Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Vascular Nox/Duox Isoforms
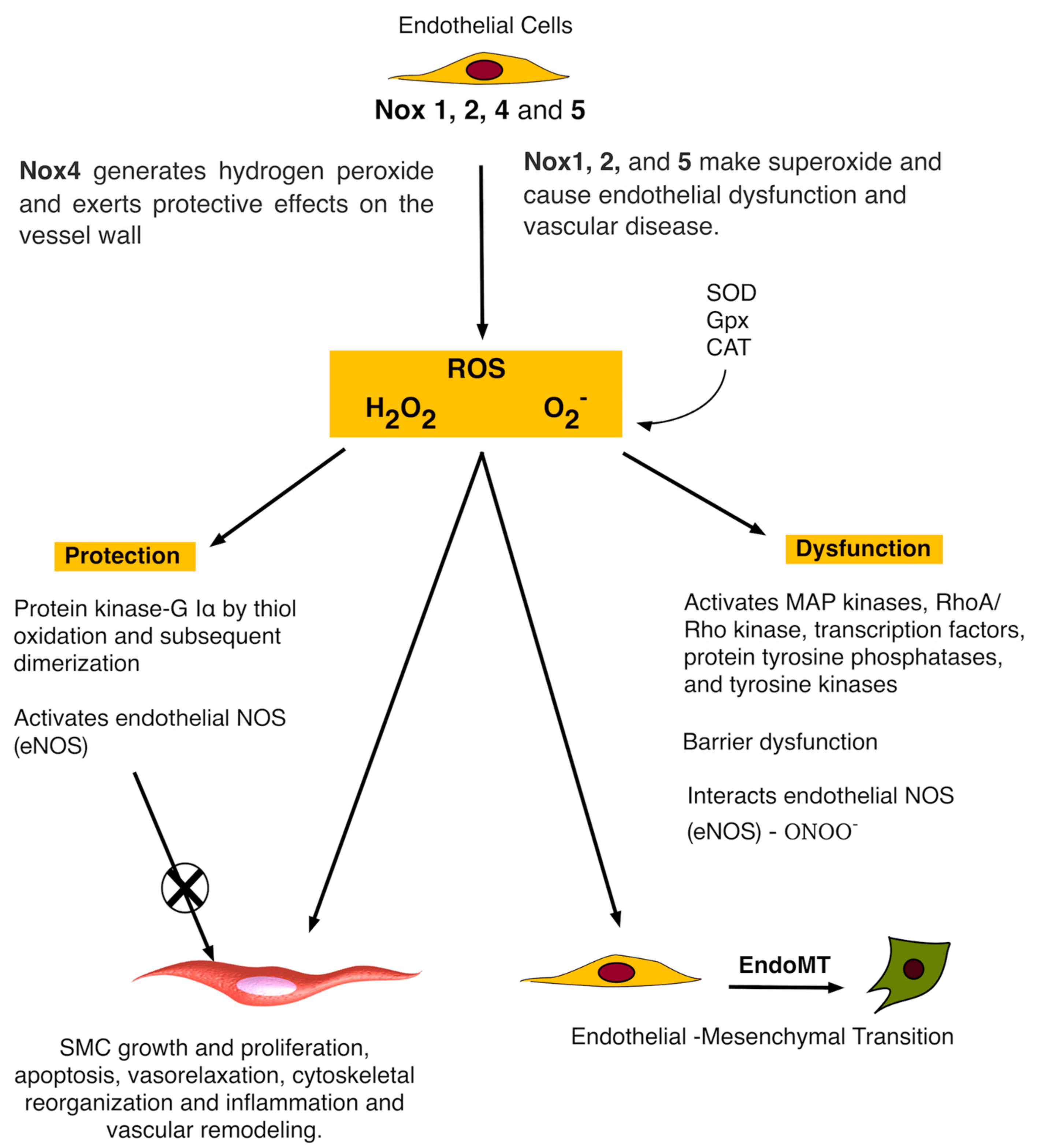
3. Nox and Vascular Endothelial Cells
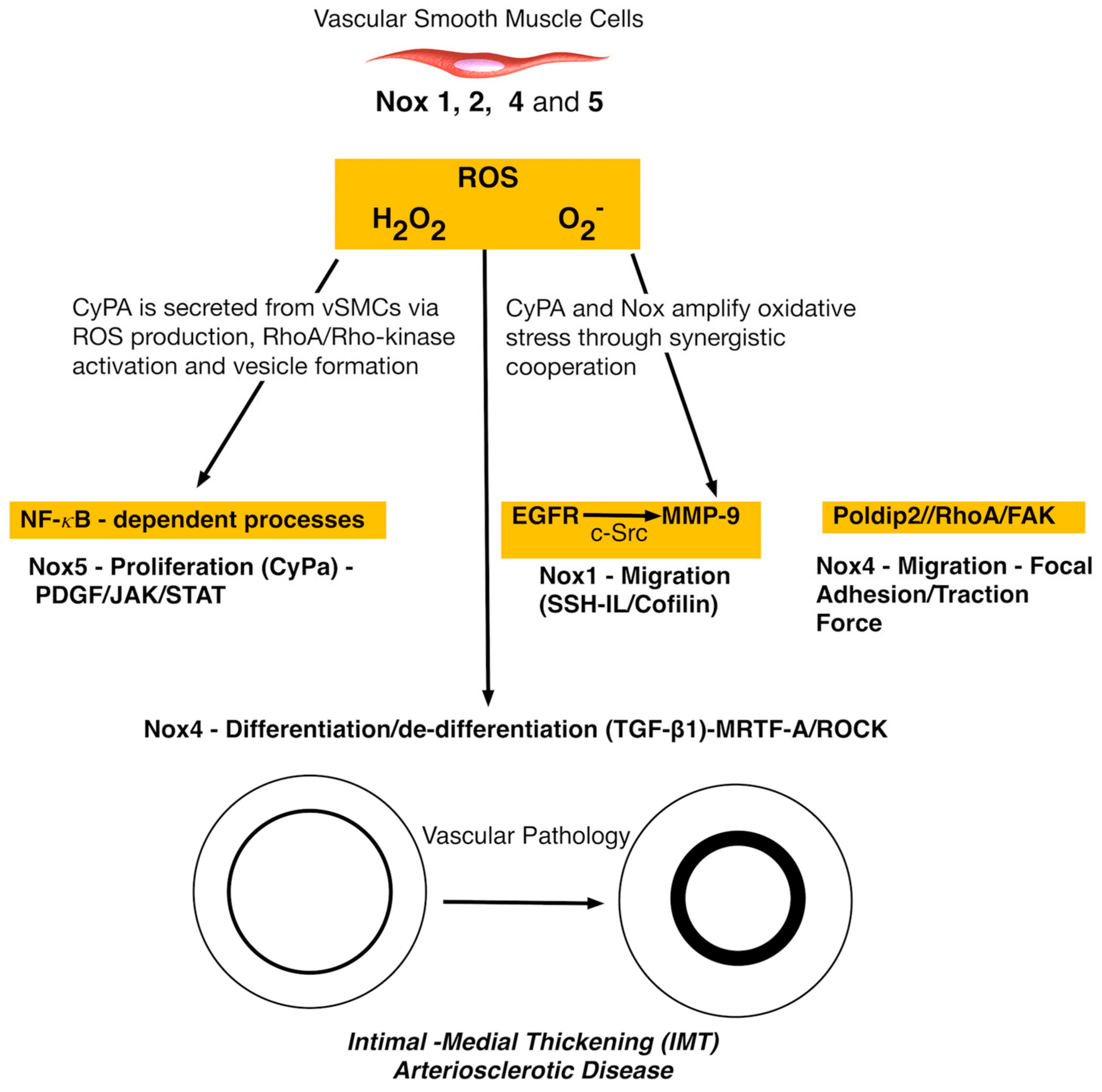
4. Nox and Vascular Smooth Muscle Cells
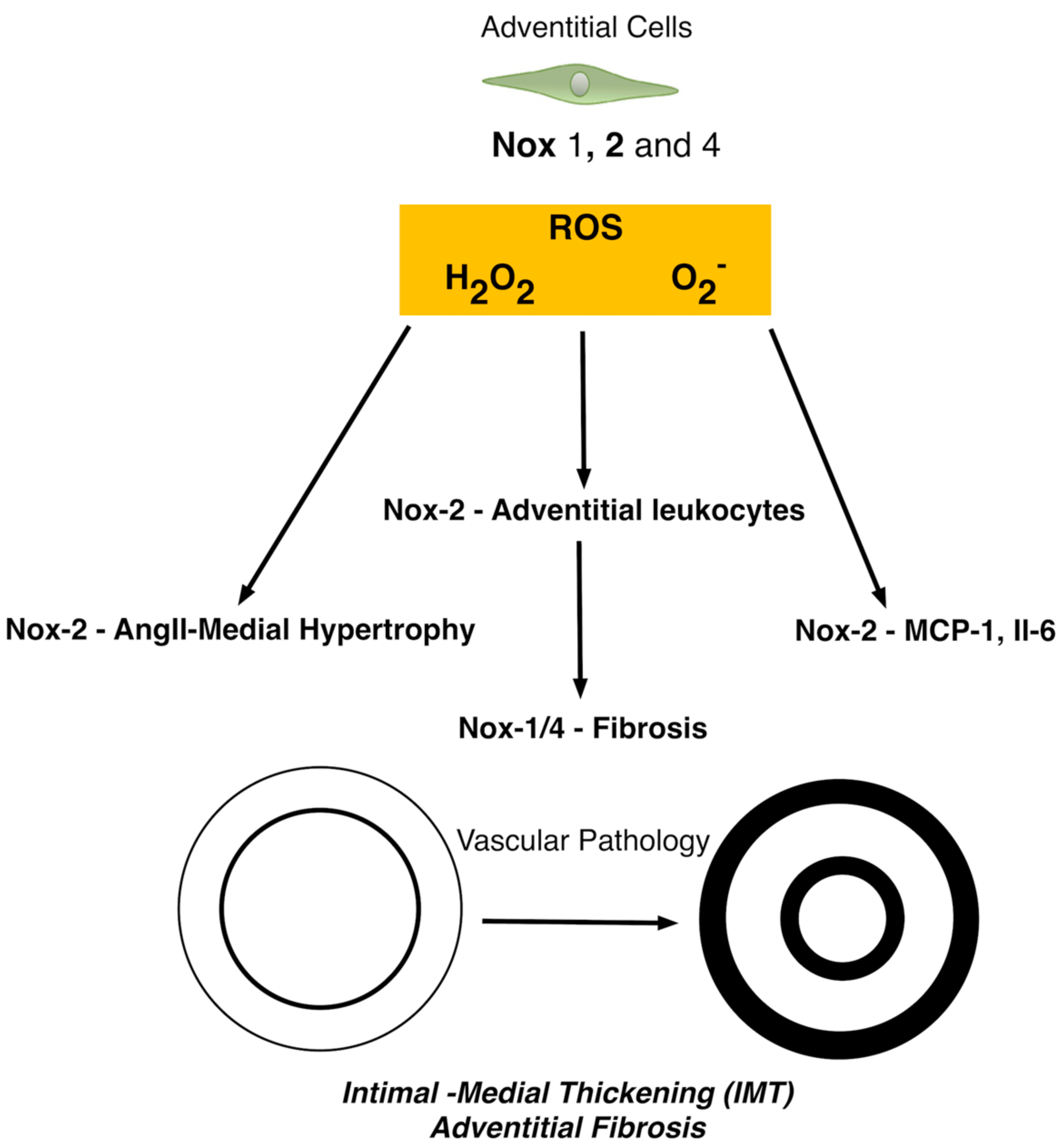
5. The Role of Nox in Adventitial Cells
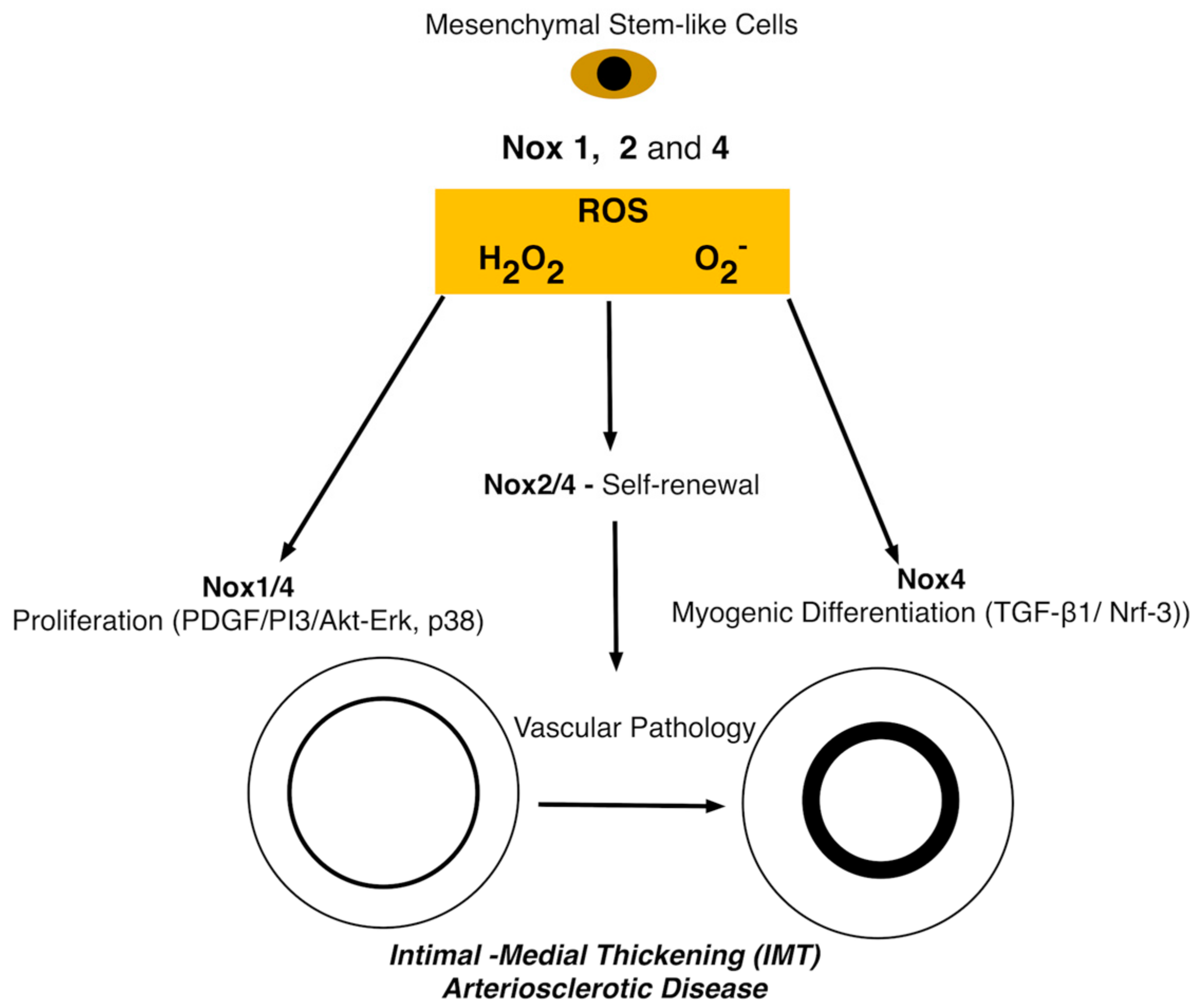
6. The Role of Nox in Stem Cells
The Role of NADPH and the ROS Pathway during Vascular Myogenic Differentiation
7. Conclusions
Author Contributions
Conflicts of Interest
References
- Lassègue, B.; San Martín, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012, 110, 1364–1390. [Google Scholar] [CrossRef] [PubMed]
- Kelley, E.E. A new paradigm for XOR-catalyzed reactive species generation in the endothelium. Pharmacol. Rep. 2015, 67, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Dolinsky, V.W.; Cole, L.K.; Sparagna, G.C.; Hatch, G.M. Cardiac mitochondrial energy metabolism in heart failure: Role of cardiolipin and sirtuins. Biochim. Biophys. Acta 2016, 1861, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function—Role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef] [PubMed]
- Ellulu, M.S.; Patimah, I.; Khaza’ai, H.; Rahmat, A.; Abed, Y.; Ali, F. Atherosclerotic cardiovascular disease: A review of initiators and protective factors. Inflammopharmacology 2016, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Loot, A.E.; Schreiber, J.G.; Fisslthaler, B.; Fleming, I. Angiotensin II impairs endothelial function via tyrosine phosphorylation of the endothelial nitric oxide synthase. J. Exp. Med. 2009, 206, 2889–2896. [Google Scholar] [CrossRef] [PubMed]
- Burgoyne, J.R.; Madhani, M.; Cuello, F.; Charles, R.L.; Brennan, J.P.; Schröder, E.; Browning, D.D.; Eaton, P. Cysteine redox sensor in PKGIa enables oxidant-induced activation. Science 2007, 317, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.R.; Sinha, S.; Owens, G.K. Vascular Smooth Muscle Cells in Atherosclerosis. Circ. Res. 2016, 118, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M.; Shimokawa, H.; Feletou, M.; Tang, E.H.C. Endothelial dysfunction and vascular disease—A 30th anniversary update. Acta Physiol. 2017, 219, 22–96. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Swiatlowska, P.; Williams, J.; Alencar, G.F.; Bevard, M.H.; Greene, E.S.; Murgai, M.; Turner, S.D.; Connelly, J.J.; Owens, G.K. Activation of the pluripotency factor OCT4 in smooth muscle cells is atheroprotective. Nat. Med. 2016, 22, 657–665. [Google Scholar]
- Tang, Z.; Wang, A.; Yuan, F.; Yan, Z.; Liu, B.; Chu, J.S.; Helms, J.A.; Li, S. Differentiation of multipotent vascular stem cells contributes to vascular diseases. Nat. Commun. 2012, 3, 875. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Cooley, B.C.; Nevado, J.; Mellad, J.; Yang, D.; St Hilaire, C.; Negro, A.; Fang, F.; Chen, G.; San, H.; Walts, A.D.; et al. TGF-β signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci. Transl. Med. 2014, 6, 227ra34. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xu, Q. Stem/Progenitor cells in vascular regeneration. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1114–1119. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Ritchie, R.P.; Huang, H.; Zhang, J.; Chen, Y.E. Smooth muscle cell differentiation in vitro: Models and underlying molecular mechanisms. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1485–1494. [Google Scholar] [CrossRef] [PubMed]
- Kurpinski, K.; Lam, H.; Chu, J.; Wang, A.; Kim, A.; Tsay, E.; Agrawal, S.; Schaffer, D.V.; Li, S. Transforming growth factor-beta and notch signaling mediate stem cell differentiation into smooth muscle cells. Stem Cells 2010, 28, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.; Wang, D.; Xu, K.; Wang, J.; Zhang, Z.; Yang, L.; Yang, G.-Y.; Li, S. Contribution of Vascular Cells to Neointimal Formation. PLoS ONE 2017, 12, e0168914. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Li, C.; Zhen, G.; Jiao, K.; He, W.; Jia, X.; Wang, W.; Shi, C.; Xing, Q.; Chen, Y.-F.; Jan De Beur, S.; Yu, B.; Cao, X. Injury-activated transforming growth factor β controls mobilization of mesenchymal stem cells for tissue remodeling. Stem Cells 2012, 30, 2498–2511. [Google Scholar] [CrossRef] [PubMed]
