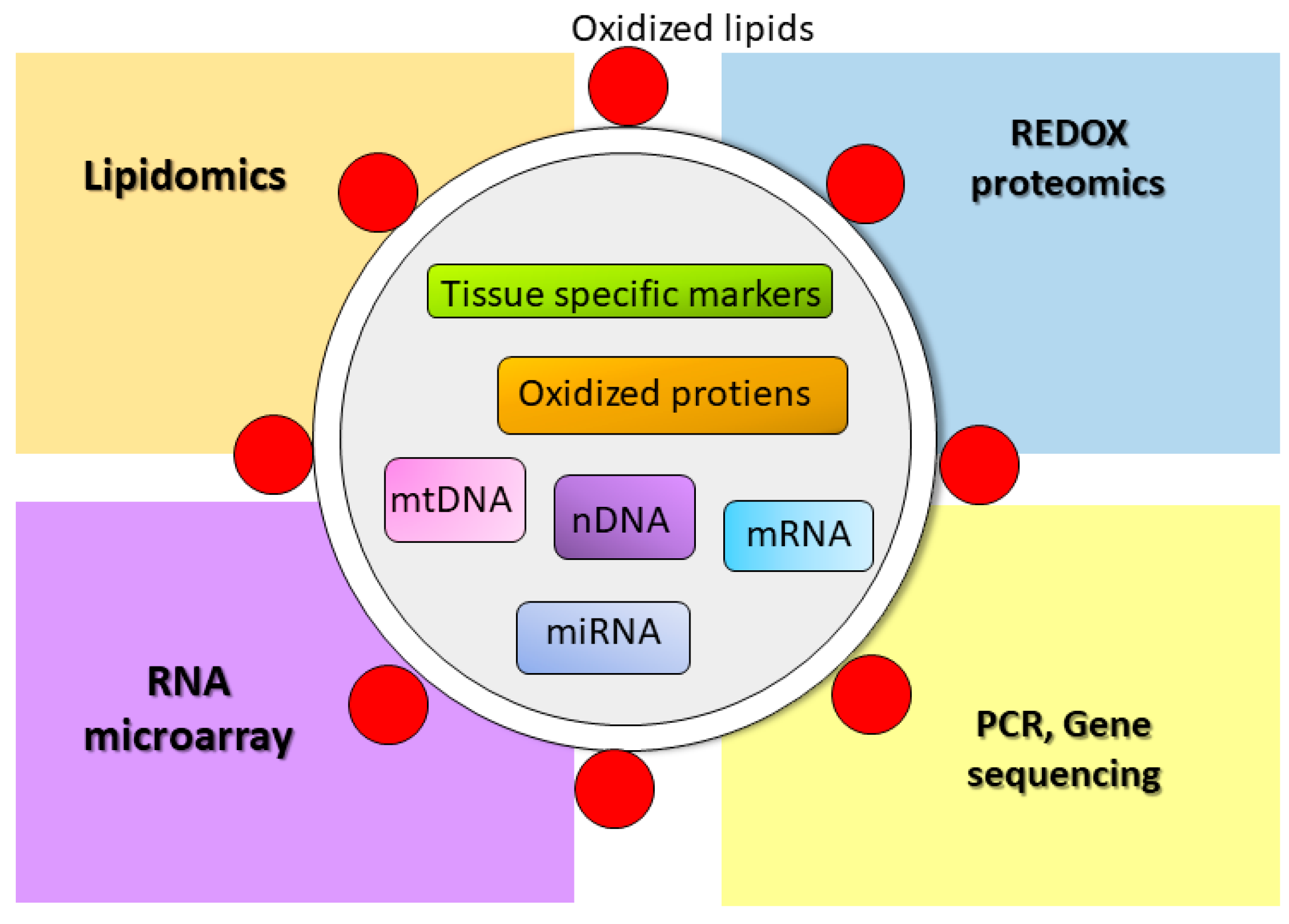
4.5. EVs as a Biomarker for Oxidative Stress
4.6. Role of EVs in Oxidative Stress Response
5. Role of Superoxide Dismutase in EV-Associated Oxidative Stress Response
5.1. SOD1
5.2. SOD2
5.3. SOD3
6. Conclusions and Future Direction
Acknowledgments
Conflicts of Interest
References
- Mertens, A.C.; Yasui, Y.; Neglia, J.P.; Potter, J.D.; Nesbit, M.E., Jr.; Ruccione, K.; Smithson, W.A.; Robison, L.L. Late mortality experience in five-year survivors of childhood and adolescent cancer: The childhood cancer survivor study. J. Clin. Oncol. 2001, 19, 3163–3172. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Jungsuwadee, P.; Vore, M.; Butterfield, D.A.; St., Clair, D.K. Collateral damage in cancer chemotherapy: Oxidative stress in nontargeted tissues. Mol. Interv. 2007, 7, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Octavia, Y.; Tocchetti, C.G.; Gabrielson, K.L.; Janssens, S.; Crijns, H.J.; Moens, A.L. Doxorubicin-induced cardiomyopathy: From molecular mechanisms to therapeutic strategies. J. Mol. Cell. Cardiol. 2012, 52, 1213–1225. [Google Scholar] [CrossRef] [PubMed]
- De Wolf, F.A. Binding of doxorubicin to cardiolipin as compared to other anionic phospholipids—An evaluation of electrostatic effects. Biosci. Rep. 1991, 11, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Davies, K.J.; Doroshow, J.H. Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J. Biol. Chem. 1986, 261, 3060–3067. [Google Scholar] [PubMed]
- Olson, R.D.; Mushlin, P.S. Doxorubicin cardiotoxicity: Analysis of prevailing hypotheses. FASEB J. 1990, 4, 3076–3086. [Google Scholar] [PubMed]
- Briere, J.J.; Favier, J.; Gimenez-Roqueplo, A.P.; Rustin, P. Tricarboxylic acid cycle dysfunction as a cause of human diseases and tumor formation. Am. J. Physiol. Cell. Physiol. 2006, 291, C1114–C1120. [Google Scholar] [CrossRef] [PubMed]
- Hausladen, A.; Fridovich, I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J. Biol. Chem. 1994, 269, 29405–29408. [Google Scholar] [PubMed]
- Minotti, G.; Cairo, G.; Monti, E. Role of iron in anthracycline cardiotoxicity: New tunes for an old song? FASEB J. 1999, 13, 199–212. [Google Scholar] [PubMed]
- Chaiswing, L.; Cole, M.P.; Ittarat, W.; Szweda, L.I.; St., Clair, D.K.; Oberley, T.D. Manganese superoxide dismutase and inducible nitric oxide synthase modify early oxidative events in acute adriamycin-induced mitochondrial toxicity. Mol. Cancer Ther. 2005, 4, 1056–1064. [Google Scholar] [CrossRef] [PubMed]
- Businaro, R.; Fioretti, E.; Fumagalli, L.; Citro, G.; De Renzis, G.; Ascoli, F. Cellular localization of bovine pancreatic trypsin inhibitor and related molecular forms in bovine lung. Histochem. J. 1988, 20, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Miriyala, S.; Thippakorn, C.; Chaiswing, L.; Xu, Y.; Noel, T.; Tovmasyan, A.; Batinic-Haberle, I.; Vander Kooi, C.W.; Chi, W.; Latif, A.A.; et al. Novel role of 4-hydroxy-2-nonenal in AIFm2-mediated mitochondrial stress signaling. Free Radic. Biol. Med. 2016, 91, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Amm, I.; Sommer, T.; Wolf, D.H. Protein quality control and elimination of protein waste: The role of the ubiquitin-proteasome system. Biochim. Biophys. Acta 2014, 1843, 182–196. [Google Scholar] [CrossRef] [PubMed]
