
On the Use of Benchmarks for Multiple Properties †

 ,
,
Abstract
:
1. Introduction
- choosing the method giving the best results for two properties, A and B;
- choosing the method giving the best results for property B, knowing that property A is well described.
2. When Condensed Information Is Not Sufficient
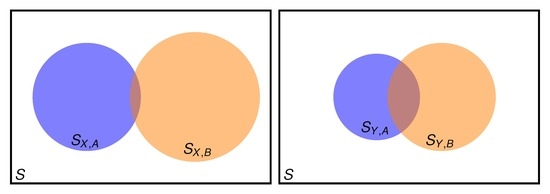
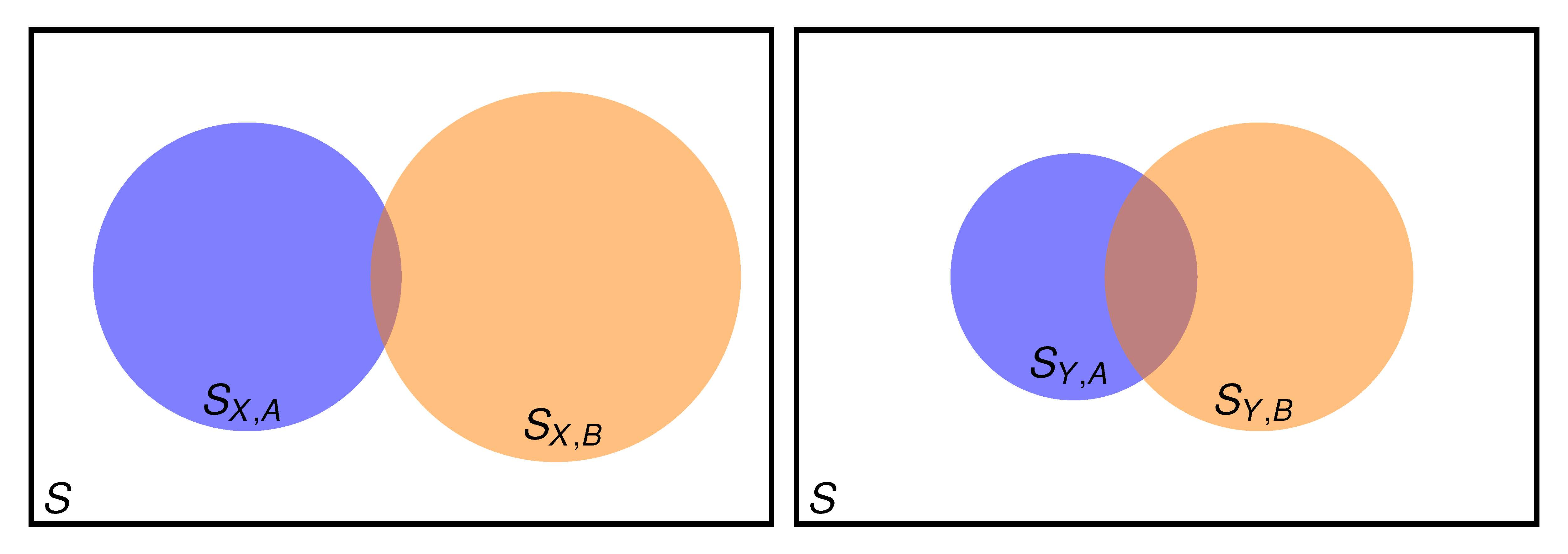
2.1. Setting the Problem
- when good results are needed for both property A and property B?
- when it is guaranteed (it can be checked) that A is well described, but good results for property B are also needed?