- Bobryshev, Y.V.; Orekhov, A.N.; Chistiakov, D.A. Vascular stem/progenitor cells: Current status of the problem. Cell Tissue Res. 2015, 362, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Yao, E.-H.; Yu, Y.; Fukuda, N. Oxidative Stress on Progenitor and Stem Cells in Cardiovascular Diseases. Curr. Pharm. Biotechnol. 2006, 7, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Nègre-Salvayre, A.; Augé, N.; Camaré, C.; Bacchetti, T.; Ferretti, G.; Salvayre, R. Dual signaling evoked by oxidized LDLs in vascular cells. Free Radic. Biol. Med. 2017, 106, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Di, A.; Mehta, D.; Malik, A.B. ROS-activated calcium signaling mechanisms regulating endothelial barrier function. Cell Calcium 2016, 60, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Gayosso, I.; Platts, S.H.; Duling, B.R. Reactive oxygen species mediate modification of glycocalyx during ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2006, 290, H2247–H2256. [Google Scholar] [CrossRef] [PubMed]
- Pervaiz, S.; Taneja, R.; Ghaffari, S. Oxidative Stress Regulation of Stem and Progenitor Cells. Antioxid. Redox Signal. 2009, 11, 2777–2789. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kröller-Schön, S.; Steven, S.; Schulz, E.; Münzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef] [PubMed]
- Ganeva, M.G.; Getova, D.P.; Gadjeva, V.G. Adverse drug reactions and reactive oxygen species. Folia Med. (Plovdiv.) 2008, 50, 5–11. [Google Scholar] [PubMed]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Kawahara, T.; Diebold, B. Regulation of Nox and Duox enzymatic activity and expression. Free Radic. Biol. Med. 2007, 43, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Skonieczna, M.; Hejmo, T.; Poterala-Hejmo, A.; Cieslar-Pobuda, A.; Buldak, R.J. NADPH Oxidases: Insights into Selected Functions and Mechanisms of Action in Cancer and Stem Cells. Oxid. Med. Cell. Longev. 2017, 2017, 9420539. [Google Scholar] [CrossRef] [PubMed]
- Siu, K.L.; Gao, L.; Cai, H. Differential Roles of Protein Complexes NOX1-NOXO1 and NOX2-p47phox in Mediating Endothelial Redox Responses to Oscillatory and Unidirectional Laminar Shear Stress. J. Biol. Chem. 2016, 291, 8653–8662. [Google Scholar] [CrossRef] [PubMed]
- Przybylska, D.; Janiszewska, D.; Goździk, A.; Bielak-Zmijewska, A.; Sunderland, P.; Sikora, E.; Mosieniak, G. NOX4 downregulation leads to senescence of human vascular smooth muscle cells. Oncotarget 2016, 7, 66429–66443. [Google Scholar] [CrossRef] [PubMed]
- Haurani, M.J.; Cifuentes, M.E.; Shepard, A.D.; Pagano, P.J. Nox4 oxidase overexpression specifically decreases endogenous Nox4 mRNA and inhibits angiotensin II-induced adventitial myofibroblast migration. Hypertension 2008, 52, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-E.; Cho, K.E.; Lee, K.E.; Kim, J.; Bae, Y.S. Nox4-mediated cell signaling regulates differentiation and survival of neural crest stem cells. Mol. Cells 2014, 37, 907–911. [Google Scholar] [CrossRef] [PubMed]
- Drummond, G.R.; Sobey, C.G. Endothelial NADPH oxidases: Which NOX to target in vascular disease? Trends Endocrinol. Metab. 2014, 25, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.-H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Annu. Rev. Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schröder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Forte, M.; Nocella, C.; De Falco, E.; Palmerio, S.; Schirone, L.; Valenti, V.; Frati, G.; Carnevale, R.; Sciarretta, S. The Pathophysiological Role of NOX2 in Hypertension and Organ Damage. High Blood Press Cardiovasc. Prev. 2016, 23, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Ueyama, T.; Nakakita, J.; Nakamura, T.; Kobayashi, T.; Kobayashi, T.; Son, J.; Sakuma, M.; Sakaguchi, H.; Leto, T.L.; Saito, N. Cooperation of p40(phox) with p47(phox) for Nox2-based NADPH oxidase activation during Fcγ receptor (FcγR)-mediated phagocytosis: Mechanism for acquisition of p40(phox) phosphatidylinositol 3-phosphate (PI(3)P) binding. J. Biol. Chem. 2011, 286, 40693–40705. [Google Scholar] [CrossRef] [PubMed]
