1. Introduction
Establishment of cellular morphology is one of the first processes in neurons that has been described to be affected by topography through contact guidance [
1]. Neurons establish cell morphology in vitro after attachment to a substrate, which initiates the process of neurite guidance and extension [
2]. During differentiation, neural progenitor cells (NPCs) form neurons, astrocytes, and oligodendrocytes [
3] with distinct morphologies. Multiple biochemical pathways come into play during the transition of a cell from a progenitor state to a differentiated state. At the mechanical level, cues, such as substrate topography, are important determinants in the differentiation of multipotent cells into either neurons or astrocytes [
4]. However, it has been a challenge to conduct a quantitative morphometric analysis of the progenitor response to varying topographical cues.
Firstly, the tracing of neurons or astrocytes on unpatterned substrates needed to shift from a manual process to a largely automated process in order to have a statistical understanding of morphology. This has been achieved with the creation of high-throughput screening tools, specifically for neurons, such as NeuronJ [
5], Metamorph [
6], algorithms developed in MATLAB [
7,
8], etc. Secondly, there have been few systematic analyses of topographical influences on differentiation conducted on an array of topographies [
9,
10,
11]. Most studies vary one topographical feature to assess the possible mechanism underlying observable cell shape changes [
4]. However, none of these studies have directly compared the response of progenitors from different origins when exposed to an array of topographical cues. Intrinsic differences exist in the neurogenecity of neural progenitors of different origins depending on the species of origin, the region of the brain, and the progenitor age at which the NPCs are isolated [
12,
13]. Thus, we hypothesized that the morphological changes of NPCs differentiated on topographies would be influenced by their origin.
In the present study, we aim to understand the complex relationship between cell morphology and differentiation in cortical and hippocampal murine NPCs (mNPCs) with the use of an array of 14 topographies. The following four morphological parameters were measured: the length of the longest extension, the number of extensions, the number of branches, and the soma area of both neurons and astrocytes derived from mNPCs. These parameters are known to be hallmarks of the impact of biochemical and mechanical perturbations on neuronal cells. For example, genetic mutations introduced into dopaminergic neurons have been shown to cause a reduction in neurite length and branching [
14]. Rett Syndrome brains (
MECP2 gene knockouts) are known to show decreased neuronal size and synapse elimination in vitro [
15]. In addition, exposure to neurite outgrowth inhibiting chemicals at various concentrations, has been reported to cause drastic increases and decreases in neurite extension in high-content screening assays [
16]. Finally, topography has been shown to be one of the biggest modulators of cellular morphology, especially in size ranges matching specific organelles of interest [
4].
The relation of topography with differentiation efficiency has been vastly explored. Studies with neural progenitors have been conducted on surfaces with features such as gratings, pillars, wells (reviewed in [
17]), or even recently, hierarchical structures [
11,
18]. Thus, how differentiation and morphology correlate with each other remains an important topic of discussion in numerous studies of neuronal differentiation on topographical substrates [
4]. This study systematically correlates the morphological changes caused by topographical cues with the differentiation efficiency of neural progenitors from different origins.
3. Discussion
The species of origin, the developmental stage of the progenitor (embryonic versus post-natal or adult), and the location from which progenitors are isolated from the nervous system have been shown in the literature to have a large impact in the outcome of neuronal and astrocyte differentiation. Neural progenitors have been shown to have varying capabilities for proliferation, differentiation, and morphology across different mammals [
20,
21,
22]. Specifically, neural stem cells have been systematically studied to be affected by developmental stage, brain region, and time in culture for their proliferation and differentiation. It has been reported that there is a developmental stage dependent decrease in the neurogenecity of cells as the population becomes more glial. Although not quantified, region-dependent differences in neuronal and astroglial morphology, independent from the donor tissue age, were shown to have region dependent intrinsic features of neuronal and glial progenitors [
12]. Seaberg et al. observed that when postnatal neural precursors from different locations of the brain (namely, the cerebral cortex, the lateral striatum, and optic nerve) were tested with varying days of isolation from the animal, the neurogenecity was shown to be different, depending on the location and age of the progenitors [
13]. Moreover, the neurogenic capacity of the cells was shown to be an intrinsic property, despite the use of different medium compositions [
13]. These studies strongly suggest that there are intrinsic differences in differentiation potential between the different types of NPCs.
