Different Effects of Metformin and A769662 on Sodium Iodate-Induced Cytotoxicity in Retinal Pigment Epithelial Cells: Distinct Actions on Mitochondrial Fission and Respiration


 , ,
, ,
Abstract
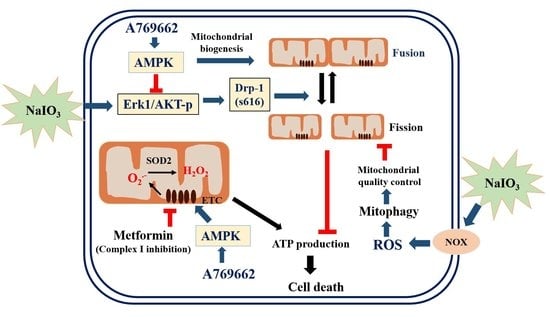
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture
2.3. Annexin V-FITC/PI Assay
2.4. Determination of Cytosolic H2O2, Cytosolic Superoxide Anion, Mitochondrial H2O2 and Mitochondrial Superoxide
2.5. Western Blot Analysis
2.6. Measurement of Mitochondrial Oxygen Consumption Rate
2.7. Mitochondrial Imaging
2.8. Statistical Analysis
3. Results
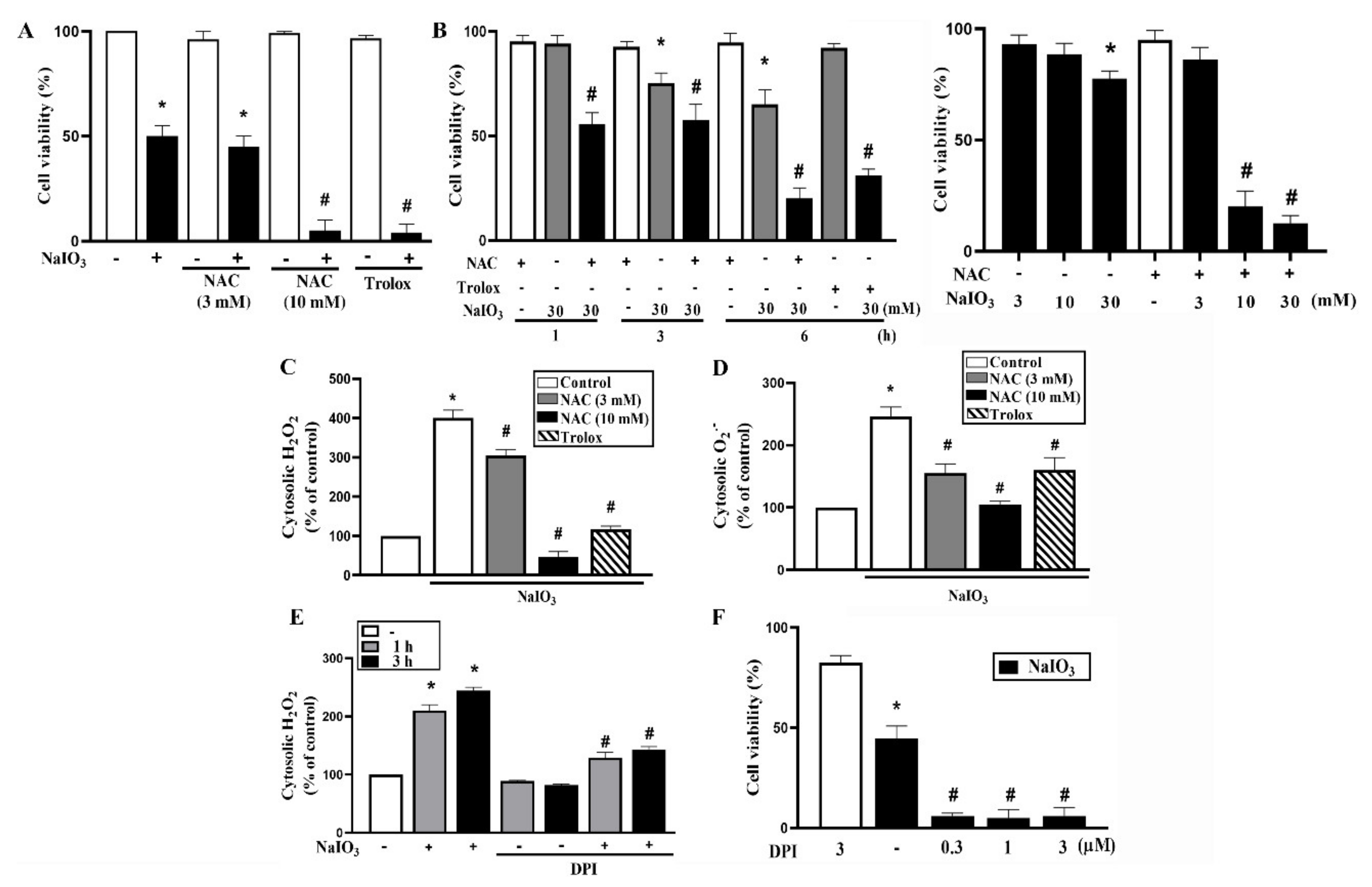
3.1. Cytosolic ROS Production Counteracts Cell Death under NaIO3 Treatment
3.2. PARP1 Is Not Involved in NaIO3-Induced Cell Death
3.3. A769662 Protection While Metformin Enhancement of NaIO3-Induced Cytotoxicity Are Unrelated to Cytosolic ROS Production
3.4. A769662 and Metformin do not Affect Mitochondrial ROS Production nor Mitochondrial Membrane Potential Under NaIO3 Stress
3.5. A769662 but not Metformin Restores Mitochondrial Respiration in NaIO3-Treated RPE Cells
3.6. Mitochondrial Complex I Inhibition Increases NaIO3-Induced Cell Death
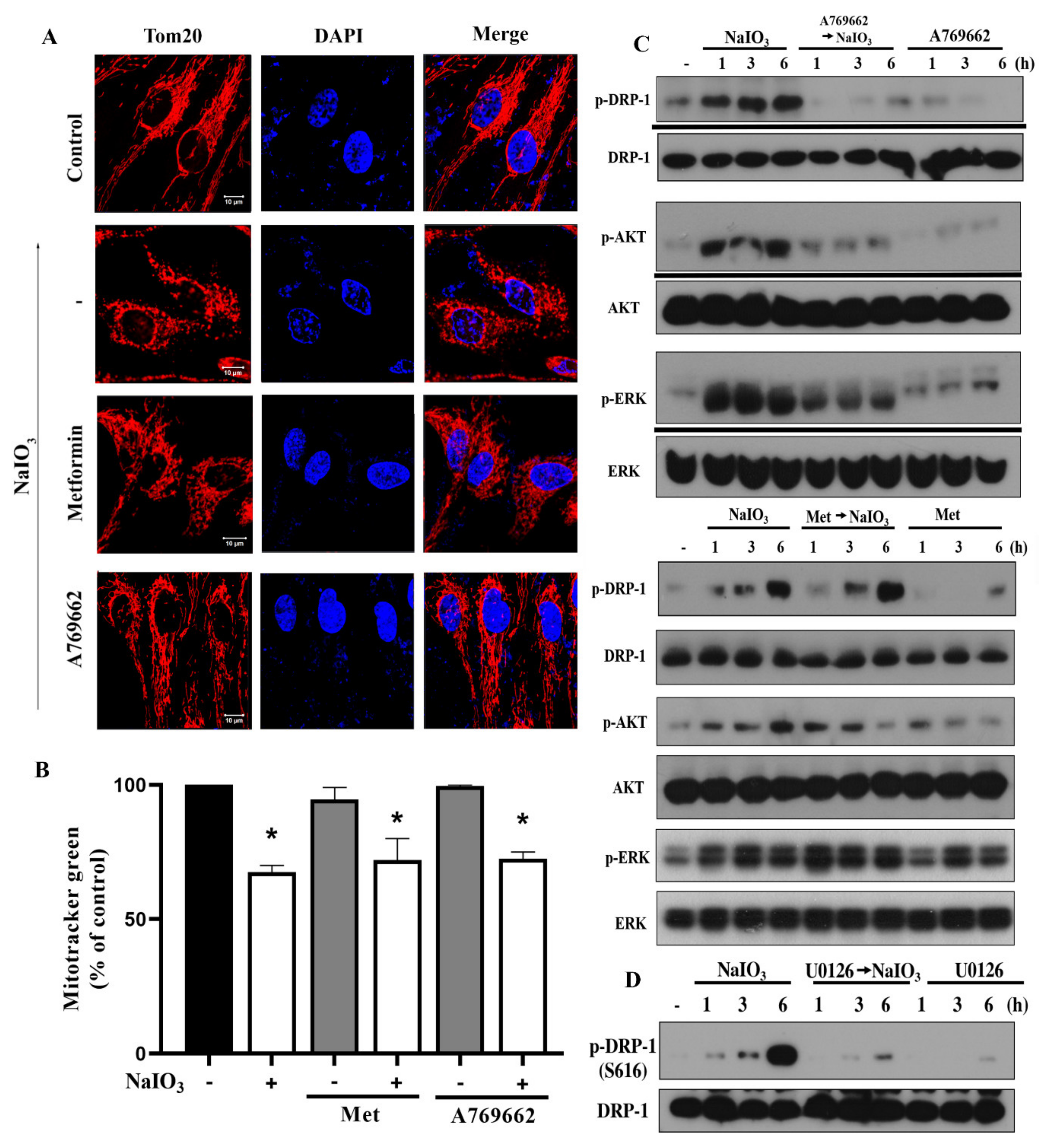
3.7. A769662, but not Metformin, Inhibits NaIO3-Induced Mitochondrial Fission via Inhibition of ERK/Akt-Dependent Drp-1 Phosphorylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bok, D. The retinal pigment epithelium: A versatile partner in vision. J. Cell Sci. Suppl. 1993, 17, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhutto, I.; Lutty, G. Understanding age-related macular degeneration (AMD): Relationships between the photoreceptor/retinal pigment epithelium/Bruch’s membrane/choriocapillaris complex. Mol. Aspects Med. 2012, 33, 295–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balmer, J.; Zulliger, R.; Roberti, S.; Enzmann, V. Retinal cell death caused by sodium iodate involves multiple caspase-dependent and caspase-independent cell-death pathways. Int. J. Mol. Sci. 2015, 16, 15086–15103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanus, J.; Anderson, C.; Sarraf, D.; Ma, J.; Wang, S. Retinal pigment epithelial cell necroptosis in response to sodium iodate. Cell Death Discov. 2016, 2, 16054. [Google Scholar] [CrossRef] [PubMed]
