Usability as the Key Factor to the Design of a Web Server for the CReF Protein Structure Predictor: The wCReF

 ,
,
Abstract
:1. Introduction
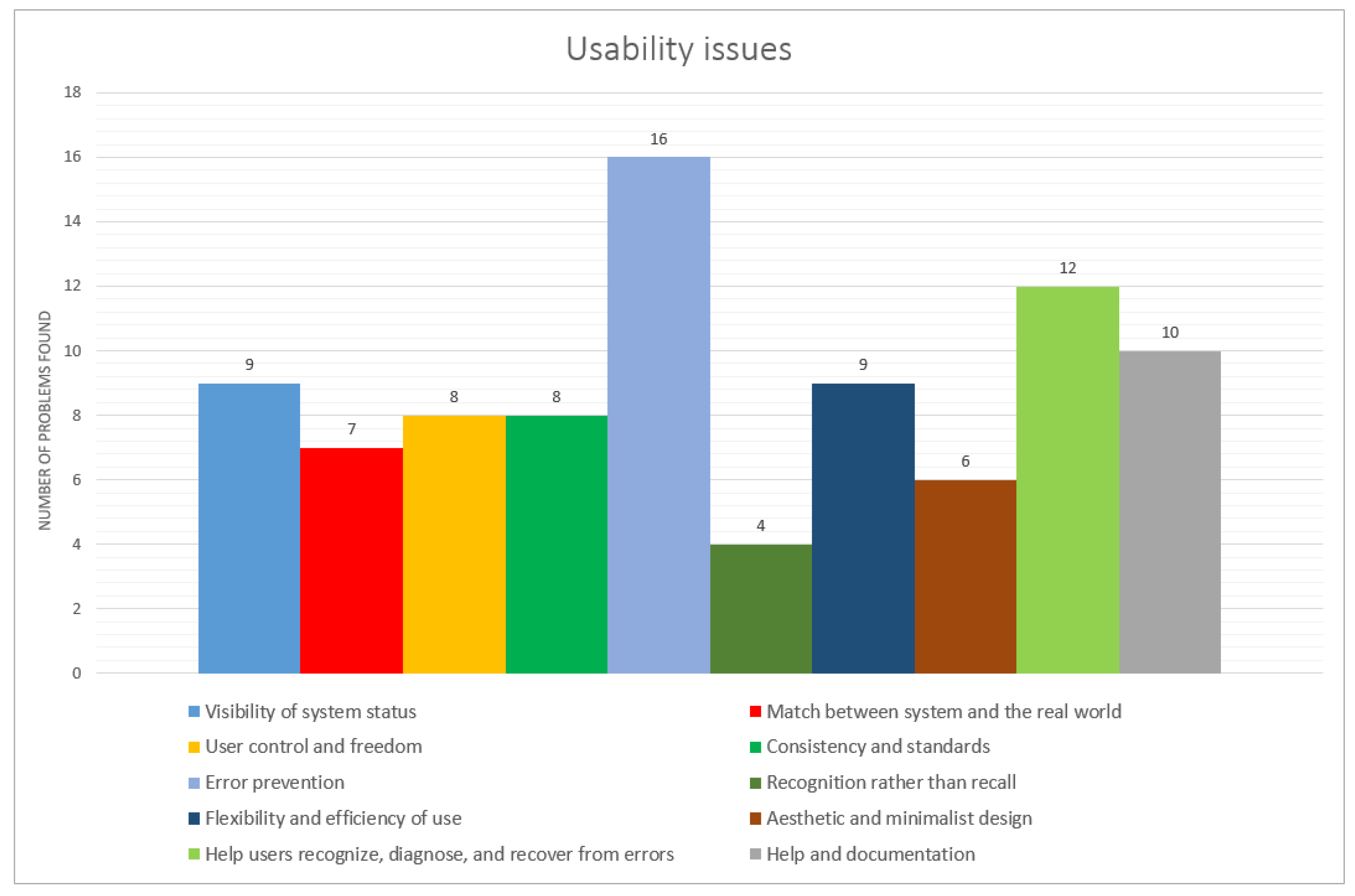
- Requirements definition: To define the requirements for its development, usability evaluations were conducted. They were guided by experts on both the human-computer interaction and bioinformatics domain areas, in three protein structure prediction servers—I-TASSER (Iterative Threading ASSEmbly Refinement) [27,28], QUARK (Computer Algorithm for Ab Initio Protein Structure Prediction and Protein Peptide Folding) [29] and Robetta [30]—all participants of the CASP (Critical Assessment of Protein Structure Prediction) competition. The inspections were conducted through the heuristic evaluation method using Nielsen’s 10 heuristics. Violations were found in all heuristics resulting in 89 usability problems. They were classified into 5 severities, 29 scored as being of high priority and 25 as problems to be solved immediately.
- Results analysis: The assessment of results, to serve as an orientation guide for the key features that wCReF, must be compiled in a software requirements document for its implementation.
- Prototyping: From this step, prototyping was carried out, which helped with the detection of new usability problems with end users by adapting the Ssemugabi satisfaction questionnaire.
2. Background
Protein Structure Prediction Servers
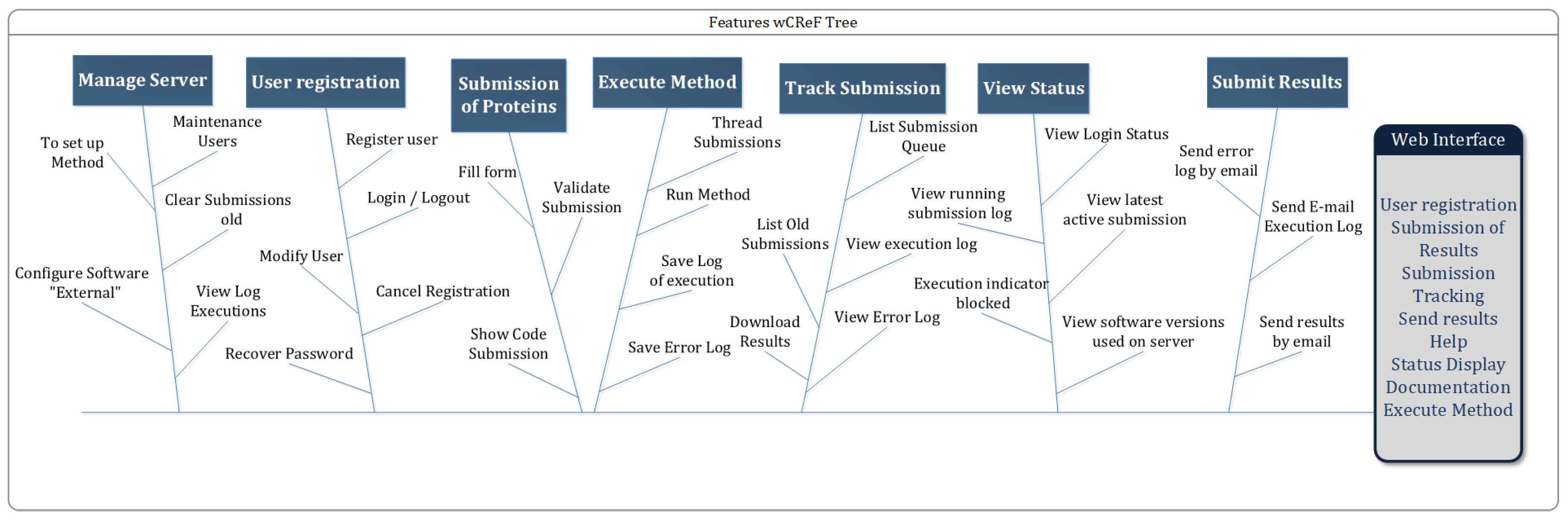
3. wCReF
3.1. Algorithm
- Fragments the target sequence using a sliding window with an odd number of residues. For each fragment, go through Steps 2 to 5 to predict its central residue dihedral angles.