- Baixauli, F.; Lopez-Otin, C.; Mittelbrunn, M. Exosomes and autophagy: Coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 2014, 5, 403. [Google Scholar] [CrossRef] [PubMed]
- Li, D.L.; Wang, Z.V.; Ding, G.; Tan, W.; Luo, X.; Criollo, A.; Xie, M.; Jiang, N.; May, H.; Kyrychenko, V.; et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation 2016, 133, 1668–1687. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.J.; Trivedi, P.C.; Pulinilkunnil, T. Autophagic dysregulation in doxorubicin cardiomyopathy. J. Mol. Cell. Cardiol. 2017, 104, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Wang, X. The ubiquitin-proteasome system in cardiac proteinopathy: A quality control perspective. Cardiovasc. Res. 2010, 85, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.U.; Gray, M.J.; Jakob, U. Protein quality control under oxidative stress conditions. J. Mol. Biol. 2015, 427, 1549–1563. [Google Scholar] [CrossRef] [PubMed]
- Plate, L.; Paxman, R.J.; Wiseman, R.L.; Kelly, J.W. Modulating protein quality control. eLife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Torti, F.M.; Bristow, M.M.; Lum, B.L.; Carter, S.K.; Howes, A.E.; Aston, D.A.; Brown, B.W., Jr.; Hannigan, J.F., Jr.; Meyers, F.J.; Mitchell, E.P.; et al. Cardiotoxicity of epirubicin and doxorubicin: Assessment by endomyocardial biopsy. Cancer Res. 1986, 46, 3722–3727. [Google Scholar] [PubMed]
- Fu, H.Y.; Sanada, S.; Matsuzaki, T.; Liao, Y.; Okuda, K.; Yamato, M.; Tsuchida, S.; Araki, R.; Asano, Y.; Asanuma, H.; et al. Chemical endoplasmic reticulum chaperone alleviates doxorubicin-induced cardiac dysfunction. Circ. Res. 2016, 118, 798–809. [Google Scholar] [CrossRef] [PubMed]
- Attar, B.M.; Levendoglu, H.; Rhee, H. Small cell carcinoma of the esophagus. Report of three cases and review of the literature. Dig. Dis. Sci. 1990, 35, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Sishi, B.J.; Loos, B.; van Rooyen, J.; Engelbrecht, A.M. Doxorubicin induces protein ubiquitination and inhibits proteasome activity during cardiotoxicity. Toxicology 2013, 309, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zheng, H.; Tang, M.; Ryu, Y.C.; Wang, X. A therapeutic dose of doxorubicin activates ubiquitin-proteasome system-mediated proteolysis by acting on both the ubiquitination apparatus and proteasome. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2541–H2550. [Google Scholar] [CrossRef] [PubMed]
- Benaroudj, N.; Zwickl, P.; Seemuller, E.; Baumeister, W.; Goldberg, A.L. ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol. Cell 2003, 11, 69–78. [Google Scholar] [CrossRef]
- Koleini, N.; Kardami, E. Autophagy and mitophagy in the context of doxorubicin-induced cardiotoxicity. Oncotarget 2017, 8, 46663–46680. [Google Scholar] [CrossRef] [PubMed]