- Hanus, J.; Anderson, C.; Wang, S. RPE necroptosis in response to oxidative stress and in AMD. Ageing Res. Rev. 2015, 24, 286–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hariri, S.; Tam, M.C.; Lee, D.; Hileeto, D.; Moayed, A.A.; Bizheva, K. Noninvasive imaging of the early effect of sodium iodate toxicity in a rat model of outer retina degeneration with spectral domain optical coherence tomography. J. Biomed. Opt. 2013, 18, 26017. [Google Scholar] [CrossRef]
- Wang, J.; Iacovelli, J.; Spencer, C.; Saint-Geniez, M. Direct effect of sodium iodate on neurosensory retina. Investig. Ophthalmol. Vis. Sci. 2014, 55, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Nadal-Nicolas, F.M.; Becerra, S.P. Pigment epithelium-derived factor protects retinal pigment epithelial cells against cytotoxicity “in vitro”. Adv. Exp. Med. Biol. 2018, 1074, 457–464. [Google Scholar] [CrossRef]
- Chan, C.M.; Huang, D.Y.; Sekar, P.; Hsu, S.H.; Lin, W.W. Reactive oxygen species-dependent mitochondrial dynamics and autophagy confer protective effects in retinal pigment epithelial cells against sodium iodate-induced cell death. J. Biomed. Sci. 2019, 26, 40. [Google Scholar] [CrossRef]
- Silwal, P.; Kim, J.K.; Yuk, J.M.; Jo, E.K. AMP-activated protein kinase and host defense against infection. Int. J. Mol. Sci. 2018, 19, 3495. [Google Scholar] [CrossRef] [Green Version]
- Thomson, D.M. The role of AMPK in the regulation of skeletal muscle size, hypertrophy, and regeneration. Int. J. Mol. Sci. 2018, 19, 3125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinberg, G.R.; Carling, D. AMP-activated protein kinase: The current landscape for drug development. Nat. Rev. Drug Discov. 2019, 18, 527–551. [Google Scholar] [CrossRef]
- Li, X.F.; Li, S.Y.; Dai, C.M.; Li, J.C.; Huang, D.R.; Wang, J.Y. PP2A inhibition by LB-100 protects retinal pigment epithelium cells from UV radiation via activation of AMPK signaling. Biochem. Biophys. Res. Commun. 2018, 506, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gaur, U.; Chong, C.M.; Lin, S.; Fang, J.; Zeng, Z.; Wang, H.; Zheng, W. Berberine protects human retinal pigment epithelial cells from hydrogen peroxide-induced oxidative damage through activation of AMPK. Int. J. Mol. Sci. 2018, 19, 1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.L.; Chen, Y.H.; Liang, C.M.; Tai, M.C.; Lu, D.W.; Chen, J.T. Glucosamine-induced autophagy through AMPK-mTOR pathway attenuates lipofuscin-like autofluorescence