- Performs a BLAST [44] search using the target fragment as the query and obtains the top scoring matches to use as templates.
- Uses the k-means algorithm to cluster the templates’ central residue torsion angles ( and ) and the secondary structure prediction of the target sequence.
- Uses the selected cluster’s centroid as the central residue torsion angles.
3.2. Implementation
3.2.1. Preparing the CReF Environment
3.2.2. Deciding the Architectural Model
3.3. The Design Process
- Identify users’ needs: to understand the features of a protein structure prediction server, the end user profiles and the commonly-performed tasks. wCReF’s interface considered both expert and novice users in the bioinformatics field.
3.3.1. Identify Users’ Needs
- Register as user (prediction servers usually require registration).
- Log in with the credentials. Change password or update personal information.
- Find out how to input data to perform the prediction.
- Send the sequence of a target protein for prediction.
- View the prediction queue.
- Modify the parameters for prediction (if available) and perform a new send.
- Check if the server returned the expected data.
- Check if it has documentation, help and bibliographical references.
- See if the results are easy to access and understand.
3.3.2. Server Usability Inspection
- Visibility of system status: There was a lack of information about where the user was in the interface. Navigation information such as the selected menu or navigation map was not available.
- Flexibility and efficiency of use: errors regarding user registration on the server, submission of a protein and also on saving the results of the prediction.
- Recognition instead of reminder: Problems occurred such as links with system instructions opened in other pages of the browser, and this was not visible on the same page.
- User control and freedom: The user must be able to return to the initial state after being in an unwanted state (undo and redo). This did not happen. Although systems present this possibility, the relative functionalities were not working or errors occurred during their execution.
- Consistency and standards: System design errors do not follow basic interface patterns following system standards. On one of the servers, for example, the menu in different parts of the system switches location (Figure 2).
- “Error messages should be highlighted in a standard format next to the error that occurred.”
- “There are red messages in the text. Although important, warnings could be presented in a different way so as not to distract users.”
- “Standardize error warnings. Errors appear in different forms in the interface, in other windows, or in text format equal to other information, such as required fields.”
- “In case of errors there is no possibility to return to the initial state.”
- “Errors do not have different colors or symbols in order to get the user’s attention.”
- “It should be possible to cancel the prediction request. In an evaluation the user sent a wrong submission, the system did not detect it and the user could not send another request until the first one was processed.”
- “Error messages do not follow a pattern. Some open in a new browser window, in text format, without buttons for confirmation or return to the previous state. The messages should appear on the same screen as the error, as it is usual on any operating system.”
- “The server does not allow the modeling of amino acid sequences smaller than 20 and greater than 200.”—Simple (Specialist A, bioinformatics)
- “When the user inputs the amino acid sequence, the server should check the input’s size and format and indicate any irregularity. Currently this verification is only performed after submission.”—Cosmetic (Specialist A, bioinformatics)
- “I changed the amino acid sequence by removing special characters and the system only validated the email. I could not perform the task.”—Catastrophic (Specialist B, HCI)
- “There are no error messages when an incorrect protein sequence is pasted.”—Catastrophic (Specialist D, HCI)
- “In the email field it is stated that an academic email must be used. However, I made up an email address and it was accepted ... I submitted it again using @hotmail email and it was detected. That is, this control seems to exist for a list of known mail servers. If there is no way to define what is academic and what is not, there should be no such restriction.”—Catastrophic (Specialist B, HCI)
- “I changed the amino acid sequence, removing some symbols, and the system simply returned information about the email.”—Catastrophic (Specialist B, HCI)
- “There is a lack of information on the limitation of generating one job at a time, until the pending one is finished.”—Catastrophic (Specialist B, HCI)
- “There is no information on why it is necessary to use an academic email. If the user does not have it, he cannot use that server.”—Catastrophic (Specialist B, HCI)
4. wCReF Usability Analysis
4.1. Software Requirements Document and Prototype
- Security requirements: Only the system administrator can configure.
- The application layer for web compatible browsers (Internet Explorer, Firefox, Internet Explorer).
- Reliability requirements: The system must be available 24 h a day, seven days a week. Because it is not a critical system, the system may be out of order until some fault that may occur is corrected.
- Performance requirements: Although not a core requirement of the system, it should be considered as a software quality factor, since bioinformatics users often need rapid results in their research.
- Hardware and software: As it is a Web application it must be accessed by any browser that is connected to the network (Internet), the necessary hardware and software not being defined here.