- El-Benna, J.; Dang, P.M.-C.; Gougerot-Pocidalo, M.-A. Priming of the neutrophil NADPH oxidase activation: Role of p47phox phosphorylation and NOX2 mobilization to the plasma membrane. Semin. Immunopathol. 2008, 30, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Lyle, A.N.; Deshpande, N.N.; Taniyama, Y.; Seidel-Rogol, B.; Pounkova, L.; Du, P.; Papaharalambus, C.; Lassègue, B.; Griendling, K.K. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009, 105, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Carrizzo, A.; Forte, M.; Lembo, M.; Formisano, L.; Puca, A.A.; Vecchione, C. Rac-1 as a new therapeutic target in cerebro- and cardio-vascular diseases. Curr. Drug Targets 2014, 15, 1231–1246. [Google Scholar] [CrossRef] [PubMed]
- Miyano, K.; Sumimoto, H. Assessment of the role for Rho family GTPases in NADPH oxidase activation. Methods Mol. Biol. 2012, 827, 195–212. [Google Scholar] [PubMed]
- Coughlin, S.R. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 2005, 3, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Bäumer, A.T.; Ten Freyhaus, H.; Sauer, H.; Wartenberg, M.; Kappert, K.; Schnabel, P.; Konkol, C.; Hescheler, J.; Vantler, M.; Rosenkranz, S. Phosphatidylinositol 3-kinase-dependent membrane recruitment of Rac-1 and p47phox is critical for alpha-platelet-derived growth factor receptor-induced production of reactive oxygen species. J. Biol. Chem. 2008, 283, 7864–7876. [Google Scholar] [CrossRef] [PubMed]
- Lien, G.-S.; Wu, M.-S.; Bien, M.-Y.; Chen, C.-H.; Lin, C.-H.; Chen, B.-C. Epidermal growth factor stimulates nuclear factor-κB activation and heme oxygenase-1 expression via c-Src, NADPH oxidase, PI3K, and Akt in human colon cancer cells. PLoS ONE 2014, 9, e104891. [Google Scholar] [CrossRef] [PubMed]
- Bondi, C.D.; Manickam, N.; Lee, D.Y.; Block, K.; Gorin, Y.; Abboud, H.E.; Barnes, J.L. NAD(P)H oxidase mediates TGF-beta1-induced activation of kidney myofibroblasts. J. Am. Soc. Nephrol. 2010, 21, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.R.; Holman, T.R.; Imai, Y.; Jadhav, A.; Kenyon, V.; Maloney, D.J.; Nadler, J.L.; Rai, G.; Simeonov, A.; Taylor-Fishwick, D.A. Integration of pro-inflammatory cytokines, 12-lipoxygenase and NOX-1 in pancreatic islet beta cell dysfunction. Mol. Cell. Endocrinol. 2012, 358, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Weissmann, N.; Schröder, K. Nox family NADPH oxidases in mechano-transduction: Mechanisms and consequences. Antioxid. Redox Signal. 2014, 20, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Gyurko, R.; Siqueira, C.C.; Caldon, N.; Gao, L.; Kantarci, A.; Van Dyke, T.E. Chronic hyperglycemia predisposes to exaggerated inflammatory response and leukocyte dysfunction in Akita mice. J. Immunol. 2006, 177, 7250–7256. [Google Scholar] [CrossRef] [PubMed]
- Ushio-Fukai, M. Vascular signaling through G protein-coupled receptors: New concepts. Curr. Opin. Nephrol. Hypertens. 2009, 18, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009, 47, 1239–1253. [Google Scholar] [CrossRef] [PubMed]
- Manea, S.-A.; Constantin, A.; Manda, G.; Sasson, S.; Manea, A. Regulation of Nox enzymes expression in vascular pathophysiology: Focusing on transcription factors and epigenetic mechanisms. Redox. Biol. 2015, 5, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Pendyala, S.; Natarajan, V. Redox regulation of Nox proteins. Respir. Physiol. Neurobiol. 2010, 174, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Guzik, T.J. Coronary Artery Superoxide Production and Nox Isoform Expression in Human Coronary Artery Disease. Arterioscler. Thromb. Vasc. Biol. 2005, 26, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; De Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar] [CrossRef] [PubMed]
