Effect of Melt-Derived Bioactive Glass Particles on the Properties of Chitosan Scaffolds



Abstract
:1. Introduction
2. Materials and Methods
2.1. Glass processing
2.2. Chitosan/glass composites
2.3. Thermal analysis
2.4. Porosity
2.5. Mechanical properties
2.6. Structural properties
2.7. In vitro dissolution
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boccaccini, A.R.; Erol, M.; Stark, W.J.; Mohn, D.; Hong, Z.; Mano, J.F. Polymer/bioactive glass nanocomposites for biomedical applications: A review. Compos. Sci. Technol. 2010, 70, 1764–1776. [Google Scholar] [CrossRef] [Green Version]
- Rahaman, M.N.; Day, D.E.; Sonny Bal, B.; Fu, Q.; Jung, S.B.; Bonewald, L.F.; Tomsia, A.P. Bioactive glass in tissue engineering. Acta Biomater. 2011, 7, 2355–2373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Gestel, N.A.P.; Geurts, J.; Hulsen, D.J.W.; van Rietbergen, B.; Hofmann, S.; Arts, J.J. Clinical applications of S53P4 bioactive glass in bone healing and osteomyelitic treatment: A literature review. Biomed. Res. Int. 2015, 2015, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lindfors, N.C.; Koski, I.; Heikkilä, J.T.; Mattila, K.; Aho, A.J. A prospective randomized 14-year follow-up study of bioactive glass and autogenous bone as bone graft substitutes in benign bone tumors. J. Biomed. Mater. Res. B 2010, 94B, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Massera, J.; Fagerlund, S.; Hupa, L.; Hupa, M. Crystallization mechanism of the bioactive glasses, 45S5 and S53P4. J. Am. Ceram. Soc. 2012, 95, 607–613. [Google Scholar] [CrossRef]
- Nommeots-Nomm, A.; Massera, J. Scaffolds in Tissue Engineering-Materials, Technologies, Clinical Applications, Chapter: Glass and Glass-Ceramic Scaffolds: Manufacturing Methods and the Impact of Crystallization on In-Vitro Dissolution; IntechOpen: London, UK, 2017. [Google Scholar]
- Lan Levengood, S.; Zhang, M. Chitosan-based scaffolds for bone tissue engineering. J. Mater. Chem. B 2014, 7, 3161–3184. [Google Scholar] [CrossRef] [PubMed]
- Muzzarelli, R.A.; Mattioli-Belmonte, M.; Tietz, C.; Biagini, R.; Ferioli, G.; Brunelli, M.A.; Fini, M.; Giardino, R.; Ilari, P.; Biagini, G. Stimulatory effect on bone formation exerted by a modified chitosan. Biomaterials 1994, 15, 1075–1081. [Google Scholar] [CrossRef]