2.2. Two Properties Simultaneously Needed
3. Improving the Quality of the Approximations Reduces the Risk of Unreliable Selection
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Curtiss, L.A.; Raghavachari, K.; Redfern, P.C.; Pople, J.A. Assessment of Gaussian-3 and density functional theories for a larger experimental test set. J. Chem. Phys. 2000, 112, 7374–7383. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Assessment of Gaussian-3 and density-functional theories on the G3/05 test set of experimental energies. J. Chem. Phys. 2005, 123. [Google Scholar] [CrossRef]
- Karton, A.; Daon, S.; Martin, J.M.L. W4-11: A high-confidence benchmark dataset for computational thermochemistry derived from first-principles {W4} Data. Chem. Phys. Lett. 2011, 510, 165–178. [Google Scholar] [CrossRef]
- Goerigk, L.; Grimme, S. Efficient and accurate double-hybrid-meta-GGA density functionals evaluation with the extended GMTKN30 database for general main group thermochemistry, kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2010, 7, 291–309. [Google Scholar] [CrossRef] [PubMed]
- Peverati, R.; Truhlar, D.G. Quest for a universal density functional: The accuracy of density functionals across a broad spectrum of databases in chemistry and physics. Philos. Trans. R. Soc. Lond. A 2014, 372. [Google Scholar] [CrossRef] [PubMed]
- Lejaeghere, K.; Van Speybroeck, V.; Van Oost, G.; Cottenier, S. Error estimates for solid-state density-functional theory predictions: An overview by means of the ground-state elemental crystals. Crit. Rev. Solid State Mater. Sci. 2014, 39, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Wellendorff, J.; Lundgaard, K.T.; Møgelhøj, A.; Petzold, V.; Landis, D.D.; Nørskov, J.K.; Bligaard, T.; Jacobsen, K.W. Density functionals for surface science: Exchange-correlation model development with Bayesian error estimation. Phys. Rev. B 2012, 85. [Google Scholar] [CrossRef]