Similarly, astrocytes have been reported to show heterogeneity that closely reflects the phenotypic regions of the brain defined by neurons [
23]. Astrocyte morphology is subject to factors such as their location in the CNS and the number of days that the cells have undergone morphological maturation [
22]. Postnatal hippocampal astrocytes have been categorized as protoplasmic astrocytes, which display ‘spongiform’ morphology. In addition, plasmodial astrocytes in the hippocampus possess a spongiform morphology [
24]. Over the course of morphological maturation, astrocytes undergo specific changes; their filopodial processes elongate and larger processes extend from their bodies. Beyond the first three weeks of maturation, astrocytes undergo a change in the heterogeneity of morphology itself, which drastically reduces as the cells mature further. The process is completed after the stabilization of competitive overlapping cell domains [
25]. Astrocytes developing in a large and diverse region of the brain, such as the cortex, display a much more heterogeneous combination of morphologically defined astrocyte categories [
26]. This could explain the spongiform versus astral morphology that was observed in the hippocampal versus the cortical mNPC-derived astrocytes on the topographical substrates in the present study. The morphological differences between cortical and hippocampal mNPCs derived neurons and astrocytes when exposed to the same topographies are, thus, inherent, as shown in the literature.
Notably, the differences in morphology could also be contributed by other factors. In the current study, hippocampal NPCs were isolated from the hippocampi of five day old mice. On the contrary, cortical NPCs were isolated from the whole cortices of 13-day old mouse embryos at the peak of their neurogenetic phase and were grown in the ideal media required for differentiation. While hippocampal cells require the use of two different media for the generation of neurons, cortical cells were differentiated in a single differentiation medium (
Figure S2). In addition, cortical cells required a period of five days for the expression of the TUJ1 marker, whereas neuronal differentiation of hippocampal cells required 14 days. Thus, the ideal cell culture conditions were chosen for each