- Brandes, R.P.; Takac, I.; Schröder, K. No superoxide—No stress?: Nox4, the good NADPH oxidase! Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1255–1257. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.; Murdoch, C.E.; Wang, M.; Santos, C.X.; Zhang, M.; Alom-Ruiz, S.; Anilkumar, N.; Ouattara, A.; Cave, A.C.; Walker, S.J.; et al. Endothelial Nox4 NADPH oxidase enhances vasodilatation and reduces blood pressure in vivo. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Dikalova, A.E.; Góngora, M.C.; Harrison, D.G.; Lambeth, J.D.; Dikalov, S.; Griendling, K.K. Upregulation of Nox1 in vascular smooth muscle leads to impaired endothelium-dependent relaxation via eNOS uncoupling. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H673–H679. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U. Janus-faced role of endothelial NO synthase in vascular disease: Uncoupling of oxygen reduction from NO synthesis and its pharmacological reversal. Biol. Chem. 2006, 387, 1521–1533. [Google Scholar] [CrossRef] [PubMed]
- Cai, H. Hydrogen peroxide regulation of endothelial function: Origins, mechanisms, and consequences. Cardiovasc. Res. 2005, 68, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Selemidis, S.; Sobey, C.G.; Wingler, K.; Schmidt, H.H.H.W.; Drummond, G.R. NADPH oxidases in the vasculature: Molecular features, roles in disease and pharmacological inhibition. Pharmacol. Ther. 2008, 120, 254–291. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zhang, Y.; Lim, P.; Miao, Y.; Tan, C.; Mckenzie, K.; Schyvens, C.; Whitworth, J. Apocynin but Not l-Arginine Prevents and Reverses Dexamethasone-Induced Hypertension in the Rat. Am. J. Hypertens. 2006, 19, 413–418. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.Y.; San Martín, A.; Mehta, P.K.; Dikalova, A.E.; Garrido, A.M.; Datla, S.R.; Lyons, E.; Krause, K.-H.; Banfi, B.; Lambeth, J.D.; et al. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-S.; Huang, T.Y.; Bokoch, G.M. Reactive oxygen species regulate a slingshot-cofilin activation pathway. Mol. Biol. Cell 2009, 20, 2650–2660. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesha, D.K.; Takapoo, M.; Banfi, B.; Bhalla, R.C.; Miller, F.J. Nox1 transactivation of epidermal growth factor receptor promotes N-cadherin shedding and smooth muscle cell migration. Cardiovasc. Res. 2012, 93, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Vendrov, A.E.; Madamanchi, N.R.; Niu, X.-L.; Molnar, K.C.; Runge, M.; Szyndralewiez, C.; Page, P.; Runge, M.S. NADPH oxidases regulate CD44 and hyaluronic acid expression in thrombin-treated vascular smooth muscle cells and in atherosclerosis. J. Biol. Chem. 2010, 285, 26545–26557. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Castresana, M.R.; Newman, W.H. Reactive oxygen species-sensitive p38 MAPK controls thrombin-induced migration of vascular smooth muscle cells. J. Mol. Cell. Cardiol. 2004, 36, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Datla, S.R.; McGrail, D.J.; Vukelic, S.; Huff, L.P.; Lyle, A.N.; Pounkova, L.; Lee, M.; Seidel-Rogol, B.; Khalil, M.K.; Hilenski, L.L.; et al. Poldip2 controls vascular smooth muscle cell migration by regulating focal adhesion turnover and force polarization. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H945–H957. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.T.; Gomez, D.; Bell, R.D.; Campbell, J.H.; Clowes, A.W.; Gabbiani, G.; Giachelli, C.M.; Parmacek, M.S.; Raines, E.W.; Rusch, N.J.; et al. Smooth muscle cell plasticity: Fact or fiction? Circ. Res. 2013, 112, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Szocs, K. Upregulation of Nox-Based NAD(P)H Oxidases in Restenosis After Carotid Injury. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Kazama, K.; Okada, M.; Yamawaki, H. A novel adipocytokine, omentin, inhibits platelet-derived growth factor-BB-induced vascular smooth muscle cell migration through antioxidative mechanism. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1714–H1719. [Google Scholar] [CrossRef] [PubMed]