- Dirks-Naylor, A.J. The role of autophagy in doxorubicin-induced cardiotoxicity. Life Sci. 2013, 93, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Chen, K.; Kobayashi, S.; Timm, D.; Liang, Q. Resveratrol attenuates doxorubicin-induced cardiomyocyte death via inhibition of p70 s6 kinase 1-mediated autophagy. J. Pharmacol. Exp. Ther. 2012, 341, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Hill, B.G.; Haberzettl, P.; Ahmed, Y.; Srivastava, S.; Bhatnagar, A. Unsaturated lipid peroxidation-derived aldehydes activate autophagy in vascular smooth-muscle cells. Biochem. J. 2008, 410, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Velez, J.M.; Miriyala, S.; Nithipongvanitch, R.; Noel, T.; Plabplueng, C.D.; Oberley, T.; Jungsuwadee, P.; Van Remmen, H.; Vore, M.; St., Clair, D.K. P53 regulates oxidative stress-mediated retrograde signaling: A novel mechanism for chemotherapy-induced cardiac injury. PLoS ONE 2011, 6, e18005. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, J.J.; Trivedi, P.C.; Yeung, P.; Kienesberger, P.C.; Pulinilkunnil, T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor eb expression and disrupting autophagy. Biochem. J. 2016, 473, 3769–3789. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.X.; Yin, X.M. Mitophagy: Mechanisms, pathophysiological roles, and analysis. Biol. Chem. 2012, 393, 547–564. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Margulets, V.; Chowdhury, S.R.; Thliveris, J.; Jassal, D.; Fernyhough, P.; Dorn, G.W., 2nd; Kirshenbaum, L.A. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E5537–E5544. [Google Scholar] [CrossRef] [PubMed]
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Sanchez-Madrid, F. Mitochondria know no boundaries: Mechanisms and functions of intercellular mitochondrial transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, K.; Wang, Z.; Wang, Y.; Liu, J.; Lin, L.; Shao, Y.; Gao, L.; Yin, H.; Cui, C.; et al. DNA in serum extracellular vesicles is stable under different storage conditions. BMC Cancer 2016, 16, 753. [Google Scholar] [CrossRef] [PubMed]
- Pieters, B.C.; Arntz, O.J.; Bennink, M.B.; Broeren, M.G.; van Caam, A.P.; Koenders, M.I.; van Lent, P.L.; van den Berg, W.B.; de Vries, M.; van der Kraan, P.M.; et al. Commercial cow milk contains physically stable extracellular vesicles expressing immunoregulatory TGF-beta. PLoS ONE 2015, 10, e0121123. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Lee, D.S. Illuminating the physiology of extracellular vesicles. Stem Cell Res. Ther. 2016, 7, 55. [Google Scholar] [CrossRef] [PubMed]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Babst, M. MVB vesicle formation: ESCRT-dependent, ESCRT-independent and everything in between. Curr. Opin. Cell Biol. 2011, 23, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brugger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Harris, S.L.; Levine, A.J. The regulation of exosome secretion: A novel function of the p53 protein. Cancer Res. 2006, 66, 4795–4801. [Google Scholar] [CrossRef] [PubMed]
- Lespagnol, A.; Duflaut, D.; Beekman, C.; Blanc, L.; Fiucci, G.; Marine, J.C.; Vidal, M.; Amson, R.; Telerman, A. Exosome secretion, including the DNA damage-induced p53-dependent secretory pathway, is severely compromised in TSAP6/Steap3-null mice. Cell. Death Differ. 2008, 15, 1723–1733. [Google Scholar] [CrossRef] [PubMed]