cell type. As a reference for comparison, morphological measurements on topographical substrates were accompanied with measurements of cells on an unpatterned control. The control allowed the observation of the intrinsic differences between the two cell types which were isolated from different regions of the brain, at different developmental stages, and cultured with different media. Together, the differences in the age of the mice, the region of the brain, and the culture conditions required could also result in the differences observed in the differentiation efficiency.
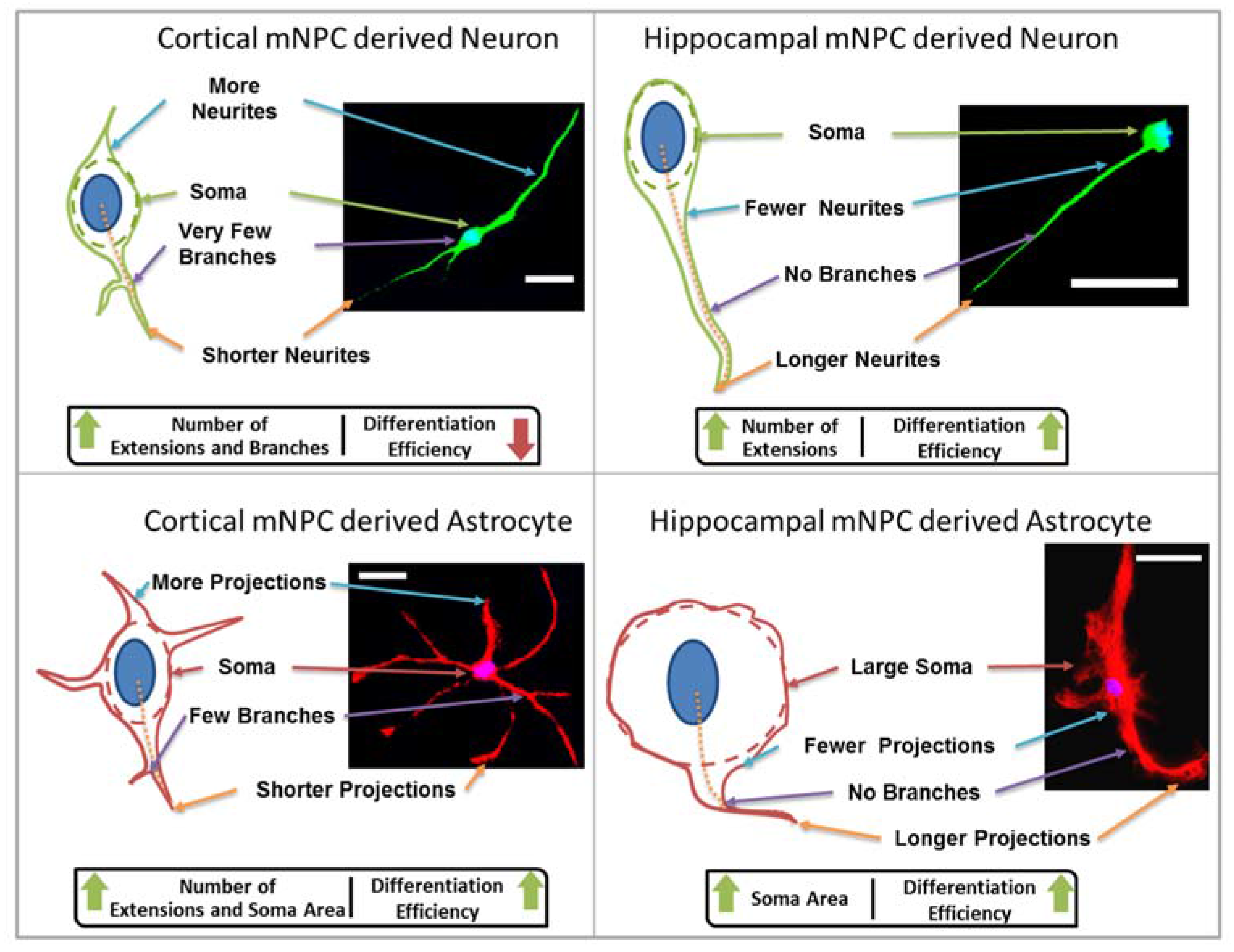
In this study, topography was used as a tool to influence NPC differentiation and establishment of cell morphology. Four morphological parameters were studied in neurons and astrocytes differentiated from cortical and hippocampal mNPCs.
Firstly, the length of the longest extension on each cell and the number of branches on each extension were measured. A change in neurite length has been shown to be associated with neural diseases such as Rett syndrome [
15] and Parkinson’s disease [