- Venkatesan, J.; Kim, S.K. Chitosan Composites for Bone Tissue Engineering—An Overview. Mar. Drugs 2010, 8, 2252–2266. [Google Scholar] [CrossRef]
- Correia, C.O.; Leite, A.J.; Mano, J.F. Chitosan/bioactive glass nanoparticles scaffolds with shape memory properties. Carbohydr. Polym. 2015, 123, 39–45. [Google Scholar] [CrossRef]
- Luz, G.M.; Mano, J.F. Chitosan/bioactive glass nanoparticles composites for biomedical applications. Biomed. Mater. 2012, 7, 054104. [Google Scholar] [CrossRef]
- Maji, K.; Dasgupta, S.; Pramanik, K.; Bissoyi, A. Preparation and Evaluation of Gelatin-Chitosan-Nanobioglass 3D Porous Scaffold for Bone Tissue Engineering. Int. J. Polym. Mater. 2016, 2016, 9825659. [Google Scholar] [CrossRef] [PubMed]
- Peter, M.; Binulal, N.S.; Soumya, S.; Nair, S.V.; Furuike, T.; Tamura, H.; Jayakumar, R. Nanocomposite scaffolds of bioactive glass ceramic nanoparticles disseminated chitosan matrix for tissue engineering applications. Carbohydr. Polym. 2010, 79, 284–289. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Doube, M.; Kłosowski, M.M.; Arganda-Carreras, I.; Cordeliéres, F.; Dougherty, R.P.; Jackson, J.; Schmid, B.; Hutchinson, J.R.; Shefelbine, S.J. BoneJ: Free and extensible bone image analysis in ImageJ. Bone 2010, 47, 1076–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orava, E.; Korventausta, J.; Rosenberg, M.; Jokinen, M.; Rosling, A. In vitro degradation of porous poly(dl-lactide-co-glycolide) (PLGA)/bioactive glass composite foams with a polar structure. Polym. Degrad. Stabil. 2007, 92, 14–23. [Google Scholar] [CrossRef]
- Kumar, S.; Koh, J. Physiochemical, Optical and Biological Activity of Chitosan-Chromone Derivative for Biomedical Applications. Int. J. Mol. Sci. 2012, 13, 6102–6116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, Y.G.; Lee, H.J.; Shin, S.S.; Choi, U.S. Dipolar-molecule complexed chitosan carboxylate, phosphate, and sulphate dispersed electrorheological suspensions. Soft Matter 2012, 8, 6273–6279. [Google Scholar] [CrossRef]
- De Britto, D.; Campana-Filho, S.P. Kinetics of the thermal degradation of chitosan. Thermochim. Acta 2007, 465, 73–82. [Google Scholar] [CrossRef]