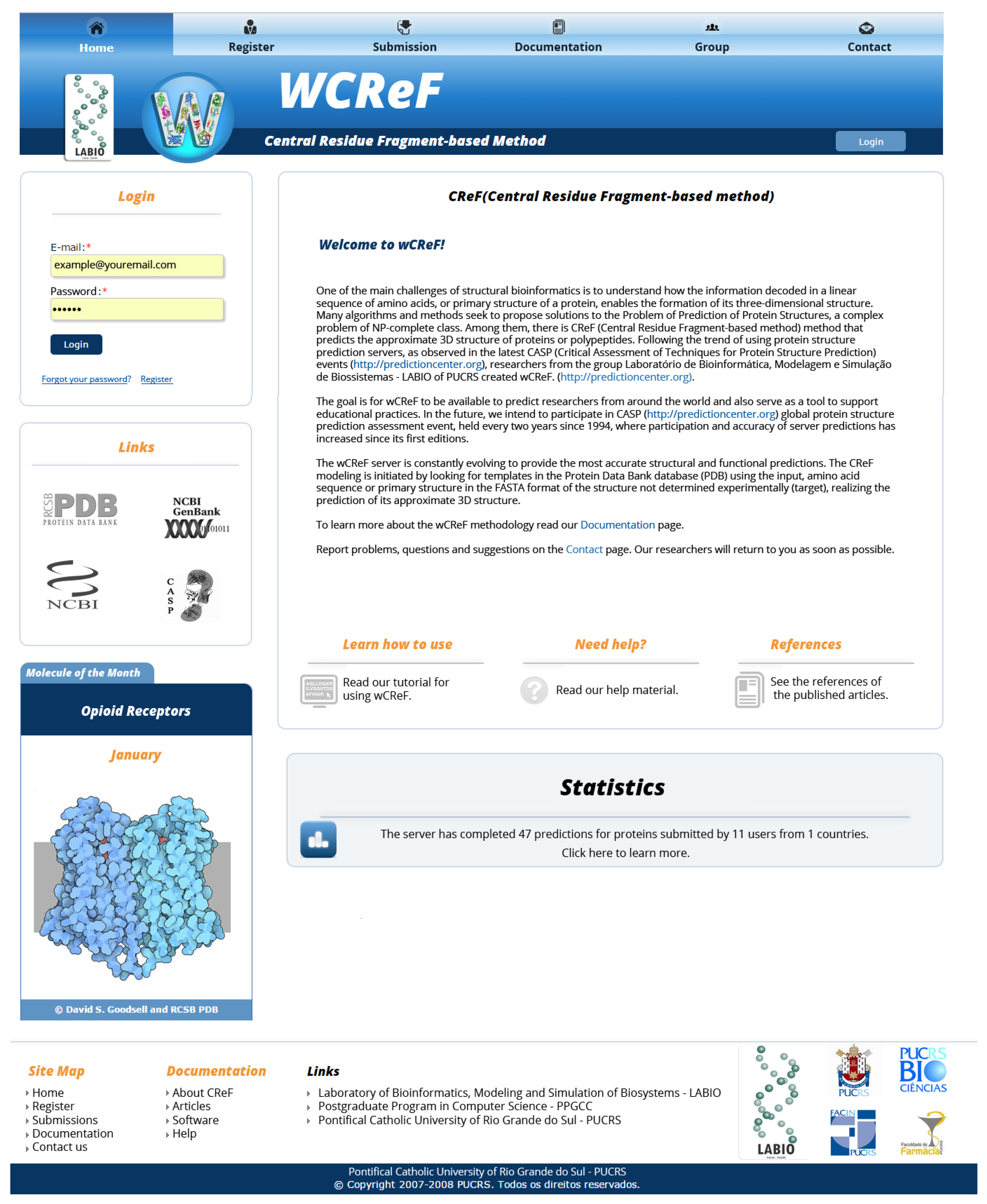
4.2. User Interface
4.3. Questionnaire Evaluation
4.3.1. Methodology
- Presentation of the research, important to motivate the user through a previous description of the research and the benefits of its contribution.
- Ethical care, through the free-informed consent term.
- Instructions on how to fill in the required information and usage scenario.
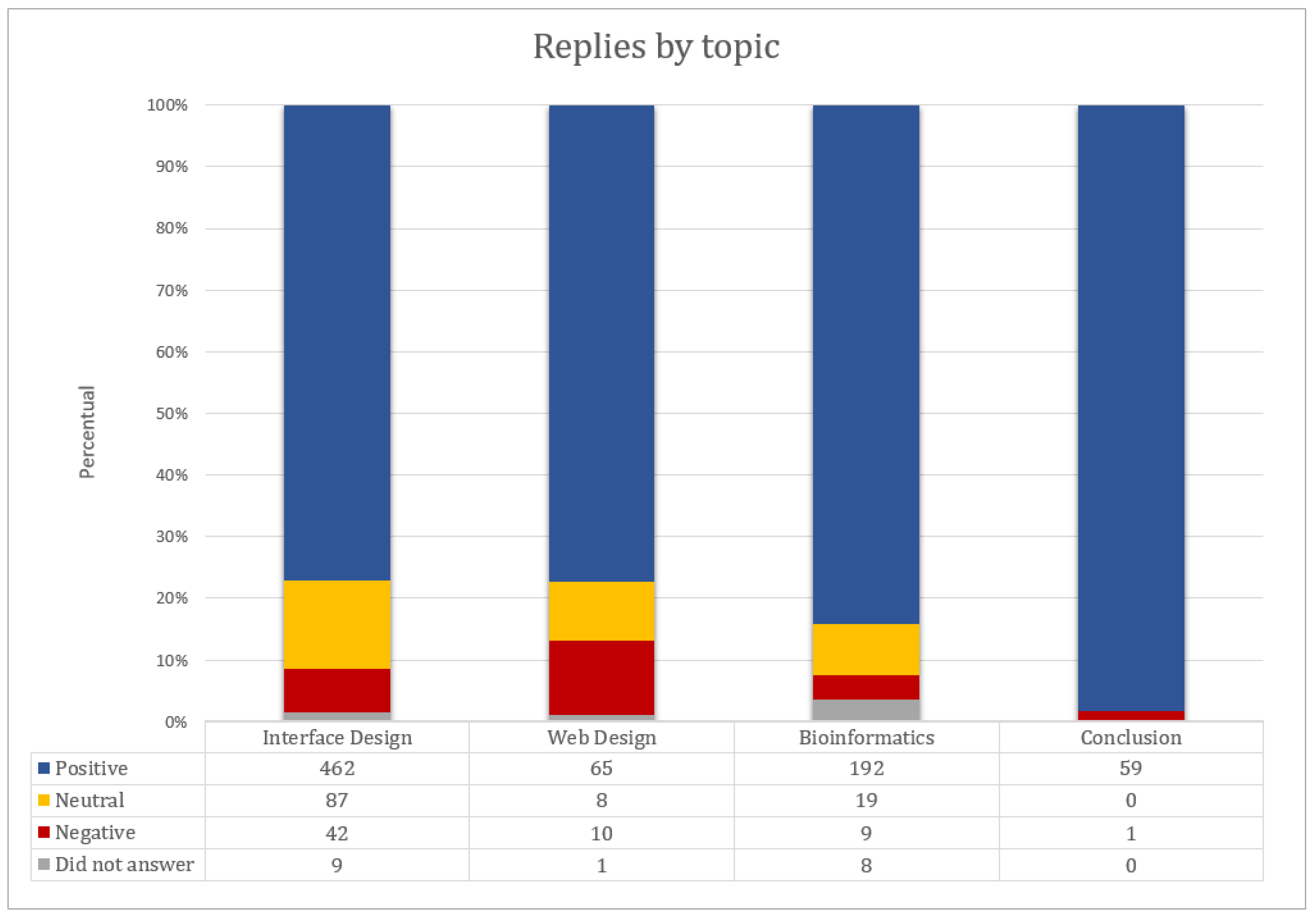
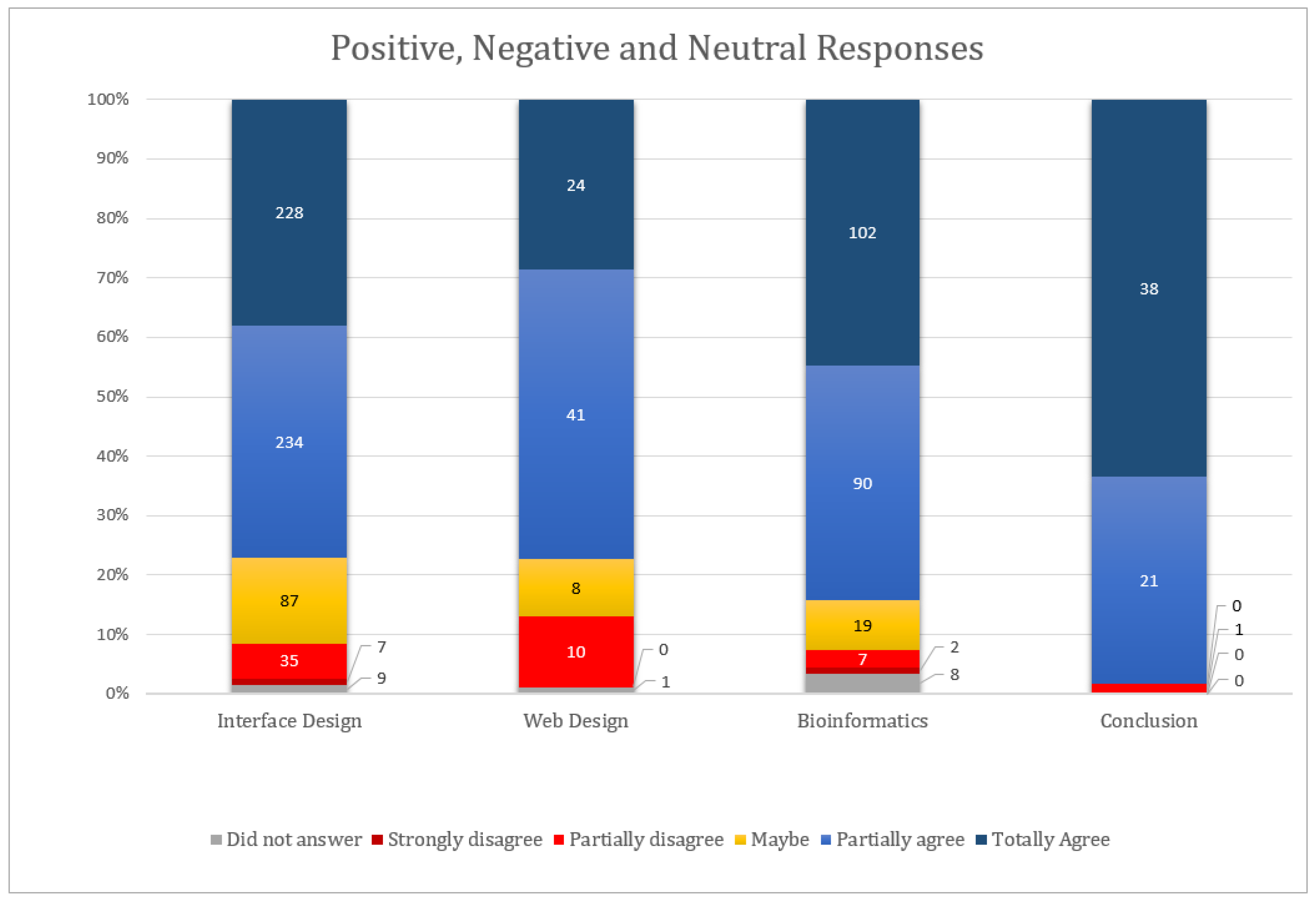
- User profile: As bioinformatics is an interdisciplinary area, we sought to know the profile of the user who performed the tests and their experience in the area, covering seven questions.
- Interface design: 50 closed questions (adapted from the Ssemugabi questionnaire), plus one question for each of the 10 heuristics, open to usability problem notes, totaling 60 questions.
- Web design: 8 issues (7 closed and 1 open) involving the simplicity of site navigation, organization and structure, also based on the Ssemugabi questionnaire.
- New questions were created to analyze the usability of bioinformatics tools, more specifically prediction servers of protein structures. This block includes 19 questions with 19 closed questions and 2 open questions. The first five closed questions refer to the relevance of the web server content for the bioinformatics area and the others about server usability: clarity of goals, objectives and results (Table 7).
- Conclusion: five research closing questions and two open questions for pointing out positive and negative aspects of the system.