14]. In addition, neurite length has been shown to be influenced by topography. In particular, differentiating hippocampal mNPCs have been reported to show a size dependent relation between neurite extension and grating depth. Neurites were observed to extend across shallow gratings where it was possible for filopodia to adhere to the bottom of the groove. In the case where grating depth was comparable to grating width, the extending neurite was no longer able to sense the presence of the substrate and did not continue extension. This led to the alignment of neurites on 1:1 aspect ratio microgrooves as reported previously [
18]. A similar observation was made in the current study (
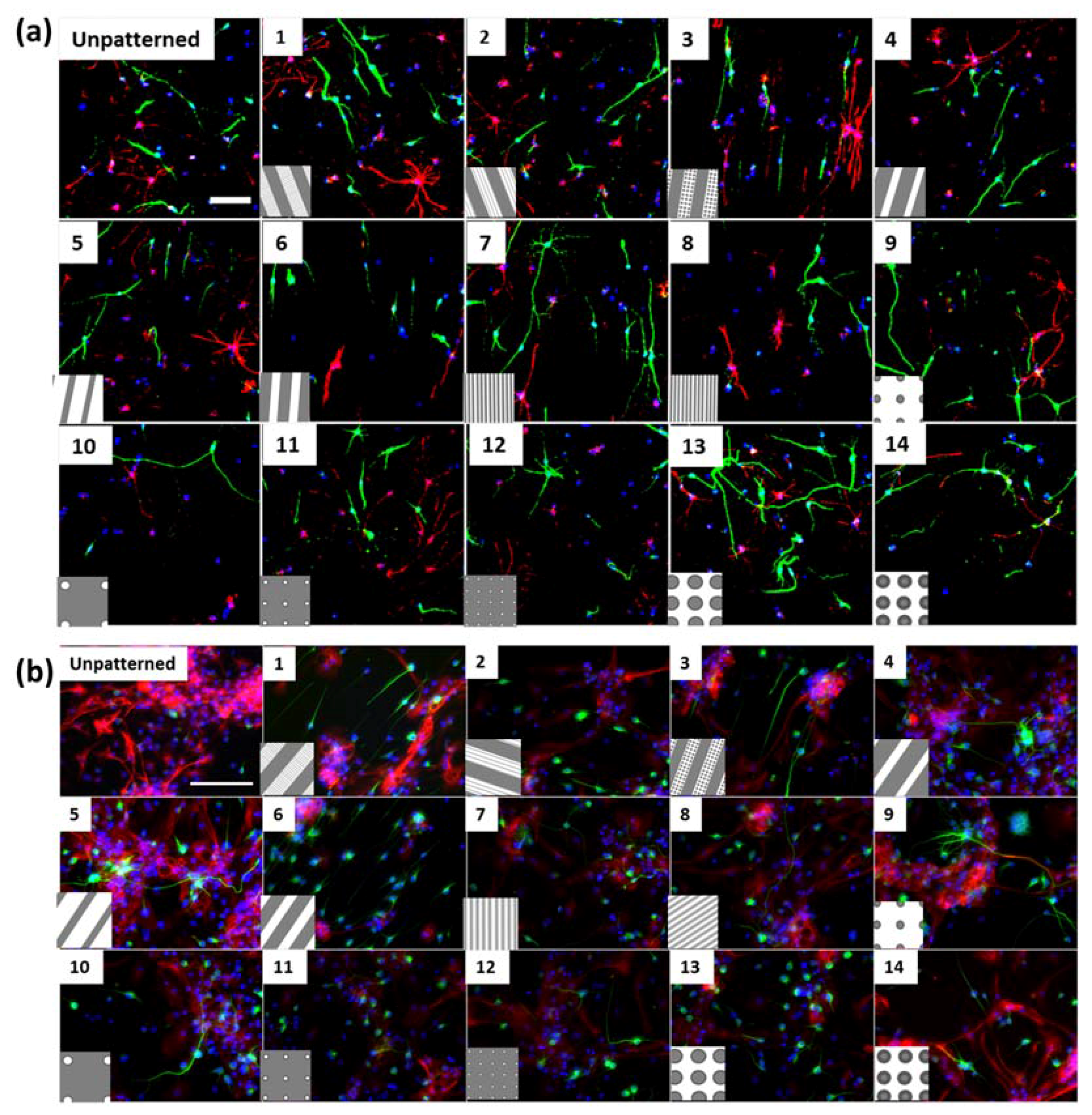
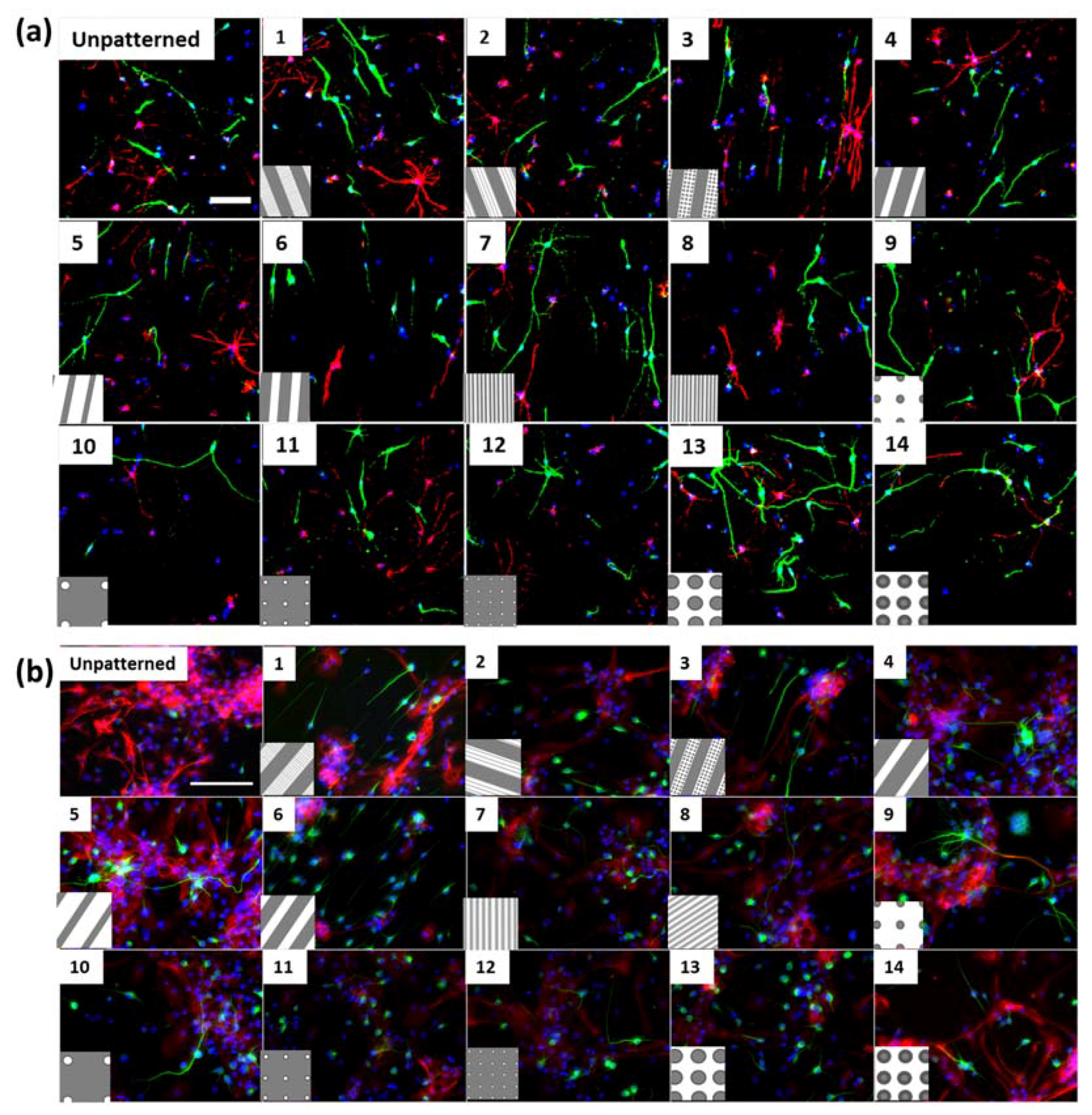
Figure S1h), where neurites were observed to extend into micron scale gratings. In addition, differentiated cortical NPCs displayed shorter extensions in comparison to hippocampal cells (
Figure 1). Hippocampal neurons specifically showed an increase in neurite length on pillars. The observation of longer neurites on isotropic topography is in contrast with the vastly reported elongation effects of anisotropic structures such as grating topographies on neuronal cells [