in human retinal pigment epithelial cells in vitro. Int. J. Mol. Sci. 2018, 19, 1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, K.R.; Zhang, Z.Q.; Yao, J.; Zhao, Y.X.; Duan, J.; Cao, C.; Jiang, Q. Ginsenoside Rg-1 protects retinal pigment epithelium (RPE) cells from cobalt chloride (CoCl2) and hypoxia assaults. PLoS ONE 2013, 8, e84171. [Google Scholar] [CrossRef]
- Xu, L.; Kong, L.; Wang, J.; Ash, J.D. Stimulation of AMPK prevents degeneration of photoreceptors and the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2018, 115, 10475–10480. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Wang, K.; Zhou, F.; Zhu, L. Paeoniflorin attenuates atRAL-induced oxidative stress, mitochondrial dysfunction and endoplasmic reticulum stress in retinal pigment epithelial cells via triggering Ca(2+)/CaMKII-dependent activation of AMPK. Arch. Pharm. Res. 2018, 41, 1009–1018. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Kajdanek, J.; Morawiec, J.; Pawlowska, E.; Blasiak, J. PGC-1alpha protects RPE cells of the aging retina against oxidative stress-induced degeneration through the regulation of senescence and mitochondrial quality control. The significance for AMD pathogenesis. Int. J. Mol. Sci. 2018, 19, 2317. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yasen, M.; Tang, D.; Ye, J.; Aisa, H.A.; Xin, X. Polyphenol-enriched extract of Rosa rugosa Thunb regulates lipid metabolism in diabetic rats by activation of AMPK pathway. Biomed. Pharmacother. 2018, 100, 29–35. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Y.Y.; Jiang, Q.; Li, K.R.; Zhao, Y.X.; Cao, C.; Yao, J. Salvianolic acid A protects RPE cells against oxidative stress through activation of Nrf2/HO-1 signaling. Free Radic. Biol. Med. 2014, 69, 219–228. [Google Scholar] [CrossRef]
- He, H.; Wei, D.; Liu, H.; Zhu, C.; Lu, Y.; Ke, Z.; Jiang, S.; Huang, J. Glycyrrhizin protects against sodium iodate-induced RPE and retinal injury though activation of AKT and Nrf2/HO-1 pathway. J. Cell. Mol. Med. 2019, 23, 3495–3504. [Google Scholar] [CrossRef] [Green Version]