- Sahu, R.; Kaushik, S.; Clement, C.C.; Cannizzo, E.S.; Scharf, B.; Follenzi, A.; Potolicchio, I.; Nieves, E.; Cuervo, A.M.; Santambrogio, L. Microautophagy of cytosolic proteins by late endosomes. Dev. Cell 2011, 20, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Odorizzi, G. Get on the exosome bus with ALIX. Nat. Cell Biol. 2012, 14, 654–655. [Google Scholar] [CrossRef] [PubMed]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Djeddi, A.; Michelet, X.; Culetto, E.; Alberti, A.; Barois, N.; Legouis, R. Induction of autophagy in ESCRT mutants is an adaptive response for cell survival in C. Elegans. J. Cell Sci. 2012, 125, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Fader, C.M.; Sanchez, D.; Furlan, M.; Colombo, M.I. Induction of autophagy promotes fusion of multivesicular bodies with autophagic vacuoles in k562 cells. Traffic 2008, 9, 230–250. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Overbye, A.; Brech, A.; Torgersen, M.L.; Jakobsen, I.S.; Sandvig, K.; Llorente, A. PIKfyve inhibition increases exosome release and induces secretory autophagy. Cell. Mol. Life Sci. CMLS 2016, 73, 4717–4737. [Google Scholar] [CrossRef] [PubMed]
- Ermak, G.; Davies, K.J. Calcium and oxidative stress: From cell signaling to cell death. Mol. Immunol. 2002, 38, 713–721. [Google Scholar] [CrossRef]
- Morel, O.; Jesel, L.; Freyssinet, J.M.; Toti, F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Barteneva, N.S.; Fasler-Kan, E.; Bernimoulin, M.; Stern, J.N.; Ponomarev, E.D.; Duckett, L.; Vorobjev, I.A. Circulating microparticles: Square the circle. BMC Cell Biol. 2013, 14, 23. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. CB 2009, 19, 1875–1885. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.L.; Sahai, E.A.; Yeo, M.; Bosch, M.; Dewar, A.; Olson, M.F. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat. Cell. Biol. 2001, 3, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.B. Adriamycin-induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc. Toxicol. 2007, 7, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Heuvingh, J.; Bonneau, S. Asymmetric oxidation of giant vesicles triggers curvature-associated shape transition and permeabilization. Biophys. J. 2009, 97, 2904–2912. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.; Adesanya, M.A.; Gardiner, E.; Sayala, H.; Madden, L.; Maraveyas, A. Chemotherapy treatment of multiple myeloma patients increases circulating levels of endothelial microvesicles. Thromb. Res. 2016, 146, 20–22. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Kohan, H.G.; Asimakopoulos, A.G.; Sudha, T.; Sell, S.; Kannan, K.; Boroujerdi, M.; Davis, P.J.; Mousa, S.A. Microvesicle removal of anticancer drugs contributes to drug resistance in human pancreatic cancer cells. Oncotarget 2016, 7, 50365–50379. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Thakur, B.K.; Weiss, J.M.; Kim, H.S.; Peinado, H.; Lyden, D. Extracellular vesicles in cancer: Cell-to-cell mediators of metastasis. Cancer Cell 2016, 30, 836–848. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Gray, E.; Heman-Ackah, S.M.; Mager, I.; Talbot, K.; Andaloussi, S.E.; Wood, M.J.; Turner, M.R. Extracellular vesicles in neurodegenerative disease—Pathogenesis to biomarkers. Nat. Rev. Neurol. 2016, 12, 346–357. [Google Scholar] [CrossRef] [PubMed]