- Wellendorff, J.; Lundgaard, K.T.; Jacobsen, K.W.; Bligaard, T. mBEEF: An accurate semi-local Bayesian error estimation density functional. J. Chem. Phys. 2014, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardirossian, N.; Head-Gordon, M. xB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Phys. Chem. Chem. Phys. 2014, 16, 9904–9924. [Google Scholar]
- Yu, H.S.; Zhang, W.; Verma, P.; Xiao Heac, X.; Truhlar, D.G. Nonseparable exchange–correlation functional for molecules, including homogeneous catalysis involving transition metals. Phys. Chem. Chem. Phys. 2015, 17, 12146–12160. [Google Scholar] [CrossRef] [PubMed]
- Goerigk, L.; Grimme, S. A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys. Chem. Chem. Phys. 2011, 13, 6670–6688. [Google Scholar] [CrossRef] [PubMed]
- Hao, P.; Sun, J.; Xiao, B.; Ruzsinszky, A.; Csonka, G.I.; Tao, J.; Glindmeyer, S.; Perdew, J.P. Performance of meta-GGA functionals on general main group thermochemistry, kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2013, 9, 355–363. [Google Scholar] [CrossRef] [PubMed]
- Civalleri, B.; Presti, D.; Dovesi, R.; Savin, A. On choosing the best density functional approximation. In Chemical Modelling: Applications and Theory; Royal Society of Chemistry: London, UK, 2012; Volume 9, pp. 168–185. [Google Scholar]
- Savin, A.; Johnson, E.R. Judging density functional approximations: Some pitfalls of statistics. Top. Curr. Chem. 2015, 365, 81–95. [Google Scholar]
- Perdew, J.P.; Sun, J.; Garza, A.J.; Scuseria, G. Intensive atomization energy: Re-thinking a metric for electronic-structure-theory methods. Z. Phys. Chem. 2016, in press. [Google Scholar] [CrossRef]
- Pernot, P.; Civalleri, B.; Presti, D.; Savin, A. Prediction uncertainty of density functional approximations for properties of crystals with cubic symmetry. J. Phys. Chem. A 2015, 119, 5288–5304. [Google Scholar] [CrossRef] [PubMed]
- Slater, J.C. A simplification of the hartree-fock method. Phys. Rev. 1951, 81, 385–390. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Csonka, G.I.; Vydrov, O.A.; Scuseria, G.E.; Constantin, L.A.; Zhou, X.; Burke, K. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 2008, 100. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.M.; Izmaylov, A.F.; Scuseria, G.E.; Savin, A. The importance of middle-range hartree-fock-type exchange for hybrid density functionals. J. Chem. Phys. 2007, 127. [Google Scholar] [CrossRef] [PubMed]