- Hou, S.; Zhang, T.; Li, Y.; Guo, F.; Jin, X. Glycyrrhizic acid prevents diabetic nephropathy by activating AMPK/SIRT1/PGC-1alpha signaling in db/db mice. J. Diabetes Res. 2017, 2017, 2865912. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Chen, C.; Zhu, X.; Li, Y.; Yu, R.; Xu, W. Glycyrrhizin suppresses RANKL-induced osteoclastogenesis and oxidative stress through inhibiting NF-kappaB and MAPK and activating AMPK/Nrf2. Calcif. Tissue Int. 2018, 103, 324–337. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Lu, Y.; Rodrigues, G.A. Resveratrol protects RPE cells from sodium iodate by modulating PPARalpha and PPARdelta. Exp. Eye Res. 2014, 118, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; An, Y.; He, X.; Zhang, D.; He, W. Protection of kaempferol on oxidative stress-induced retinal pigment epithelial cell damage. Oxid. Med. Cell. Longev. 2018, 2018, 1610751. [Google Scholar] [CrossRef]
- Mortezaee, K.; Shabeeb, D.; Musa, A.E.; Najafi, M.; Farhood, B. Metformin as a radiation modifier; implications to normal tissue protection and tumor sensitization. Curr. Clin. Pharmacol. 2019, 14, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Iranshahy, M.; Rezaee, R.; Karimi, G. Hepatoprotective activity of metformin: A new mission for an old drug? Eur. J. Pharmacol. 2019, 850, 1–7. [Google Scholar] [CrossRef]
- Rehman, R.; Abidi, S.H.; Alam, F. Metformin, oxidative stress, and infertility: A way forward. Front. Physiol. 2018, 9, 1722. [Google Scholar] [CrossRef]
- He, K.; Hu, H.; Ye, S.; Wang, H.; Cui, R.; Yi, L. The effect of metformin therapy on incidence and prognosis in prostate cancer: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 2218. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Jiang, D.; Wang, J.; Wang, R.; Chen, T.; Wang, K.; Durgahee, M.S.A.; Wei, X.; Cao, S. Metformin prescription and aortic aneurysm: Systematic review and meta-analysis. Heart 2019, 105, 1351–1357. [Google Scholar] [CrossRef]
- Ren, J.; Zhang, Y. Targeting autophagy in aging and aging-related cardiovascular diseases. Trends Pharmacol. Sci. 2018, 39, 1064–1076. [Google Scholar] [CrossRef] [PubMed]
- Kanigur Sultuybek, G.; Soydas, T.; Yenmis, G. NF-kappaB as the mediator of metformin’s effect on ageing and ageing-related diseases. Clin. Exp. Pharmacol. Physiol. 2019, 46, 413–422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvioli, S.; Ardizzoni, A.; Franceschi, C.; Cossarizza, A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: Implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997, 411, 77–82. [Google Scholar] [CrossRef] [Green Version]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Dong, Q.; He, J.; Wang, X.; Xing, J.; Wang, E.; Qiu, X.; Li, Q. SIRT4 inhibits malignancy progression of NSCLCs, through mitochondrial dynamics mediated by the ERK-Drp1 pathway. Oncogene 2017, 36, 2724–2736. [Google Scholar] [CrossRef]