- Georgieva, V.; Zvezdova, D.; Vlaev, L. Non-isothermal kinetics of thermal degradation of chitosan. Chem. Cent. J. 2012, 6, 81. [Google Scholar] [CrossRef]
- Zawadzki, J.; Kaczmarek, H. Thermal treatment of chitosan in various conditions. Carbohydr. Polym. 2010, 80, 395–401. [Google Scholar] [CrossRef]
- Caridade, S.G.; Merino, E.G.; Alves, N.M.; Bermudez, V.D.Z.; Boccaccini, A.R.; Mano, J.F. Chitosan membranes containing micro or nanO–Size bioactive glass particles: Evolution of biomineralization followed by in situ dynamic mechanical analysis. J. Mech. Behav. Biomed. Mater. 2013, 20, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Bui, X.; Oudadesse, H.; Le Gal, Y.; Mostafa, A.; Cathelineau, G. Microspheres of Chitosan-Bioactive Glass for Application in Orthopedic Surgery. In vitro experiment. Recent Res. Modern Med. 2011, 359–367. [Google Scholar]
- Yao, Q.; Nooeaid, P.; Detsch, R.; Roether, J.; Dong, Y.; Goudouri, O.; Schubert, D.; Boccaccini, A. Bioglass/chitosan–polycaprolactone bilayered composite scaffolds intended for osteochondral tissue engineering. J. Biomed. Mater. Res. A 2014, 102, 4510–4518. [Google Scholar] [CrossRef] [PubMed]
- Efimov, A.M.; Pogareva, V.G. IR absorption spectra of vitreous silica and silicate glasses: The nature of bands in the 1300 to 5000 cm−1 region. Chem. Geol. 2006, 229, 198–217. [Google Scholar] [CrossRef]
- Cerruti, M.; Greenspan, D.; Powers, K. Effect of pH and ionic strength on the reactivity of Bioglass® 45S5. Biomaterials 2005, 26, 1665–1674. [Google Scholar] [CrossRef] [PubMed]
- Tainio, J.; Paakinaho, K.; Ahola, N.; Hannula, M.; Hyttinen, J.; Kellomäki, M.; Massera, J. In vitro degradation of borosilicate bioactive glass and poly(l-lactide-co-ε-caprolactone) composite scaffolds. Materials 2017, 10, 1274. [Google Scholar] [CrossRef] [PubMed]
- Madihally, S.V.; Matthew, H.W.T. Porous chitosan scaffolds for tissue engineering. Biomaterials 1999, 20, 1133–1142. [Google Scholar] [CrossRef]
- Dash, M.; Chiellini, F.; Ottenbrite, R.M.; Chiellini, E. Chitosan—A versatile semi-synthetic polymer in biomedical applications. Prog. Polym. Sci. 2011, 36, 981–1014. [Google Scholar] [CrossRef]
- Teng, S.H.; Lee, E.J.; Yoon, B.H.; Shin, D.S.; Kim, H.E.; Oh, J.S. Chitosan/nanohydroxyapatite composite membranes via dynamic filtration for guided bone regeneration. J. Biomed. Mater. Res. A 2009, 88, 569–580. [Google Scholar] [CrossRef]