4.3.2. Results
- Presentation of a course map: A map with the same functionality as the menu has been added, besides indicating the place where the user is in the interface.
- External links: should point to corresponding web pages. In this way, a revision of the interface links was performed to ensure that there were no errors in this regard.
- Internal links: The creation of links to a given section on a page, mainly on pages that have an extensive content, was suggested, facilitating the navigation.
- Color patterns for links: All interface links have been adjusted to match web standards: unvisited links in blue and visited links in green or purple.
- Copyright statement: Because the wCReF interface and the CReF method are the results of scientific research, within an academic environment, there is a concern about the attribution of copyright of any material, source or software that is used. Although all references were cited, some links with the full reference were not working. A review of the references made available was then performed in order to solve the problem.
- Highlight buttons/icons: It was suggested to highlight the save button by using metaphors, such as a floppy disk icon.
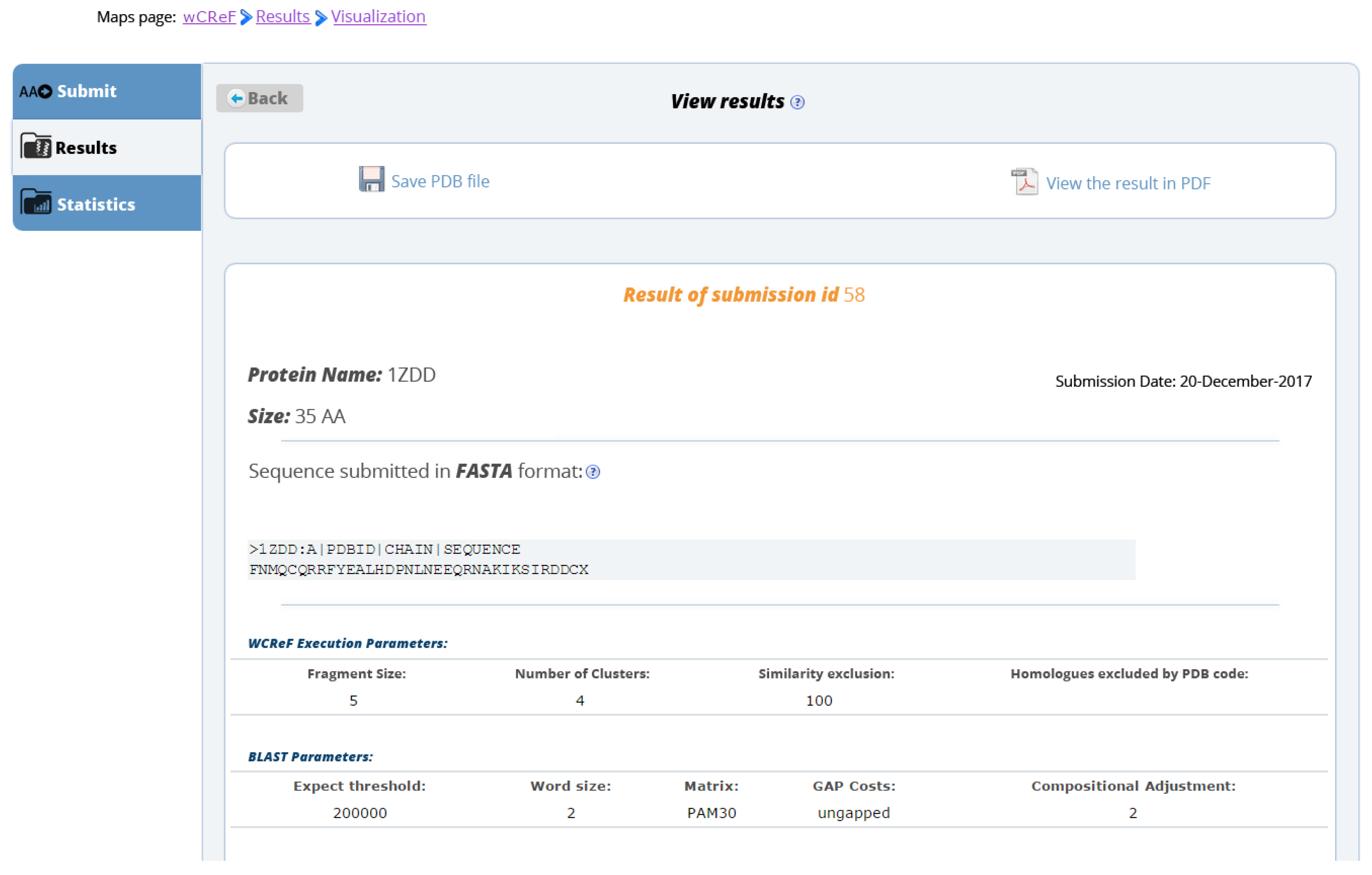
- Results presentation: Users suggested that after submitting a protein sequence, they would be forwarded to a page to track their submission instead of remaining on the prediction submission page.