- Tanner, C.; Wang, G.; Liu, N.; Andrikopoulos, S.; Zajac, J.D.; Ekinci, E.I. Metformin: Time to review its role and safety in chronic kidney disease. Med. J. Aust. 2019, 211, 37–42. [Google Scholar] [CrossRef]
- Fujita, Y.; Inagaki, N. Metformin: New preparations and nonglycemic benefits. Curr. Diabetes Rep. 2017, 17, 5. [Google Scholar] [CrossRef]
- Wang, Y.W.; He, S.J.; Feng, X.; Cheng, J.; Luo, Y.T.; Tian, L.; Huang, Q. Metformin: A review of its potential indications. Drug Des. Dev. Ther. 2017, 11, 2421–2429. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, E. Metformin-induced mitochondrial complex I inhibition: Facts, uncertainties, and consequences. Front. Endocrinol. (Lausanne) 2018, 9, 753. [Google Scholar] [CrossRef]
- Hyttinen, J.M.; Petrovski, G.; Salminen, A.; Kaarniranta, K. 5’-Adenosine monophosphate-activated protein kinase—Mammalian target of rapamycin axis as therapeutic target for age-related macular degeneration. Rejuvenation Res. 2011, 14, 651–660. [Google Scholar] [CrossRef]
- Ducommun, S.; Ford, R.J.; Bultot, L.; Deak, M.; Bertrand, L.; Kemp, B.E.; Steinberg, G.R.; Sakamoto, K. Enhanced activation of cellular AMPK by dual-small molecule treatment: AICAR and A769662. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E688–E696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, N.N.; Zhang, Y.; Ren, J. Mitophagy, mitochondrial dynamics, and homeostasis in cardiovascular aging. Oxid. Med. Cell. Longev. 2019, 2019, 9825061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jang, K.H.; Do, Y.J.; Son, D.; Son, E.; Choi, J.S.; Kim, E. AIF-independent parthanatos in the pathogenesis of dry age-related macular degeneration. Cell Death Discov. 2017, 8, e2526. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Abeta-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 2016, 1863, 2820–2834. [Google Scholar] [CrossRef] [PubMed]