- Jay, D.B.; Papaharalambus, C.A.; Seidel-Rogol, B.; Dikalova, A.E.; Lassègue, B.; Griendling, K.K. Nox5 mediates PDGF-induced proliferation in human aortic smooth muscle cells. Free Radic. Biol. Med. 2008, 45, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Liu, Z.; Xu, Q.; Peng, H.; Chen, L.; Huang, X.; Yang, T.; Yu, Z.; Cheng, G.; Zhang, G.; et al. Involvement of vascular peroxidase 1 in angiotensin II-induced hypertrophy of H9c2 cells. J. Am. Soc. Hypertens. 2017, 11, 519–529.e1. [Google Scholar] [CrossRef] [PubMed]
- Manea, A.; Manea, S.-A.; Todirita, A.; Albulescu, I.C.; Raicu, M.; Sasson, S.; Simionescu, M. High-glucose-increased expression and activation of NADPH oxidase in human vascular smooth muscle cells is mediated by 4-hydroxynonenal-activated PPARα and PPARβ/δ. Cell Tissue Res. 2015, 361, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Gole, H.K.A.; Tharp, D.L.; Bowles, D.K. Upregulation of intermediate-conductance Ca2+-activated K+ channels (KCNN4) in porcine coronary smooth muscle requires NADPH oxidase 5 (NOX5). PLoS ONE 2014, 9, e105337. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.I.; Csányi, G.; Ranayhossaini, D.J.; Feck, D.M.; Blose, K.J.; Assatourian, L.; Vorp, D.A.; Pagano, P.J. MEF2B-Nox1 signaling is critical for stretch-induced phenotypic modulation of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Haigh, S.; Barman, S.; Fulton, D.J.R. From form to function: The role of Nox4 in the cardiovascular system. Front. Physiol. 2012, 3, 412. [Google Scholar] [CrossRef] [PubMed]
- Coen, M.; Gabbiani, G.; Bochaton-Piallat, M.-L. Myofibroblast-mediated adventitial remodeling: An underestimated player in arterial pathology. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2391–2396. [Google Scholar] [CrossRef] [PubMed]
- Majesky, M.W. Adventitia and perivascular cells. Arterioscler. Thromb. Vasc. Biol. 2015, 35, e31–e35. [Google Scholar] [CrossRef] [PubMed]
- Csányi, G.; Taylor, W.R.; Pagano, P.J. NOX and inflammation in the vascular adventitia. Free Radic. Biol. Med. 2009, 47, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Meijles, D.N.; Pagano, P.J. Nox and Inflammation in the Vascular Adventitia. Hypertension 2016, 67, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Pagano, P.J.; Clark, J.K.; Cifuentes-Pagano, M.E.; Clark, S.M.; Callis, G.M.; Quinn, M.T. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: Enhancement by angiotensin II. Proc. Natl. Acad. Sci. USA 1997, 94, 14483–14488. [Google Scholar] [CrossRef] [PubMed]
- Tieu, B.C.; Lee, C.; Sun, H.; Lejeune, W.; Recinos, A.; Ju, X.; Spratt, H.; Guo, D.-C.; Milewicz, D.; Tilton, R.G.; et al. An adventitial IL-6/MCP1 amplification loop accelerates macrophage-mediated vascular inflammation leading to aortic dissection in mice. J. Clin. Investig. 2009, 119, 3637–3651. [Google Scholar] [CrossRef] [PubMed]
- Cifuentes, M.E.; Rey, F.E.; Carretero, O.A.; Pagano, P.J. Upregulation of p67(phox) and gp91(phox) in aortas from angiotensin II-infused mice. Am. J. Physiol. Heart Circ. Physiol. 2000, 279, H2234–H2240. [Google Scholar] [PubMed]
- Al Ghouleh, I.; Frazziano, G.; Rodriguez, A.I.; Csányi, G.; Maniar, S.; St Croix, C.M.; Kelley, E.E.; Egaña, L.A.; Song, G.J.; Bisello, A.; et al. Aquaporin 1, Nox1, and Ask1 mediate oxidant-induced smooth muscle cell hypertrophy. Cardiovasc. Res. 2013, 97, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Gabbiani, G. Cell-matrix and cell-cell contacts of myofibroblasts: Role in connective tissue remodeling. Thromb. Haemost. 2003, 90, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Clempus, R.E. Vascular NAD(P)H oxidases: Specific features, expression, and regulation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R277–R297. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.L.; Gorin, Y. Myofibroblast differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney Int. 2011, 79, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol. 2008, 8, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Valle-Prieto, A.; Conget, P.A. Human mesenchymal stem cells efficiently manage oxidative stress. Stem Cells Dev. 2010, 19, 1885–1893. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Galderisi, U.; Marino, I.R. From the laboratory bench to the patient’s bedside: An update on clinical trials with mesenchymal stem cells. J. Cell. Physiol. 2007, 211, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Nesti, C.; Pasquali, L.; Vaglini, F.; Siciliano, G.; Murri, L. The role of mitochondria in stem cell biology. Biosci. Rep. 2007, 27, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-T.; Shih, Y.-R.V.; Kuo, T.K.; Lee, O.K.; Wei, Y.-H. Coordinated changes of mitochondrial biogenesis and antioxidant enzymes during osteogenic differentiation of human mesenchymal stem cells. Stem Cells 2008, 26, 960–968. [Google Scholar] [CrossRef] [PubMed]