- When the user submits a protein to the server, he/she receives in his/her e-mail a summary of his/her request, with the name of the protein submitted, the sequence given, the optional parameters and the date it was sent to the system together with the place it is in the queue.
- When the prediction of the target protein is completed, another email is sent to inform that the prediction is finished, along with the link to view the results and the protein in the PDB format as an attachment.
5. Discussion
- “Very good visualization interface of the predicted structure with all the data we have set for the algorithm execution.” (User A, Computer Science)
- “Easy to use, very simple.” (User A, Computer Science)
- “The system is easy for beginners”, “the results are presented in a consistent way” and “the help is available at every step.” (User B, Biology)
- “The system has a simple and clear interface.” “The understanding of the method and submissions are easy.” (User C, Computer Science)
- “The interface is easy to use, beautiful and simple.” (User D, Information Systems)
- “Easy to use interface.” (User E, Mathematics, and User G, Pharmacy)
- “wCReF is a Web system to predict the 3D structure of proteins. The system follows the basic standards required for usability to new and advanced users responding satisfactorily to the purpose for which it was developed.” (User F, Biological Sciences)
- “The interface is simple and lightweight (no overload of colors, images) and has objective menus.” (User H, Biological Sciences)
- System visibility: viewing submissions made and their status, updated every 30 s.
- Clear information about the need to sign in: displays a visible information that the user is required to register on the server for sending jobs and viewing the job queue and sent jobs.
- Minimalist design: reduced menu and priority to the sequence submission area. The menu options are shown only upon request.
- Warning and error messages: displayed in windows on the same page where the error occurred and requiring user confirmation; attention to colors and symbols to highlight information.
- Preventing mistakes: indication of what data are required and which are optional. The function of clearing a field is only performed through user confirmation.
- Error handling: if we enter incorrect data, such as an amino acid sequence in incompatible format, the server displays a warning notice.
- Delete a job or prediction directly: use of a button, without the need for various actions, with user confirmation and the option of sending more than one job at a time to the server.
- Interface consistency: menus, icons and interface buttons are unique and standardized.
- Prediction result not only sent by email: this makes it difficult to use the server as part of a workflow. There is a page with the results that can be read from time to time to determine whether the prediction was complete.
Availability and Requirements
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bruhn, R.; Jennings, S.F. A multidisciplinary bioinformatics minor. ACM SIGCSE Bull. 2007, 39, 348–352. [Google Scholar] [CrossRef]
- Ranganathan, S. Bioinformatics education—Perspectives and challenges. PLoS Comput. Biol. 2005, 1, e52. [Google Scholar] [CrossRef] [PubMed]
- Goujon, M.; McWilliam, H.; Li, W.; Valentin, F.; Squizzato, S.; Paern, J.; Lopez, R. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 2010, 38, W695–W699. [Google Scholar] [CrossRef] [PubMed]
- Fulekar, M. Bioinformatics: Applications in Life and Environmental Sciences; Springer Science & Business Media: New York, NY, USA, 2009. [Google Scholar]
- Luscombe, N.M.; Greenbaum, D.; Gerstein, M. What is bioinformatics? An introduction and overview. Yearb. Med. Inform. 2001, 1, 2. [Google Scholar]
- Gibas, C.; Jambeck, P. Developing Bioinformatics Computer Skills; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2001. [Google Scholar]
- Magana, A.J.; Taleyarkhan, M.; Alvarado, D.R.; Kane, M.; Springer, J.; Clase, K. A survey of scholarly literature describing the field of bioinformatics education and bioinformatics educational research. CBE-Life Sci. Educ. 2014, 13, 607–623. [Google Scholar] [CrossRef] [PubMed]
- Lesk, A. Introduction to Bioinformatics; Oxford University Press: Oxford, UK, 2013. [Google Scholar]
- Al-Ageel, N.; Al-Wabil, A.; Badr, G.; AlOmar, N. Human factors in the design and evaluation of bioinformatics tools. Procedia Manuf. 2015, 3, 2003–2010. [Google Scholar] [CrossRef]
- Acland, A.; Agarwala, R.; Barrett, T.; Beck, J.; Benson, D.A.; Bollin, C.; Bolton, E.; Bryant, S.H.; Canese, K.; Church, D.M.; et al. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2014, 42, D13–D21. [Google Scholar]
- Douglas, C.; Goulding, R.; Farris, L.; Atkinson-Grosjean, J. Socio-Cultural characteristics of usability of bioinformatics databases and tools. Interdiscip. Sci. Rev. 2011, 36, 55–71. [Google Scholar] [CrossRef]
- Bolchini, D.; Finkelstein, A.; Perrone, V.; Nagl, S. Better bioinformatics through usability analysis. Bioinformatics 2008, 25, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Bolchini, D.; Finkestein, A.; Paolini, P. Designing usable bio-information architectures. In Human-Computer Interaction. Interacting in Various Application Domains; Springer: Berlin/Heidelberg, Germany, 2009; pp. 653–662. [Google Scholar]
- Mirel, B. Usability and usefulness in bioinformatics: Evaluating a tool for querying and analyzing protein interactions based on scientists’ actual research questions. In Proceedings of the IEEE International Professional Communication Conference (IPCC 2007), Seattle, WA, USA, 1–3 October 2007; pp. 1–8. [Google Scholar]
- Mirel, B.; Wright, Z. Heuristic evaluations of bioinformatics tools: A development case. In Human-Computer Interaction. New Trends; Springer: Berlin/Heidelberg, Germany, 2009; pp. 329–338. [Google Scholar]
- Shaer, O.; Kol, G.; Strait, M.; Fan, C.; Grevet, C.; Elfenbein, S. G-nome surfer: A tabletop interface for collaborative exploration of genomic data. In Proceedings of the SIGCHI Conference on Human Factors in Computing Systems, Atlanta, GA, USA, 10–15 April 2010; pp. 1427–1436. [Google Scholar]
- Machado, V.S.; Paixão-Cortes, W.R.; de Souza, O.N.; de Borba Campos, M. Decision-Making for Interactive Systems: A Case Study for Teaching and Learning in Bioinformatics. In Proceedings of the International Conference on Learning and Collaboration Technologies, Vancouver, BC, Canada, 9–14 July 2017; Springer: New York, NY, USA, 2017; pp. 90–109. [Google Scholar]
- Veretnik, S.; Fink, J.L.; Bourne, P.E. Computational biology resources lack persistence and usability. PLoS Comput. Biol. 2008, 4, e1000136. [Google Scholar] [CrossRef] [PubMed]
- Javahery, H.; Seffah, A.; Radhakrishnan, T. Beyond power: Making bioinformatics tools user-centered. Commun. ACM 2004, 47, 58–63. [Google Scholar] [CrossRef]
- Seemann, T. Ten recommendations for creating usable bioinformatics command line software. GigaScience 2013, 2, 15. [Google Scholar] [CrossRef] [PubMed]