11]. However, previous studies have also reported that rat hippocampal neurons on pillar topographies have increasing extension lengths with increasing width. It was found that extension lengths were the longest on pillars with a 2 μm width [
27]. In addition, E18.5 mouse hippocampal neurons have been shown to have faster neurite elongation on nanopillars with specifically 500 nm spacing. Neurites were reported to extend on top of the pillars, which provide optimally spaced points for substrate adhesion [
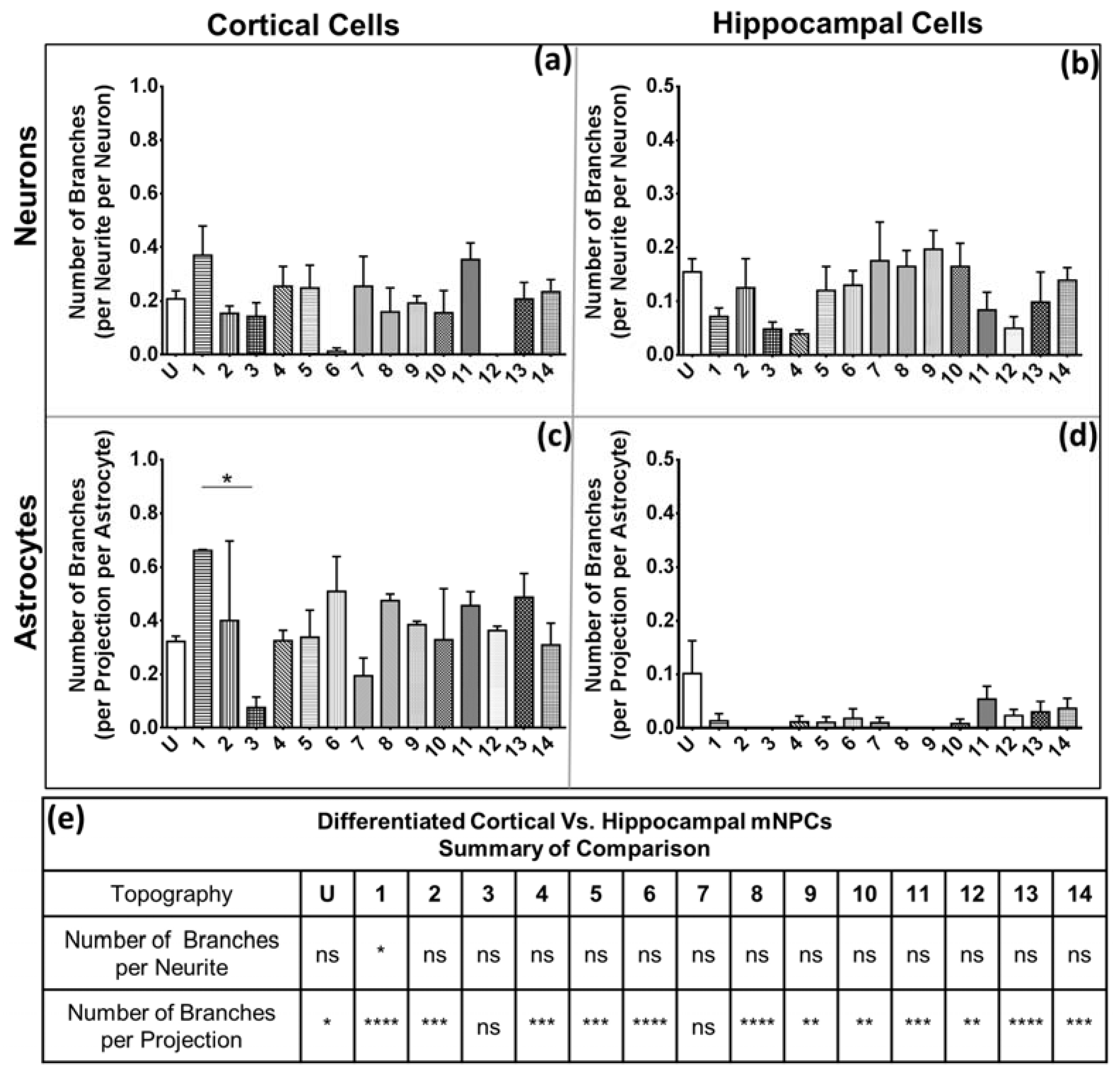
28]. Hippocampal cells also possessed fewer branches on both neurons and astrocytes. The branching is independent of the expression of projections, as was observed in the quantification of the average number of projections and branches per projection (
Figure 4 and
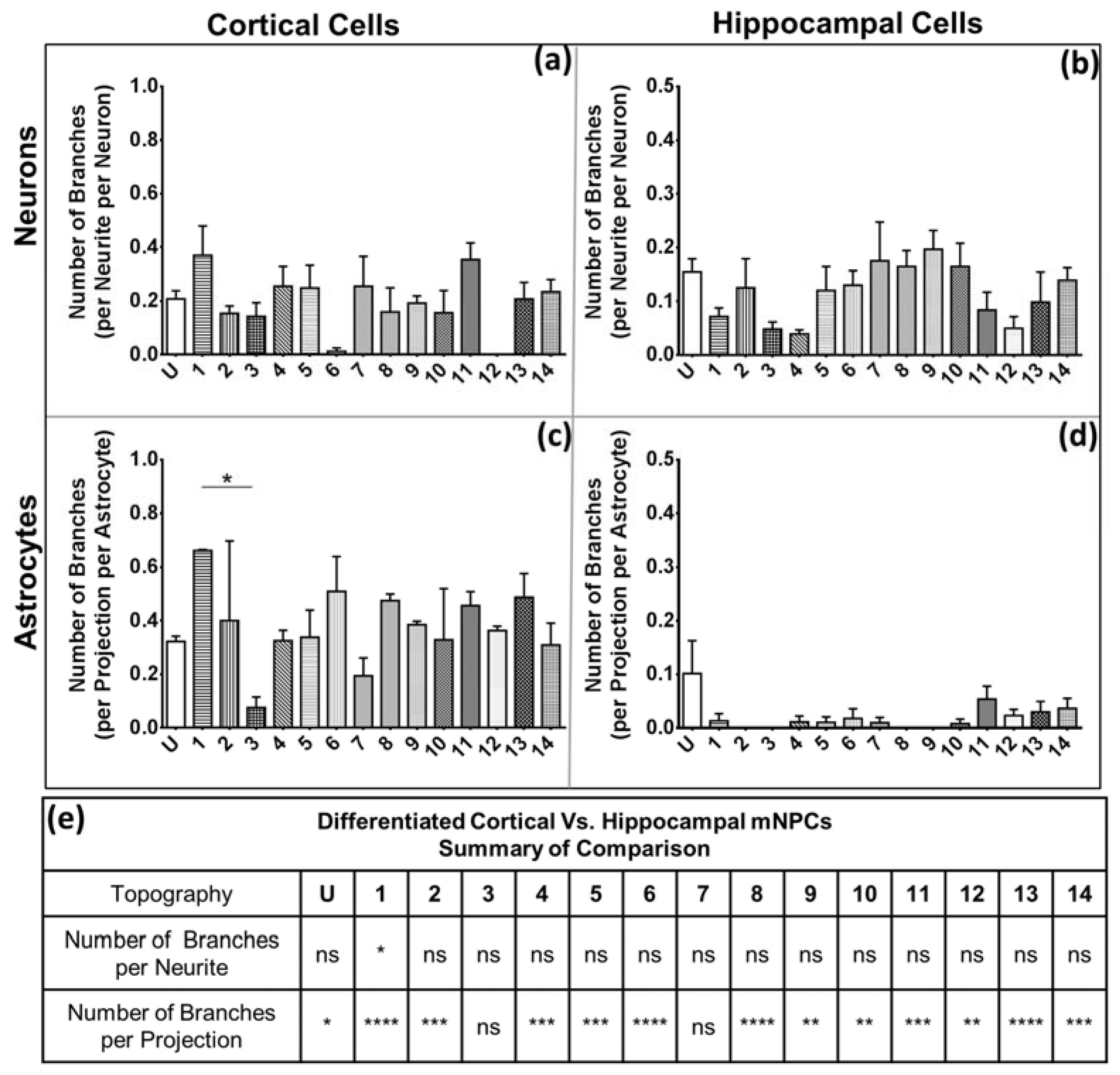
Figure 5). Thus, hippocampal mNPC-derived astrocytes not only expressed few projections from the cell center, but also a very small degree of branching on the cells that did possess projections. These properties seem to be unique to hippocampal cells.