- Suda, T.; Takubo, K.; Semenza, G.L. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 2011, 9, 298–310. [Google Scholar] [CrossRef] [PubMed]
- Imhoff, B.R.; Hansen, J.M. Differential redox potential profiles during adipogenesis and osteogenesis. Cell. Mol. Biol. Lett. 2011, 16, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. (Lond.) 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Park, S.G.; Kim, J.H.; Xia, Y.; Sung, J.-H. Generation of reactive oxygen species in adipose-derived stem cells: Friend or foe? Expert Opin. Ther. Targets 2011, 15, 1297–1306. [Google Scholar] [CrossRef] [PubMed]
- Sart, S.; Song, L.; Li, Y. Controlling Redox Status for Stem Cell Survival, Expansion, and Differentiation. Oxid. Med. Cell. Longev. 2015, 2015, 105135. [Google Scholar] [CrossRef] [PubMed]
- Tormos, K.V.; Anso, E.; Hamanaka, R.B.; Eisenbart, J.; Joseph, J.; Kalyanaraman, B.; Chandel, N.S. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab. 2011, 14, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Carrière, A.; Ebrahimian, T.G.; Dehez, S.; Augé, N.; Joffre, C.; André, M.; Arnal, S.; Duriez, M.; Barreau, C.; Arnaud, E.; et al. Preconditioning by mitochondrial reactive oxygen species improves the proangiogenic potential of adipose-derived cells-based therapy. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar] [PubMed]
- Lemasters, J.J.; Holmuhamedov, E. Voltage-dependent anion channel (VDAC) as mitochondrial governator—Thinking outside the box. Biochim. Biophys. Acta 2006, 1762, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Atashi, F.; Modarressi, A.; Pepper, M.S. The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: A review. Stem Cells Dev. 2015, 24, 1150–1163. [Google Scholar] [CrossRef] [PubMed]
- Vlasits, J.; Jakopitsch, C.; Bernroitner, M.; Zamocky, M.; Furtmüller, P.G.; Obinger, C. Mechanisms of catalase activity of heme peroxidases. Arch. Biochem. Biophys. 2010, 500, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Duan, Q.; Wang, T.; Ahmed, M.; Zhang, N.; Li, Y.; Li, L.; Yao, X. Mitochondrial Respiratory Chain Inhibitors Involved in ROS Production Induced by Acute High Concentrations of Iodide and the Effects of SOD as a Protective Factor. Oxid. Med. Cell. Longev. 2015, 2015, 217670. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Kim, H.J.; Chang, E.-J.; Lee, Z.H.; Hwang, S.J.; Kim, H.-M.; Lee, Y.; Kim, H.-H. IL-17 stimulates the proliferation and differentiation of human mesenchymal stem cells: Implications for bone remodeling. Cell Death Differ. 2009, 16, 1332–1343. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Song, S.-Y.; Park, S.G.; Song, S.U.; Xia, Y.; Sung, J.-H. Primary involvement of NADPH oxidase 4 in hypoxia-induced generation of reactive oxygen species in adipose-derived stem cells. Stem Cells Dev. 2012, 21, 2212–2221. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Turner, O.; Stolz, D.; Griffith, L.G.; Wells, A. Production of reactive oxygen species by multipotent stromal cells/mesenchymal stem cells upon exposure to fas ligand. Cell Transplant. 2012, 21, 2171–2187. [Google Scholar] [CrossRef] [PubMed]