- Pavelin, K.; Cham, J.A.; de Matos, P.; Brooksbank, C.; Cameron, G.; Steinbeck, C. Bioinformatics meets user-centred design: A perspective. PLoS Comput. Biol. 2012, 8, e1002554. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, P.; Abell, W.; Churcher, C.; McKinnon, A.; McCallum, J. Usability of navigation tools for browsing genetic sequences. In Proceedings of the Eleventh Australasian Conference on User Interface, Brisbane, QD, Australia, 1 January 2010; Australian Computer Society, Inc.: Darlinghurst, NSW, Australia, 2010; Volume 106, pp. 33–41. [Google Scholar]
- Dorn, M.; Norberto de Souza, O. CReF: A central-residue-fragment-based method for predicting approximate 3-D polypeptides structures. In Proceedings of the 2008 ACM Symposium on Applied Computing, Fortaleza, Brazil, 16–20 March 2008; ACM: New York, NY, USA, 2008; pp. 1261–1267. [Google Scholar]
- Dorn, M.; Breda, A.; Norberto de Souza, O. A hybrid method for the protein structure prediction problem. In Proceedings of the Brazilian Symposium on Bioinformatics, Santo André, Brazil, 28–30 August 2008; Springer: New York, NY, USA, 2008; pp. 47–56. [Google Scholar]
- Dorn, M.; Norberto de Souza, O. Mining the Protein Data Bank with CReF to predict approximate 3-D structures of polypeptides. Int. J. Data Min. Bioinform. 2010, 4, 281–299. [Google Scholar] [CrossRef] [PubMed]
- Machado, V.S.; da Silva Tanus, M.D.S.; Paixão-Cortes, W.R.; Norberto de Souza, O.; de Borba Campos, M.; Silveira, M.S. wCReF–A Web Server for the CReF Protein Structure Predictor. In Information Technology-New Generations; Springer: New York, NY, USA, 2018; pp. 831–838. [Google Scholar]
- Wu, S.; Skolnick, J.; Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007, 5, 17. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Interplay of I-TASSER and QUARK for template-based and ab initio protein structure prediction in CASP10. Proteins Struct. Funct. Bioinform. 2014, 82, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [PubMed]
- Anfinsen, C.B. Principles that govern the folding of protein chains. Science 1973, 181, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Kozma, D.; Tusnády, G.E. TMFoldWeb: A web server for predicting transmembrane protein fold class. Biol. Direct 2015, 10, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marks, D.S.; Hopf, T.A.; Sander, C. Protein structure prediction from sequence variation. Nat. Biotechnol. 2012, 30, 1072–1080. [Google Scholar] [CrossRef] [PubMed]
- Santos Filho, O.A.; Alencastro, R.D. Modelagem de proteínas por homologia. Química Nova 2003, 26, 253–259. [Google Scholar] [CrossRef]
- Dill, K.A.; MacCallum, J.L. The protein-folding problem, 50 years on. Science 2012, 338, 1042–1046. [Google Scholar] [CrossRef] [PubMed]
- Roche, D.B.; Buenavista, M.T.; Tetchner, S.J.; McGuffin, L.J. The IntFOLD server: An integrated web resource for protein fold recognition, 3D model quality assessment, intrinsic disorder prediction, domain prediction and ligand binding site prediction. Nucleic Acids Res. 2011, 39, W171–W176. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Wu, S.; Zhang, Y. Ab Initio Protein Structure Prediction. In From Protein Structure to Function with Bioinformatics; Springer: Dordrecht, The Netherlands, 2009; pp. 3–25. [Google Scholar]
- Cao, R.; Freitas, C.; Chan, L.; Sun, M.; Jiang, H.; Chen, Z. ProLanGO: Protein Function Prediction Using Neural Machine Translation Based on a Recurrent Neural Network. Molecules 2017, 22, 1732. [Google Scholar] [CrossRef] [PubMed]
- Dall’Agno, K.C.; Norberto de Souza, O. An expert protein loop refinement protocol by molecular dynamics simulations with restraints. Expert Syst. Appl. 2013, 40, 2568–2574. [Google Scholar] [CrossRef]
- Moult, J.; Fidelis, K.; Kryshtafovych, A.; Schwede, T.; Tramontano, A. Critical Assessment of Methods of Protein Structure Prediction (CASP)–Round XII. Proteins Struct. Funct. Bioinform. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Oron, T.R.; Clark, W.T.; Bankapur, A.R.; D’Andrea, D.; Lepore, R.; Funk, C.S.; Kahanda, I.; Verspoor, K.M.; Ben-Hur, A.; et al. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol. 2016, 17, 184. [Google Scholar] [CrossRef] [PubMed]
- Zardecki, C.; Dutta, S.; Goodsell, D.S.; Voigt, M.; Burley, S.K. RCSB Protein Data Bank: A Resource for Chemical, Biochemical, and Structural Explorations of Large and Small Biomolecules. Chem. Educ. 2016, 93, 569–575. [Google Scholar] [CrossRef]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Shindyalov, I.; Bourne, P. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Hartson, H.R. Human–computer interaction: Interdisciplinary roots and trends. J. Syst. Softw. 1998, 43, 103–118. [Google Scholar] [CrossRef]
- Sharp, H.; Rogers, Y.; Preece, J. Interaction Design: Beyond Human-Computer Interaction; Wiley: New York, NY, USA, 2007. [Google Scholar]
- Nielsen, J. Usability inspection methods. In Proceedings of the Conference Companion on Human Factors in Computing Systems, Boston, MA, USA, 24–28 April 1994; ACM: New York, NY, USA, 1994; pp. 413–414. [Google Scholar]
- Nielsen, J. Usability Engineering; Elsevier: Amsterdam, The Netherlands, 1994. [Google Scholar]
- Winckler, M.A.A.; Farenc, C.; Palanque, P.; Pimenta, M.S. Avaliação da Navegação de Interfaces Web a partir de Modelos. In Proceedings of the Workshop Sobre Fatores Humanos Em Sistemas Computacionais, Florianópolis, Brazil, 15–17 October 2001; Volume 4. [Google Scholar]
- Oliveira, E.S.; Lima, C.R.B. Realce das normas e padrões: A usabilidade como fator primordial para a boa interatividade do usuário. Cad. Ciênc. Hum. Soc. Apl. 2013, 1, 1–17. [Google Scholar]
- Starovasnik, M.A.; Braisted, A.C.; Wells, J.A. Structural mimicry of a native protein by a minimized binding domain. Proc. Natl. Acad. Sci. USA 1997, 94, 10080–10085. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Leung, K.S.; Nakane, T.; Wong, M.H. iview: An interactive WebGL visualizer for protein-ligand complex. BMC Bioinform. 2014, 15, 56. [Google Scholar] [CrossRef] [PubMed]
- Porollo, A.A.; Adamczak, R.; Meller, J. POLYVIEW: A flexible visualization tool for structural and functional annotations of proteins. Bioinformatics 2004, 20, 2460–2462. [Google Scholar] [CrossRef] [PubMed]
- Ssemugabi, S. Usability Evaluation of a Web-Based E-Learning Application: A Study of Two Evaluation Methods. Ph.D. Thesis, University of South Africa (UNISA), Pretoria, South Africa, 2009. [Google Scholar]
- Ssemugabi, S.; De Villiers, R. A comparative study of two usability evaluation methods using a web-based e-learning application. In Proceedings of the 2007 Annual Research Conference of the South African Institute of Computer Scientists and Information Technologists on IT Research in Developing Countries, Port Elizabeth, South Africa, 2–3 October 2007; ACM: New York, NY, USA, 2007; pp. 132–142. [Google Scholar]
- Likert, R. Likert technique for attitude measurement. In Social Psychology: Experimentation, Theory, Research; Sahakian, W.S., Ed.; Intext Educational Publishers: Scranton, PA, USA, 1972; pp. 101–119. [Google Scholar]