- Liverani, C.; Mercatali, L.; Cristofolini, L.; Giordano, E.; Minardi, S.; Della Porta, G.; De Vita, A.; Miserocchi, G.; Spadazzi, C.; Tasciotti, E.; et al. Investigating the Mechanobiology of Cancer Cell–ECM Interaction Through Collagen-Based 3D Scaffolds. Cell. Mol. Bioeng. 2017, 10, 223–234. [Google Scholar] [CrossRef]
- Szymanska, E.; Winnicka, K. Stability of Chitosan—A Challenge for Pharmaceutical and Biomedical Applications. Mar. Drugs 2015, 13, 1819–1846. [Google Scholar] [CrossRef] [PubMed]
- Vergnol, G.; Ginsac, N.; Rivory, P.; Meille, S.; Chenal, J.M.; Balvay, S.; Chevalier, J.; Hartmann, D.J. In vitro and in vivo evaluation of a polylactic acid-bioactive glass composite for bone fixation devices. J. Biomed. Mater. Res. B 2016, 104, 180–191. [Google Scholar] [CrossRef] [PubMed]
- Hench, L.L. The Story of Bioglass. J. Mater. Sci. Mater. Med. 2006, 17, 967–978. [Google Scholar] [CrossRef] [PubMed]
- Quasim, S.B.; Husain, S.; Huang, Y.; Pogorielov, M.; Dreineka, V.; Lymdin, M.; Rawlinson, A.; Ur Rehman, I. In-vitro and in-vivo degradation studies of freeze gelated porous chitosan composite scaffolds for tissue engineering applications. Polym. Degrad. Stab. 2017, 136, 31–38. [Google Scholar] [CrossRef]
- Massera, J.; Hupa, L.; Hupa, M. Influence of partial substitution of CaO with MgO on the thermal properties and in vitro reactivity of the bioactive glass S53P4. J. Non-Cryst. Solids 2012, 358, 2701–2707. [Google Scholar] [CrossRef]
- Massera, J.; Hupa, L. Influence of SrO substitution for CaO on the properties of bioactive glass S53P4. J. Mater. Sci. Mater. Med. 2014, 25, 657–668. [Google Scholar] [CrossRef] [PubMed]
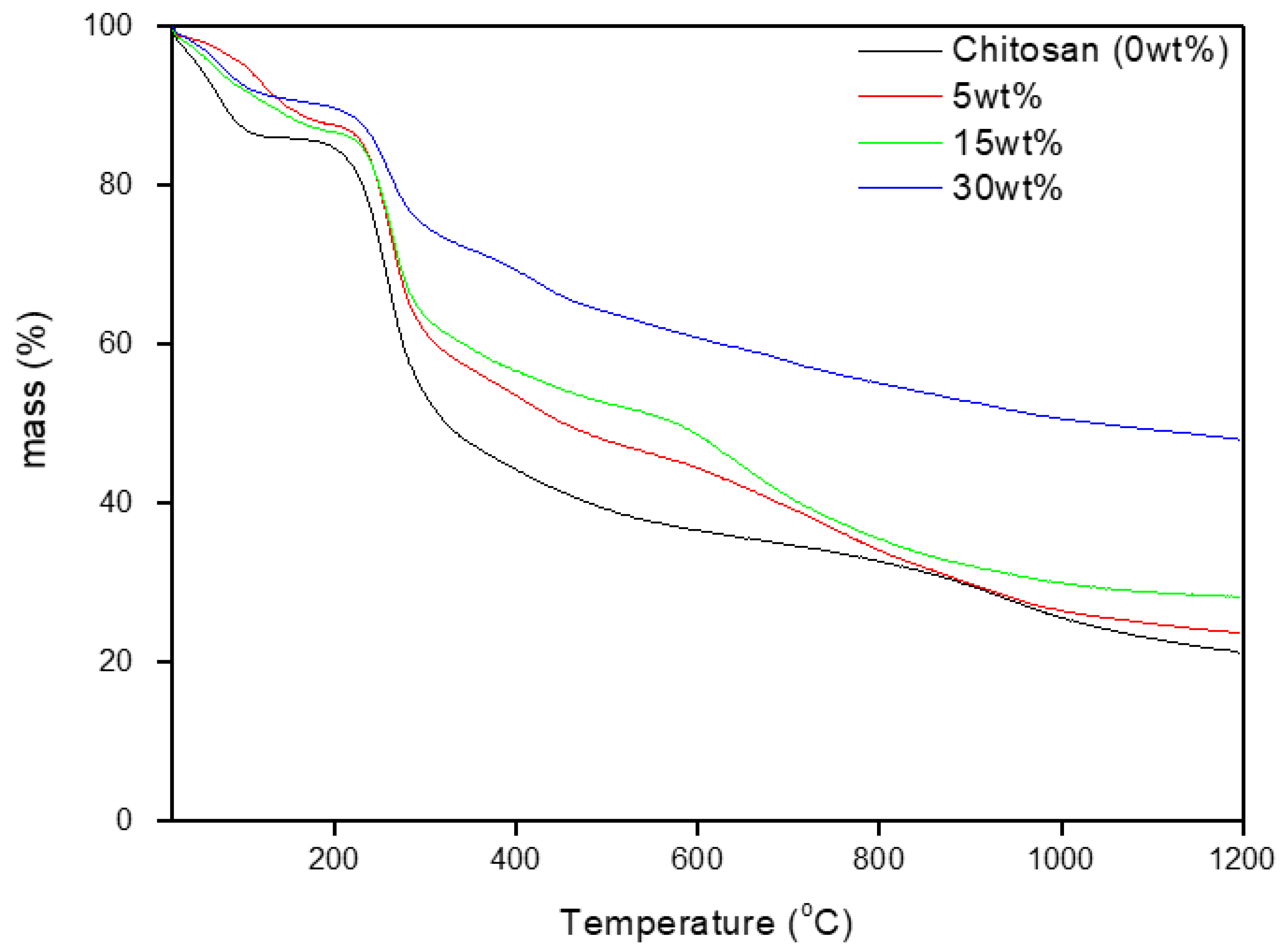






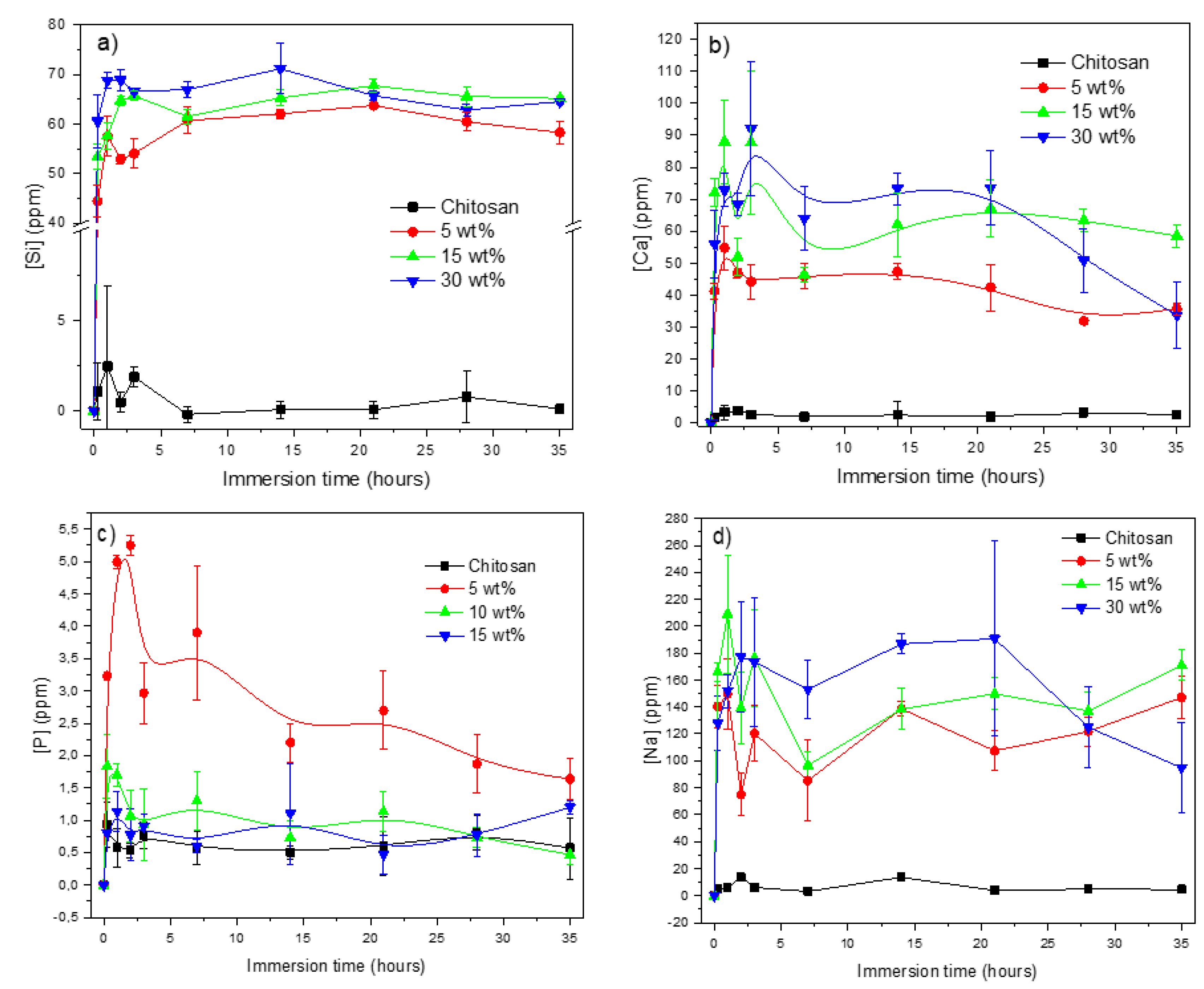


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Wavenumber (cm−1) | Identification of Absorption Bond |
|---|---|
| 3496–3440 | OH group |
| 1377 | O–H and N–H axial stretching, NH group-stretching vibration |
| 3345, 3370 | O–H band that overlaps N–H band, –NH2 and –OH groups |
| 2910, 2913, 2859 | C–H stretching |
| 1300, 2926, 2880, 665 | C–H bending vibration |
| 1421, 1322 | OH, CH vibration in the ring |
| 2877,1421, 1322, 1249 | CH2 in pyranose ring |