- Buzas, E.I.; Gyorgy, B.; Nagy, G.; Falus, A.; Gay, S. Emerging role of extracellular vesicles in inflammatory diseases. Nat. Rev. Rheumatol. 2014, 10, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Malik, Z.A.; Kott, K.S.; Poe, A.J.; Kuo, T.; Chen, L.; Ferrara, K.W.; Knowlton, A.A. Cardiac myocyte exosomes: Stability, HSP60, and proteomics. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H954–H965. [Google Scholar] [CrossRef] [PubMed]
- Heiserman, J.P.; Chen, L.; Kim, B.S.; Kim, S.C.; Tran, A.L.; Siebenborn, N.; Knowlton, A.A. TLR4 mutation and HSP60-induced cell death in adult mouse cardiac myocytes. Cell Stress Chaperones 2015, 20, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Eldh, M.; Ekstrom, K.; Valadi, H.; Sjostrand, M.; Olsson, B.; Jernas, M.; Lotvall, J. Exosomes communicate protective messages during oxidative stress; possible role of exosomal shuttle RNA. PLoS ONE 2010, 5, e15353. [Google Scholar] [CrossRef] [PubMed]
- Atienzar-Aroca, S.; Flores-Bellver, M.; Serrano-Heras, G.; Martinez-Gil, N.; Barcia, J.M.; Aparicio, S.; Perez-Cremades, D.; Garcia-Verdugo, J.M.; Diaz-Llopis, M.; Romero, F.J.; et al. Oxidative stress in retinal pigment epithelium cells increases exosome secretion and promotes angiogenesis in endothelial cells. J. Cell. Mol. Med. 2016, 20, 1457–1466. [Google Scholar] [CrossRef] [PubMed]
- Mancek-Keber, M.; Frank-Bertoncelj, M.; Hafner-Bratkovic, I.; Smole, A.; Zorko, M.; Pirher, N.; Hayer, S.; Kralj-Iglic, V.; Rozman, B.; Ilc, N.; et al. Toll-like receptor 4 senses oxidative stress mediated by the oxidation of phospholipids in extracellular vesicles. Sci. Signal. 2015, 8, ra60. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, I.; Santoro, N.; Chen, Y.; Hoque, R.; Ouyang, X.; Caprio, S.; Shlomchik, M.J.; Coffman, R.L.; Candia, A.; Mehal, W.Z. Hepatocyte mitochondrial DNA drives nonalcoholic steatohepatitis by activation of TLR9. J. Clin. Investig. 2016, 126, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Xu, M.J.; Koritzinsky, E.H.; Zhou, Z.; Wang, W.; Cao, H.; Yuen, P.S.; Ross, R.A.; Star, R.A.; Liangpunsakul, S.; et al. Mitochondrial DNA-enriched microparticles promote acute-on-chronic alcoholic neutrophilia and hepatotoxicity. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Momen-Heravi, F.; Bala, S.; Kodys, K.; Szabo, G. Exosomes derived from alcohol-treated hepatocytes horizontally transfer liver specific miRNA-122 and sensitize monocytes to LPS. Sci. Rep. 2015, 5, 9991. [Google Scholar] [CrossRef] [PubMed]
- Hirsova, P.; Ibrahim, S.H.; Krishnan, A.; Verma, V.K.; Bronk, S.F.; Werneburg, N.W.; Charlton, M.R.; Shah, V.H.; Malhi, H.; Gores, G.J. Lipid-induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology 2016, 150, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, C.; Liu, L.; A, X.; Chen, B.; Li, Y.; Du, J. Macrophage-derived mir-155-containing exosomes suppress fibroblast proliferation and promote fibroblast inflammation during cardiac injury. Mol. Ther. 2017, 25, 192–204. [Google Scholar] [CrossRef] [PubMed]