- Campos, P.; Matias, M. Avaliação de usabilidade de sites web. Rev. Caminhos 2012, 3, 189–203. [Google Scholar]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Edition | Year | Number of Registered Prediction Servers |
|---|---|---|
| CASP12 | 2016 | 80 |
| CASP11 | 2014 | 84 |
| CASP10 | 2012 | 122 |
| CASP9 | 2010 | 139 |
| CASP8 | 2008 | 122 |
| CASP7 | 2006 | 98 |
| CASP6 | 2004 | 65 |
| CASP5 | 2002 | 72 |
| Prediction Server | Available at: | Reference |
|---|---|---|
| QUARK | http://zhanglab.ccmb.med.umich.edu/QUARK/ | [29] |
| I-TASSER | http://zhanglab.ccmb.med.umich.edu/I-TASSER/ | [27,28] |
| Robetta | http://robetta.bakerlab.org/ | [30] |
| Participant | Education | Expertise |
|---|---|---|
| A | PhD in Physical and Biomolecular Sciences | Bioinformatics |
| B | PhD in Informatics in Education | Computer Science, Informatics in Education and HCI |
| C | PhD student in Computer Science | Bioinformatics and Software Development |
| D | PhD in Design | HCI, Design and Assistive Technologies |
| Severity | Type | Meaning |
|---|---|---|
| 0 | Insignificant | It is not fully considered an usability problem. |
| 1 | Cosmetic | Only an aesthetic problem: it does not need to be repaired unless there is extra time available in the project. |
| 2 | Simple | Minor usability issue: fixing this issue should have low priority. |
| Serious | Greater usability problem: it is important to fix it, so it should be given high priority. | |
| 4 | Catastrophic | Usability catastrophe: it is mandatory to fix it before the product is released. |
| Inspector | A | B | C | D | Total |
|---|---|---|---|---|---|
| I-TASSER | 3 | 12 | 3 | X | 18 |
| QUARK | 3 | 16 | 5 | 21 | 24 + 21 = 45 |
| Robetta | 2 | 13 | 11 | X | 26 |
| TOTAL | 8 | 41 | 19 | 21 | 89 |
| Type of Parameters | Parameter | Definition |
|---|---|---|
| CReF | Fragment size | size of amino acids in a fragment: 5, 7 or 9 |
| Number of clusters for and angles | Define the number of clusters created by the k-means algorithm | |
| Exclude homologues with similarity over | Threshold to exclude fragments from the template search: 100%, 90%, 80% or 70% | |
| Exclude homologues based on PDB code | Exclude from the PDB template search the proteins informed with PDB code | |
| BLAST general | Expect threshold | Probability to find this sequence in a random model |
| Word size | Sequence length for alignment: the default values are 2 or 3 | |
| Matrix | Associate the score that will be applied to aligned sequences. Select according to sequence type | |
| Gap costs | The cost of a gap in the aligned sequence, according to the selected matrix |
| Number | Question |
|---|---|
| 1 | The content of the site follows the application patterns in bioinformatics. |
| 2 | The information is relevant to the development of activities on the server. |
| 3 | The content is at the appropriate level for my understanding. |
| 4 | It is clear which materials are copyrighted and which ones are not. |
| 5 | The server content cites the appropriate references of the material that is used. |
| 5 | I understood what the server objectives are. |
| 7 | I understood the methodology used in wCReF. |
| 8 | I can perform a submission of a target protein satisfactorily. |
| 9 | If I am an advanced user, I can understand the optional parameters in the wCReF submission. |
| 10 | The optional wCReF parameters are in accordance with those used by related applications that I know of. |
| 11 | I can clearly find information about the results. |
| 12 | The response time for my submission is in line with what I expected. |
| 13 | The results are presented in a clear way. |
| 14 | The results are presented in a concise manner. |
| 15 | The results are in line with what I expected. |
| 16 | I can save my results. |
| 17 | The results of the submission are communicated to me without being connected to the server by other means, such as email. |
| 18 | Statistics give me useful information. |
| 19 | As a researcher of the area I can send suggestions, complaints and ask questions to the server’s responsible. |
| Participant | Study Area |
|---|---|
| A | Post-graduation in Medicine and Health Sciences; Graduation in Pharmacy. |
| B | Post-Graduation in Computer Science; Graduation in Computer Engineering. |
| C | Post-graduation in Cellular and Molecular Biology; Degree in biology. |
| D | Post-Graduation in Computer Science; Graduation in Computer Science. |
| E | Post-Graduation in Computer Science; Bachelor in Information Systems. |
| F | Post-Graduate in Zoology; |
| G | Graduate in Pediatrics and Child Health Undergraduate in Biological Sciences. |
| H | Post-Graduation in Computer Science; Bachelor in Mathematics. |
| I | Postgraduate in Zoology; Graduation in biological sciences. |
| J | Master’s Degree in Pharmaceutical Biotechnology; Graduation in Pharmacy. |
| K | Undergraduate Degree in Biological Sciences. |
| L | Undergraduate Degree in Biological Sciences. |
| Heuristic | Guideline |
|---|---|
| Visibility of system status | Provide the result of the prediction in different ways, not just by e-mail. Example: Offer a page with results that can be constantly updated to check if the prediction has been completed. |
| The server needs to inform the user that the protein structure is being predicted. | |
| The system must inform the user of the time required for prediction. | |
| The system should show the user the list of prediction jobs. | |
| The system must inform about how the requested prediction will be delivered. | |
| The message that the protein sequence was sent must be highlighted so that the user is sure to have successfully submitted the prediction request. | |
| The system should make it clear that the user needs to log in to submit a prediction. | |
| Match between system and the real world | Include buttons or icons symbolizing actions familiar to the user such as back, forward, home, save, etc. |
| Provide the option of sending more than one prediction at once. | |
| Focusing the area of prediction submission, avoiding the excess of menus and information, without categorization. | |
| User control and freedom | Provide standard forward, back, undo, redo (undo and redo) buttons available on all interface screens. |
| Allow users to update their password and personal information. | |
| There must be an option to exit the system (log-out). | |
| Prioritize warning messages when performing actions. Example: when the “clean form” button is clicked, it must confirm the user’s intent, as it erases the information previously entered. | |
| Consistency and standards | Standardize the error messages. Avoid error messages in a new browser window, in text format, without confirmation buttons and returning to the previous state. |
| The messages should appear on the same screen as the error, as is the default for any operating system. | |
| Standardize the buttons and differentiate buttons with different functions. | |