- Dickinson, B.C.; Peltier, J.; Stone, D.; Schaffer, D.V.; Chang, C.J. Nox2 redox signaling maintains essential cell populations in the brain. Nat. Chem. Biol. 2011, 7, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Wei, X.; Jiang, L.; Niu, C.; Zhang, J.; Chen, S.; Meng, D. Nox2 and Nox4 regulate self-renewal of murine induced-pluripotent stem cells. IUBMB Life 2016, 68, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Kanda, Y.; Hinata, T.; Kang, S.W.; Watanabe, Y. Reactive oxygen species mediate adipocyte differentiation in mesenchymal stem cells. Life Sci. 2011, 89, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, S.-H.; Park, S.G.; Choi, J.-S.; Xia, Y.; Sung, J.-H. The pivotal role of reactive oxygen species generation in the hypoxia-induced stimulation of adipose-derived stem cells. Stem Cells Dev. 2011, 20, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Schröder, K.; Wandzioch, K.; Helmcke, I.; Brandes, R.P. Nox4 acts as a switch between differentiation and proliferation in preadipocytes. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.; Mooney, C.J.; Hakimjavadi, R.; Fitzpatrick, E.; Guha, S.; Collins, L.E.; Loscher, C.E.; Morrow, D.; Redmond, E.M.; Cahill, P.A. Adult vascular smooth muscle cells in culture express neural stem cell markers typical of resident multipotent vascular stem cells. Cell Tissue Res. 2014, 358, 203–216. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.; Hakimjavadi, R.; Greene, C.; Mooney, C.J.; Fitzpatrick, E.; Collins, L.E.; Loscher, C.E.; Guha, S.; Morrow, D.; Redmond, E.M.; et al. Embryonic rat vascular smooth muscle cells revisited—A model for neonatal, neointimal SMC or differentiated vascular stem cells? Vasc. Cell 2014, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Dan, P.; Velot, É.; Decot, V.; Menu, P. The role of mechanical stimuli in the vascular differentiation of mesenchymal stem cells. J. Cell Sci. 2015, 128, 2415–2422. [Google Scholar] [CrossRef] [PubMed]
- Khani, M.-M.; Tafazzoli-Shadpour, M.; Goli-Malekabadi, Z.; Haghighipour, N. Mechanical characterization of human mesenchymal stem cells subjected to cyclic uniaxial strain and TGF-β1. J. Mech. Behav. Biomed. Mater. 2015, 43, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.S.; Lee, J.-C. β-Catenin mediates cyclic strain-stimulated cardiomyogenesis in mouse embryonic stem cells through ROS-dependent and integrin-mediated PI3K/Akt pathways. J. Cell. Biochem. 2011, 112, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Chen, B.; Wang, G.; Yu, B.; Ren, G.; Ni, G. Effects of mechanical strain on oxygen free radical system in bone marrow mesenchymal stem cells from children. Injury 2011, 42, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Goettsch, C.; Goettsch, W.; Arsov, A.; Hofbauer, L.C.; Bornstein, S.R.; Morawietz, H. Long-term cyclic strain downregulates endothelial Nox4. Antioxid. Redox Signal. 2009, 11, 2385–2397. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Luo, Z.; Pepe, A.E.; Margariti, A.; Zeng, L.; Xu, Q. Embryonic stem cell differentiation into smooth muscle cells is mediated by Nox4-produced H2O2. Am. J. Physiol. Cell Physiol. 2009, 296, C711–C723. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.; Lee, K.P.; Jung, S.H.; Cui, L.; Ko, K.; Kim, B.; Won, K.J. DJ-1 Regulates Differentiation of Human Mesenchymal Stem Cells into Smooth Muscle-like Cells in Response to Sphingosylphosphorylcholine. Proteomics 2017. [Google Scholar] [CrossRef] [PubMed]
- Mahadev, K.; Motoshima, H.; Wu, X.; Ruddy, J.M.; Arnold, R.S.; Cheng, G.; Lambeth, J.D.; Goldstein, B.J. The NAD(P)H oxidase homolog Nox4 modulates insulin-stimulated generation of H2O2 and plays an integral role in insulin signal transduction. Mol. Cell. Biol. 2004, 24, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Pepe, A.E.; Xiao, Q.; Zampetaki, A.; Zhang, Z.; Kobayashi, A.; Hu, Y.; Xu, Q. Crucial role of nrf3 in smooth muscle cell differentiation from stem cells. Circ. Res. 2010, 106, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Q.; Pepe, A.E.; Wang, G.; Luo, Z.; Zhang, L.; Zeng, L.; Zhang, Z.; Hu, Y.; Ye, S.; Xu, Q. Nrf3-Pla2g7 interaction plays an essential role in smooth muscle differentiation from stem cells. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 730–744. [Google Scholar] [CrossRef] [PubMed]
- Coant, N.; Ben Mkaddem, S.; Pedruzzi, E.; Guichard, C.; Tréton, X.; Ducroc, R.; Freund, J.-N.; Cazals-Hatem, D.; Bouhnik, Y.; Woerther, P.-L.; et al. NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate of proliferative progenitor cells in the colon. Mol. Cell. Biol. 2010, 30, 2636–2650. [Google Scholar] [CrossRef] [PubMed]