- Henderson, T.M.; Izmaylov, A.F.; Scuseria, G.E.; Savin, A. Assessment of a middle-range hybrid functional. J. Chem. Theory Comput. 2008, 4, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.D.; Cai, T.T.; DasGupta, A. Interval estimation for a binomial proportion. Stat. Sci. 2001, 16, 101–133. [Google Scholar]
- Lejaeghere, K.; Vanduyfhuys, L.; Verstraelen, T.; Speybroeck, V.V.; Cottenier, S. Is the error on first-principles volume predictions absolute or relative? Comput. Mater. Sci. 2016, 117, 390–396. [Google Scholar] [CrossRef]
- Duan, X.M.; Song, G.L.; Li, Z.H.; Wang, X.J.; Chen, G.H.; Fan, K.N. Accurate prediction of heat of formation by combining Hartree-Fock/density functional theory calculation with linear regression correction approach. J. Chem. Phys. 2004, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lejaeghere, K.; Jaeken, J.; Speybroeck, V.V.; Cottenier, S. Ab initio based thermal property predictions at a low cost: An error analysis. Phys. Rev. B 2014, 89. [Google Scholar] [CrossRef]
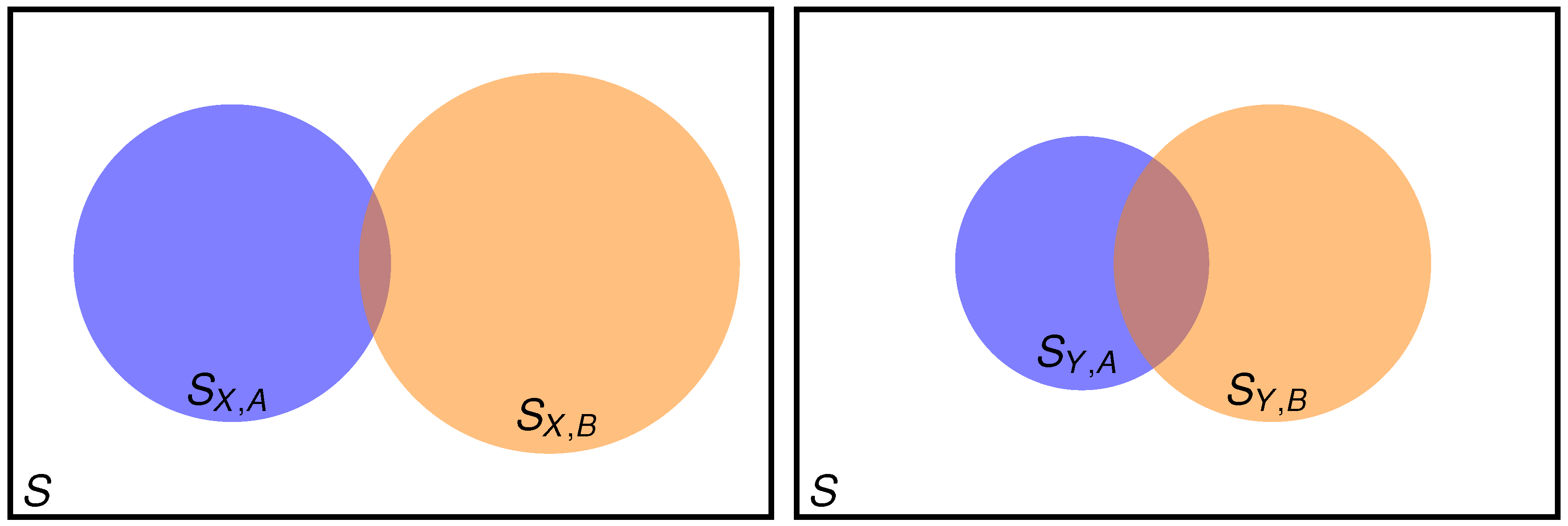

{kind=link}
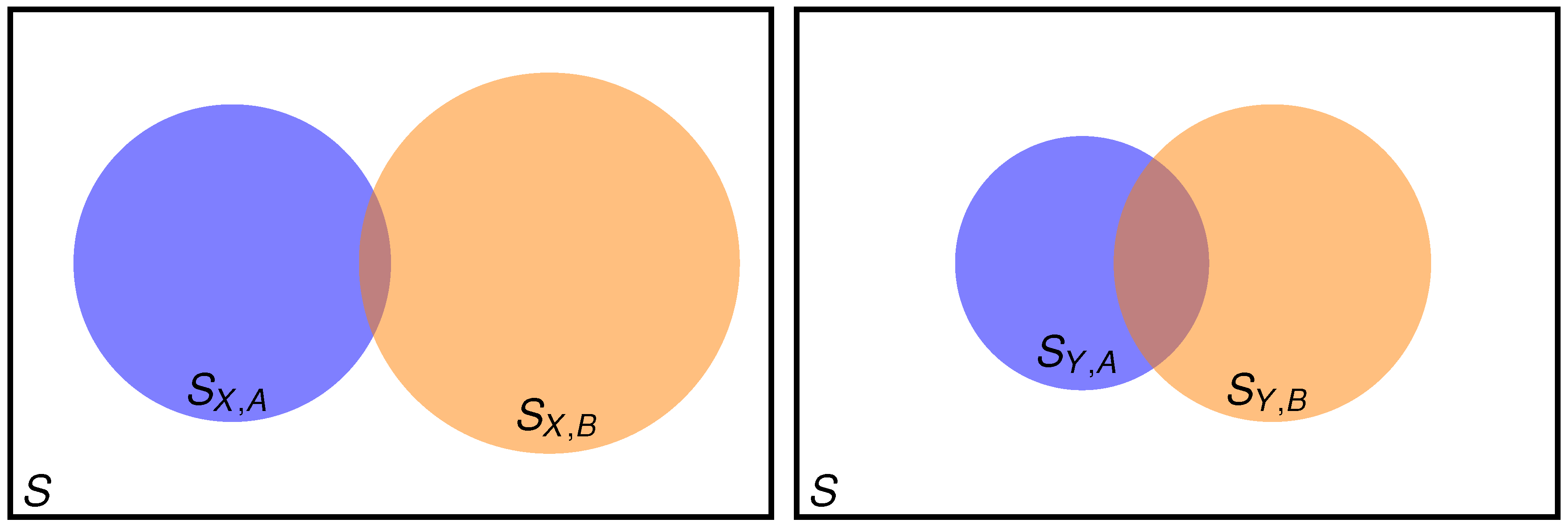{kind=link}
| Method | ||||
|---|---|---|---|---|
| LDA | ||||
| PBEsol | ||||
| HISS |
| Corrected Method | ||||
|---|---|---|---|---|
| LDA | ||||
| PBEsol | ||||
| HISS |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Civalleri, B.; Dovesi, R.; Pernot, P.; Presti, D.; Savin, A. On the Use of Benchmarks for Multiple Properties. Computation 2016, 4, 20. https://doi.org/10.3390/computation4020020
Civalleri B, Dovesi R, Pernot P, Presti D, Savin A. On the Use of Benchmarks for Multiple Properties. Computation. 2016; 4(2):20. https://doi.org/10.3390/computation4020020
Chicago/Turabian StyleCivalleri, Bartolomeo, Roberto Dovesi, Pascal Pernot, Davide Presti, and Andreas Savin. 2016. "On the Use of Benchmarks for Multiple Properties" Computation 4, no. 2: 20. https://doi.org/10.3390/computation4020020