- Tao, A.; Xu, X.; Kvietys, P.; Kao, R.; Martin, C.; Rui, T. Experimental diabetes mellitus exacerbates ischemia/reperfusion-induced myocardial injury by promoting mitochondrial fission: Role of down-regulation of myocardial Sirt1 and subsequent Akt/Drp1 interaction. Int. J. Biochem. Cell. Biol. 2018, 105, 94–103. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chiang, S.F.; Chen, W.T.; Ke, T.W.; Chen, T.W.; You, Y.S.; Lin, C.Y.; Chao, K.S.C.; Huang, C.Y. HMGB1 promotes ERK-mediated mitochondrial Drp1 phosphorylation for chemoresistance through RAGE in colorectal cancer. Cell Death Discov. 2018, 9, 1004. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.B.; Wu, Y.T.; Wu, T.P.; Wei, Y.H. Role of AMPK-mediated adaptive responses in human cells with mitochondrial dysfunction to oxidative stress. Biochim. Biophys. Acta 2014, 1840, 1331–1344. [Google Scholar] [CrossRef]
- Kullmann, F.A.; McDonnell, B.M.; Wolf-Johnston, A.S.; Kanai, A.J.; Shiva, S.; Chelimsky, T.; Rodriguez, L.; Birder, L.A. Stress-induced autonomic dysregulation of mitochondrial function in the rat urothelium. Neurourol. Urodyn. 2019, 38, 572–581. [Google Scholar] [CrossRef]
- Zhang, X.; Feng, Y.; Wang, Y.; Wang, J.; Xiang, D.; Niu, W.; Yuan, F. Resveratrol ameliorates disorders of mitochondrial biogenesis and dynamics in a rat chronic ocular hypertension model. Life Sci. 2018, 207, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.I.; Lee, J.S.; Kwak, B.K.; Hwang, W.M.; Kim, M.J.; Kim, Y.B.; Chung, S.S.; Park, K.S. Metformin ameliorates lipotoxic beta-cell dysfunction through a concentration-dependent dual mechanism of action. Diabetes Metab. J. 2019, 43, 854–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chan, C.-M.; Sekar, P.; Huang, D.-Y.; Hsu, S.-H.; Lin, W.-W. Different Effects of Metformin and A769662 on Sodium Iodate-Induced Cytotoxicity in Retinal Pigment Epithelial Cells: Distinct Actions on Mitochondrial Fission and Respiration. Antioxidants 2020, 9, 1057. https://doi.org/10.3390/antiox9111057
Chan C-M, Sekar P, Huang D-Y, Hsu S-H, Lin W-W. Different Effects of Metformin and A769662 on Sodium Iodate-Induced Cytotoxicity in Retinal Pigment Epithelial Cells: Distinct Actions on Mitochondrial Fission and Respiration. Antioxidants. 2020; 9(11):1057. https://doi.org/10.3390/antiox9111057
Chicago/Turabian StyleChan, Chi-Ming, Ponarulselvam Sekar, Duen-Yi Huang, Shu-Hao Hsu, and Wan-Wan Lin. 2020. "Different Effects of Metformin and A769662 on Sodium Iodate-Induced Cytotoxicity in Retinal Pigment Epithelial Cells: Distinct Actions on Mitochondrial Fission and Respiration" Antioxidants 9, no. 11: 1057. https://doi.org/10.3390/antiox9111057