- Urao, N.; Inomata, H.; Razvi, M.; Kim, H.W.; Wary, K.; McKinney, R.; Fukai, T.; Ushio-Fukai, M. Role of nox2-based NADPH oxidase in bone marrow and progenitor cell function involved in neovascularization induced by hindlimb ischemia. Circ. Res. 2008, 103, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Goettsch, C.; Wongboonsin, J.; Iwata, H.; Schneider, R.K.; Kuppe, C.; Kaesler, N.; Chang-Panesso, M.; Machado, F.G.; Gratwohl, S.; et al. Adventitial MSC-like Cells Are Progenitors of Vascular Smooth Muscle Cells and Drive Vascular Calcification in Chronic Kidney Disease. Cell Stem Cell 2016, 19, 628–642. [Google Scholar] [CrossRef] [PubMed]
- Nemenoff, R.A.; Horita, H.; Ostriker, A.C.; Furgeson, S.B.; Simpson, P.A.; VanPutten, V.; Crossno, J.; Offermanns, S.; Weiser-Evans, M.C.M. SDF-1α induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Herring, B.P.; Hoggatt, A.M.; Burlak, C.; Offermanns, S. Previously differentiated medial vascular smooth muscle cells contribute to neointima formation following vascular injury. Vasc. Cell 2014, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Chappell, J.; Harman, J.L.; Narasimhan, V.M.; Yu, H.; Foote, K.; Simons, B.D.; Bennett, M.R.; Jorgensen, H.F. Extensive Proliferation of a Subset of Differentiated, yet Plastic, Medial Vascular Smooth Muscle Cells Contributes to Neointimal Formation in Mouse Injury and Atherosclerosis Models. Circ. Res. 2016, 119, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; San Martín, A.; Valdivia, A.; Martin-Garrido, A.; Griendling, K.K. Redox-Sensitive Regulation of Myocardin-Related Transcription Factor (MRTF-A) Phosphorylation via Palladin in Vascular Smooth Muscle Cell Differentiation Marker Gene Expression. PLoS ONE 2016, 11, e0153199. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-S.; Chen, B.; Huang, C.-K.; Zhou, J.-J.; Huang, X.; Wang, A.-J.; Li, B.-M.; He, W.-H.; Zhu, X. Ursolic acid suppresses TGF-β1-induced quiescent HSC activation and transformation by inhibiting NADPH oxidase expression and Hedgehog signaling. Exp. Ther. Med. 2017, 14, 3577–3582. [Google Scholar] [CrossRef] [PubMed]
- Redmond, E.M.; Hamm, K.; Cullen, J.P.; Hatch, E.; Cahill, P.A.; Morrow, D. Inhibition of patched-1 prevents injury-induced neointimal hyperplasia. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, S.A.; Piera-Velazquez, S. Endothelial to mesenchymal transition (EndoMT) in the pathogenesis of Systemic Sclerosis-associated pulmonary fibrosis and pulmonary arterial hypertension. Myth or reality? Matrix Biol. 2016, 51, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Kang, X.; Wei, X.; Wang, X.; Jiang, L.; Niu, C.; Zhang, J.; Chen, S.; Meng, D. Nox2 contributes to the arterial endothelial specification of mouse induced pluripotent stem cells by upregulating Notch signaling. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.-S.; Aly, Z.A.; Lai, C.-F.; Cheng, S.-L.; Cai, J.; Huang, E.; Behrmann, A.; Towler, D.A. Vascular Bmp Msx2 Wnt signaling and oxidative stress in arterial calcification. Ann. N. Y. Acd. Sci. 2007, 1117, 40–50. [Google Scholar] [CrossRef] [PubMed]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burtenshaw, D.; Hakimjavadi, R.; Redmond, E.M.; Cahill, P.A. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants 2017, 6, 90. https://doi.org/10.3390/antiox6040090
Burtenshaw D, Hakimjavadi R, Redmond EM, Cahill PA. Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants. 2017; 6(4):90. https://doi.org/10.3390/antiox6040090
Chicago/Turabian StyleBurtenshaw, Denise, Roya Hakimjavadi, Eileen M. Redmond, and Paul A. Cahill. 2017. "Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate" Antioxidants 6, no. 4: 90. https://doi.org/10.3390/antiox6040090
APA StyleBurtenshaw, D., Hakimjavadi, R., Redmond, E. M., & Cahill, P. A. (2017). Nox, Reactive Oxygen Species and Regulation of Vascular Cell Fate. Antioxidants, 6(4), 90. https://doi.org/10.3390/antiox6040090