| Maintain a standard menu across all interface screens. Preferably use menus in a single format, in the same location and standardized across the entire interface. | |
| Mandatory fields must use the usual symbology that indicates the field of this type | |
| This speeds up user interaction. | |
| Standardize behaviors for similar actions. | |
| Define a pattern of what the user needs to report prior to submitting his/her job; for example, email verification, password and information related to the amino acid sequence. Required fields must be flagged. | |
| Error prevention | Inform the user about the type of input for submission in the system. Example: FASTA format. Do not allow the submission or loading of a file that is not compatible with the one indicated. |
| Confirm and advise the user if the protein sequence is according to the required standards for submission. | |
| When the user inputs data incorrectly, the server should check the size and formatting of the input and indicate any irregularity. | |
| Error prevention | If the user is required to log-in before submitting a prediction request, she/he must be informed in advance. |
| The system should allow the user to choose the type of structure prediction to be performed and allow the modification of input parameters. | |
| User registration must be unique for every system. For example, the registration must be the same to send the prediction and to participate in the discussion forum. | |
| Allow the registration of different types of e-mail, not just academic accounts. | |
| This allows the use of the system by other students or teachers and can also be used as an educational tool. | |
| The system should provide an example of the correct FASTA sequence to be used on the main page to guide the user. | |
| The error messages should be highlighted in the standard format in the same window, next to the error. What often happens is a message in red, but one that can be confused with other prominent messages on the page. | |
| Recognition rather than recall | The system should indicate a marked path to know where the user is at any given time. |
| Describe the features that are not familiar language to the users, that is providing a glossary of technical terms, to facilitate the use of less experienced users. Example: features such as fragment libraries or options like specify secondary structure for specific resides or assign additional restraints and templates to guide modeling. | |
| Provide explanatory terms, with associated links, close to their functionality. A quick help can be included with the mouse or a help symbol that explains the data in a line. | |
| Flexibility and efficiency of use | The action of deleting a job in the system must be unique, preventing the user from performing several activities to complete the process. |
| The user should have the option to cancel the prediction sent, since many servers only accept one prediction per user. | |
| The system must allow customization of the submission process, distinguishing between experienced and inexperienced users. | |
| Aesthetic and minimalist design | Avoid too much text to pass information to users, differentiated only by titles. |
| This makes the content confusing and have too much information. Prioritize the standardization and use of shortcuts, buttons, messages and the distribution of information according to the user’s interest, assembled in blocks or new screens. | |
| Present the menus in a minimalist way. Highlight the functionality menu of the application, leaving less prominently those that are from other pages or other tools. | |
| The actions of similar functionalities should be gathered and presented in a simplified way, without an excess or redundancy of options. For example, actions could be shown only when clicked or when the mouse pointer was placed over items. | |
| Interface warnings and important information should be presented in a clear and prominent format, avoiding red information in the middle of the text, so as not to distract and confuse users. | |
| Help users recognize, diagnose and recover errors | Standardize error warnings, preventing them from appearing in different forms in the interface, as in other windows or in text format equal to other information, as required fields. |
| In case of errors, allow the user to return to the initial state. | |
| Help users recognize, diagnose and recover errors | Again, the prediction submitted should have the possibility of cancellation. In case of sending the wrong submission, the system must detect and inform the user. |
| Enable the user to recover password and username. | |
| Identify if the user is already registered in the system. | |
| Indicate errors with different colors or symbols in order to draw the user’s attention. | |
| Avoid losing forms’ data when the user accidentally clicks the back button. | |
| Help and documentation | The documentation on the server functionalities, the possible inputs and outputs should be clear and available in a simplified way. The user must be informed about the entries that the server accepts, as well as the output files. |
| Quick and contextual help should be made available so that the user does not need to read all the documentation, especially for those who are interested only in configuring and visualizing, such as quick help in each entry field on the submission page. | |
| Clarify to the user essential information, such as the reason for the need to log-in with an academic e-mail or sending a single job at a time in the documentation, in cases where it applies. | |
| The parameters presented in the results must be explained through links and appropriate documentation, such as S (Sheet), H (Helix) or C (Coil) in the prediction of the secondary structure, or what SignalP (Signal Peptide), C-score, S-score or Y-score mean, or the TM-score (TM-score is a metric for measuring the structural similarity of two protein models) parameter. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Machado Paixão-Cortes, V.S.; Dos Santos da Silva Tanus, M.; Paixão-Cortes, W.R.; De Souza, O.N.; De Borba Campos, M.; Silveira, M.S. Usability as the Key Factor to the Design of a Web Server for the CReF Protein Structure Predictor: The wCReF. Information 2018, 9, 20. https://doi.org/10.3390/info9010020
Machado Paixão-Cortes VS, Dos Santos da Silva Tanus M, Paixão-Cortes WR, De Souza ON, De Borba Campos M, Silveira MS. Usability as the Key Factor to the Design of a Web Server for the CReF Protein Structure Predictor: The wCReF. Information. 2018; 9(1):20. https://doi.org/10.3390/info9010020
Chicago/Turabian StyleMachado Paixão-Cortes, Vanessa Stangherlin, Michele Dos Santos da Silva Tanus, Walter Ritzel Paixão-Cortes, Osmar Norberto De Souza, Marcia De Borba Campos, and Milene Selbach Silveira. 2018. "Usability as the Key Factor to the Design of a Web Server for the CReF Protein Structure Predictor: The wCReF" Information 9, no. 1: 20. https://doi.org/10.3390/info9010020