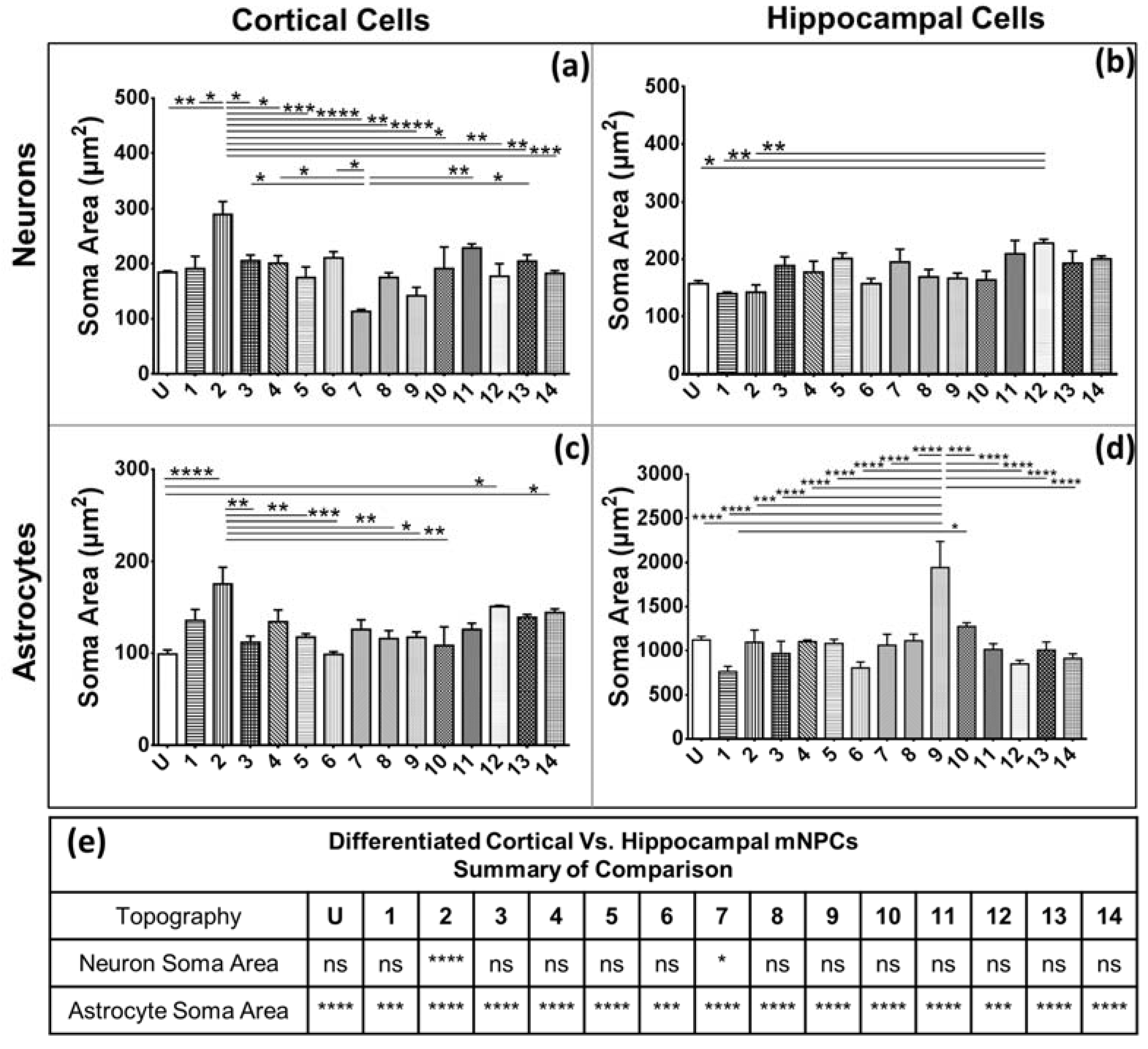