| 1422 | Vibration of C–OH group |
| 1724, 1580, 1395 | C=O, Stretching vibration C=O |
| 1646, 1642 | C=O in amide I group |
| 1653, 1657 | Amide I |
| 1650 | Stretching vibration of amide I |
| 1381 | CH3 in amide group |
| 1096, 1030 | C–O group in amide group |
| 1580 | Amide II |
| 1562, 1552 | Amide II band due to N–H bending (Amide II) |
| 1320 | Amide III |
| 1320, 1590 | Amino characteristic peaks |
| 1593 | NH2 bending vibration in amino group |
| 1417 | Coupling C–N axial stretching |
| 1249, 1075, 1033 | C–O group, C–O vibration stretching |
| 1152, 1153 | –C–O–C– bridge |
| 1153–897 | Polysaccharide, C–O and C–O–C |
| 1085 | C–O–C bond |
| 1065, 1150, 1024 | C–O–C symmetric, C–O–C asymmetric vibration |
| 1380 | Stretching vibration of methyl group |
| 893, 1153 | Saccharide structure |
| Sample Name | Measured Inorganic Mass (wt%) | Porosity | Tensile Strength at 50% Deformation (MPa) | Young’s Modulus (MPa) |
|---|---|---|---|---|
| Chitosan (0 wt%) | 88% | 0.12 ± 0.06 | 0.23 ± 0.05 | |
| 5 wt% | 5 ± 3 | 75% | 0.4 ± 0.1 | 0.78 ± 0.01 |
| 15 wt% | 11 ± 5 | 66% | 0.29 ± 0.05 | 0.44 ± 0.09 |
| 30 wt% | 36 ± 12 | 75% | 0.2 ± 0.1 | 0.25 ± 0.16 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faqhiri, H.; Hannula, M.; Kellomäki, M.; Calejo, M.T.; Massera, J. Effect of Melt-Derived Bioactive Glass Particles on the Properties of Chitosan Scaffolds. J. Funct. Biomater. 2019, 10, 38. https://doi.org/10.3390/jfb10030038
Faqhiri H, Hannula M, Kellomäki M, Calejo MT, Massera J. Effect of Melt-Derived Bioactive Glass Particles on the Properties of Chitosan Scaffolds. Journal of Functional Biomaterials. 2019; 10(3):38. https://doi.org/10.3390/jfb10030038
Chicago/Turabian StyleFaqhiri, Hamasa, Markus Hannula, Minna Kellomäki, Maria Teresa Calejo, and Jonathan Massera. 2019. "Effect of Melt-Derived Bioactive Glass Particles on the Properties of Chitosan Scaffolds" Journal of Functional Biomaterials 10, no. 3: 38. https://doi.org/10.3390/jfb10030038
APA StyleFaqhiri, H., Hannula, M., Kellomäki, M., Calejo, M. T., & Massera, J. (2019). Effect of Melt-Derived Bioactive Glass Particles on the Properties of Chitosan Scaffolds. Journal of Functional Biomaterials, 10(3), 38. https://doi.org/10.3390/jfb10030038