- Cambier, L.; de Couto, G.; Ibrahim, A.; Echavez, A.K.; Valle, J.; Liu, W.; Kreke, M.; Smith, R.R.; Marban, L.; Marban, E. Y RNA fragment in extracellular vesicles confers cardioprotection via modulation of IL-10 expression and secretion. EMBO Mol. Med. 2017, 9, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Fridovich, I. Superoxide dismutases. An adaptation to a paramagnetic gas. J. Biol. Chem. 1989, 264, 7761–7764. [Google Scholar] [PubMed]
- Petersen, S.V.; Oury, T.D.; Ostergaard, L.; Valnickova, Z.; Wegrzyn, J.; Thogersen, I.B.; Jacobsen, C.; Bowler, R.P.; Fattman, C.L.; Crapo, J.D.; et al. Extracellular superoxide dismutase (EC-SOD) binds to type i collagen and protects against oxidative fragmentation. J. Biol. Chem. 2004, 279, 13705–13710. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Marklund, S.L.; Holme, E.; Hellner, L. Superoxide dismutase in extracellular fluids. Clin. Chim. Acta Int. J. Clin. Chem. 1982, 126, 41–51. [Google Scholar] [CrossRef]
- Marklund, S.L.; Bjelle, A.; Elmqvist, L.G. Superoxide dismutase isoenzymes of the synovial fluid in rheumatoid arthritis and in reactive arthritides. Ann. Rheum. Dis. 1986, 45, 847–851. [Google Scholar] [CrossRef] [PubMed]
- De Haan, J.B.; Cristiano, F.; Iannello, R.; Bladier, C.; Kelner, M.J.; Kola, I. Elevation in the ratio of Cu/Zn-superoxide dismutase to glutathione peroxidase activity induces features of cellular senescence and this effect is mediated by hydrogen peroxide. Hum. Mol. Genet. 1996, 5, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sayana, P.; Zhang, X.; Le, W. Genetics of amyotrophic lateral sclerosis: An update. Mol. Neurodegener. 2013, 8, 28. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 mediated pathogenesis of amyotrophic lateral sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Ravits, J. Focality, stochasticity and neuroanatomic propagation in ALS pathogenesis. Exp. Neurol. 2014, 262(Pt. B), 121–126. [Google Scholar] [CrossRef] [PubMed]
- Holley, A.K.; Bakthavatchalu, V.; Velez-Roman, J.M.; St., Clair, D.K. Manganese superoxide dismutase: Guardian of the powerhouse. Int. J. Mol. Sci. 2011, 12, 7114–7162. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Yerbury, J.J.; Turner, B.J.; Guest, W.C.; Pokrishevsky, E.; O’Neill, M.A.; Yanai, A.; Silverman, J.M.; Zeineddine, R.; Corcoran, L.; et al. Intercellular propagated misfolding of wild-type Cu/Zn superoxide dismutase occurs via exosome-dependent and -independent mechanisms. Proc. Natl. Acad. Sci. USA 2014, 111, 3620–3625. [Google Scholar] [CrossRef] [PubMed]
- Van Remmen, H.; Ikeno, Y.; Hamilton, M.; Pahlavani, M.; Wolf, N.; Thorpe, S.R.; Alderson, N.L.; Baynes, J.W.; Epstein, C.J.; Huang, T.T.; et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol. Genom. 2003, 16, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.C.; Oberley, T.D.; Vichitbandha, S.; Ho, Y.S.; St., Clair, D.K. The protective role of manganese superoxide dismutase against adriamycin-induced acute cardiac toxicity in transgenic mice. J. Clin. Investig. 1996, 98, 1253–1260. [Google Scholar] [CrossRef] [PubMed]
- Yen, H.C.; Oberley, T.D.; Gairola, C.G.; Szweda, L.I.; St., Clair, D.K. Manganese superoxide dismutase protects mitochondrial complex I against adriamycin-induced cardiomyopathy in transgenic mice. Arch. Biochem. Biophys. 1999, 362, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.S.; Rebel, V.I.; Derby, R.; Bell, K.; Huang, T.T.; Kuypers, F.A.; Epstein, C.J.; Burakoff, S.J. Absence of mitochondrial superoxide dismutase results in a murine hemolytic anemia responsive to therapy with a catalytic antioxidant. J. Exp. Med. 2001, 193, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Hendrickson, D.J.; Fisher, J.H.; Jones, C.; Ho, Y.S. Regional localization of human extracellular superoxide dismutase gene to 4pter-q21. Genomics 1990, 8, 736–738. [Google Scholar] [CrossRef]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Iversen, M.B.; Gottfredsen, R.H.; Larsen, U.G.; Enghild, J.J.; Praetorius, J.; Borregaard, N.; Petersen, S.V. Extracellular superoxide dismutase is present in secretory vesicles of human neutrophils and released upon stimulation. Free Radic. Biol. Med. 2016, 97, 478–488. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Cell/Tissue Type of Origin | EV Type | Oxidative Stress Condition | Oxidative Stress-Related Cargo | Effect | Reference |
|---|---|---|---|---|---|
| Cardiomyocytes | Exosomes | Ethanol, hypoxia/reoxy-genation | HSP60 | TLR4-mediated apoptosis | Heiserman et al. [67] |
| Mast cells | Exosomes | H2O2 | mRNA | H2O2 tolerance | Eldh et al. [68] |
| Retinal pigment epithelial cells | Exosomes | Ethanol | VEGF protein and mRNA | Angiogenesis | Atienzar-Aroca et al. [69] |
| HEK293 cells | Exosomes + MVs | Ca2+ ionophore (Lipoxygenase stimulator) | Oxidized phospholipids | TLR4-mediated NFκB activation | Mancek-Keber et al. [70] |
| Liver | MVs | High fat diet treated mice (NASH model) | Oxidized mtDNA | TLR9-induced TNFα, IL-6 production | Garcia-Martinez et al. [71] |
| Liver | MVs | Chronic-plus-binge alcohol drinking | mtDNA | TLR9-mediated neutrophilic inflammation | Cai et al. [72] |
| Liver | Exosomes | Alcoholic hepatitis | miR-122 | Sensitize monocytes to LPS | Momen-Heravi et al. [73] |
| Liver | MVs | Saturated fatty acid-induced lipotoxicity | TRAIL | DR5-dependent macrophage activation | Hirsova et al. [74] |
| Macrophage | Exosomes | Myocardial infarction | miR-155 | Fibroblast inflammation | Wang et al. [75] |
| Cardiosphere-derived cells | EVs | Myocardial infarction | Y RNA fragment | IL-10 expression and secretion | Cambier et al. [76] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yarana, C.; St. Clair, D.K. Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses. Antioxidants 2017, 6, 75. https://doi.org/10.3390/antiox6040075
Yarana C, St. Clair DK. Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses. Antioxidants. 2017; 6(4):75. https://doi.org/10.3390/antiox6040075
Chicago/Turabian StyleYarana, Chontida, and Daret K. St. Clair. 2017. "Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses" Antioxidants 6, no. 4: 75. https://doi.org/10.3390/antiox6040075
APA StyleYarana, C., & St. Clair, D. K. (2017). Chemotherapy-Induced Tissue Injury: An Insight into the Role of Extracellular Vesicles-Mediated Oxidative Stress Responses. Antioxidants, 6(4), 75. https://doi.org/10.3390/antiox6040075