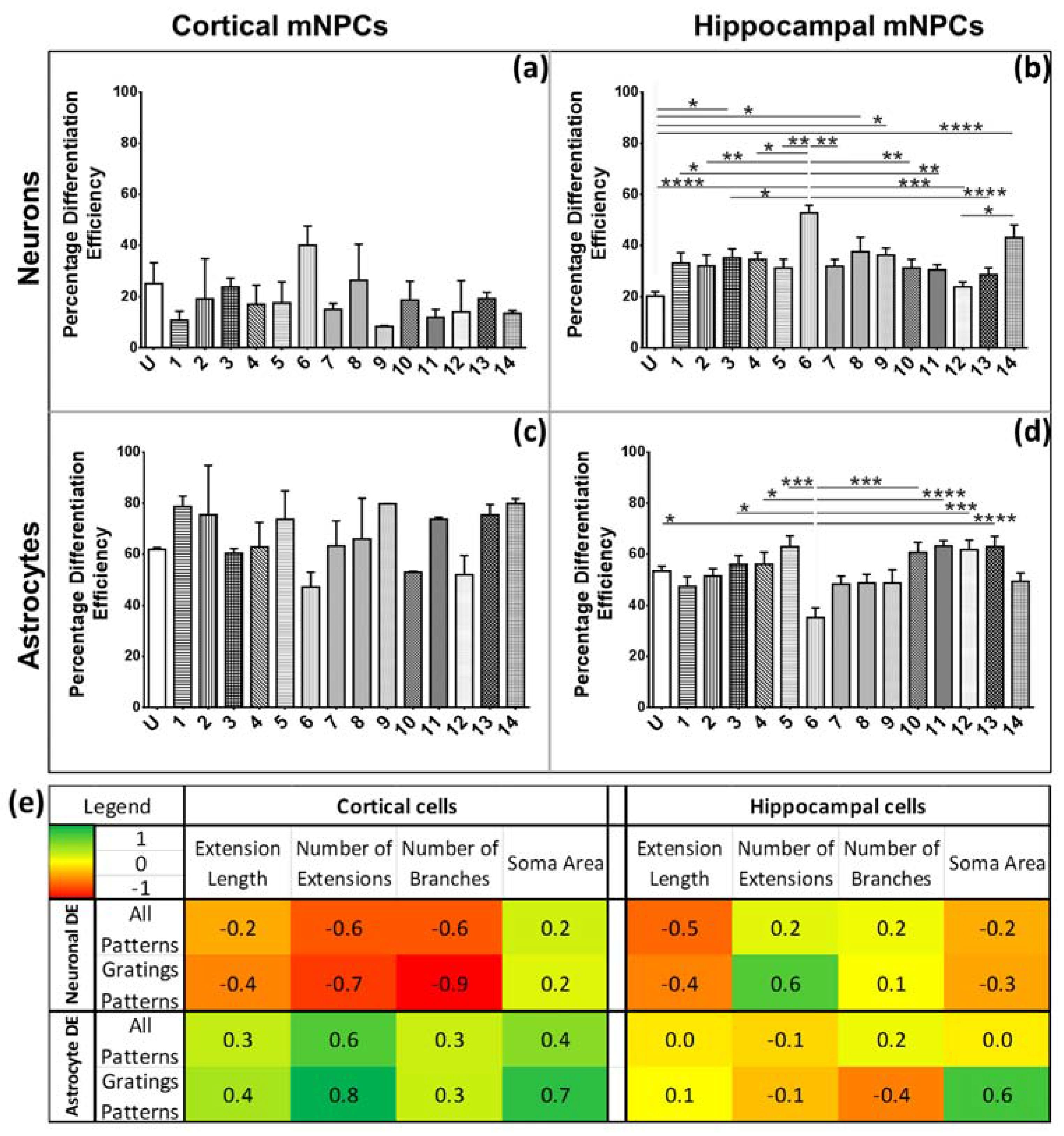
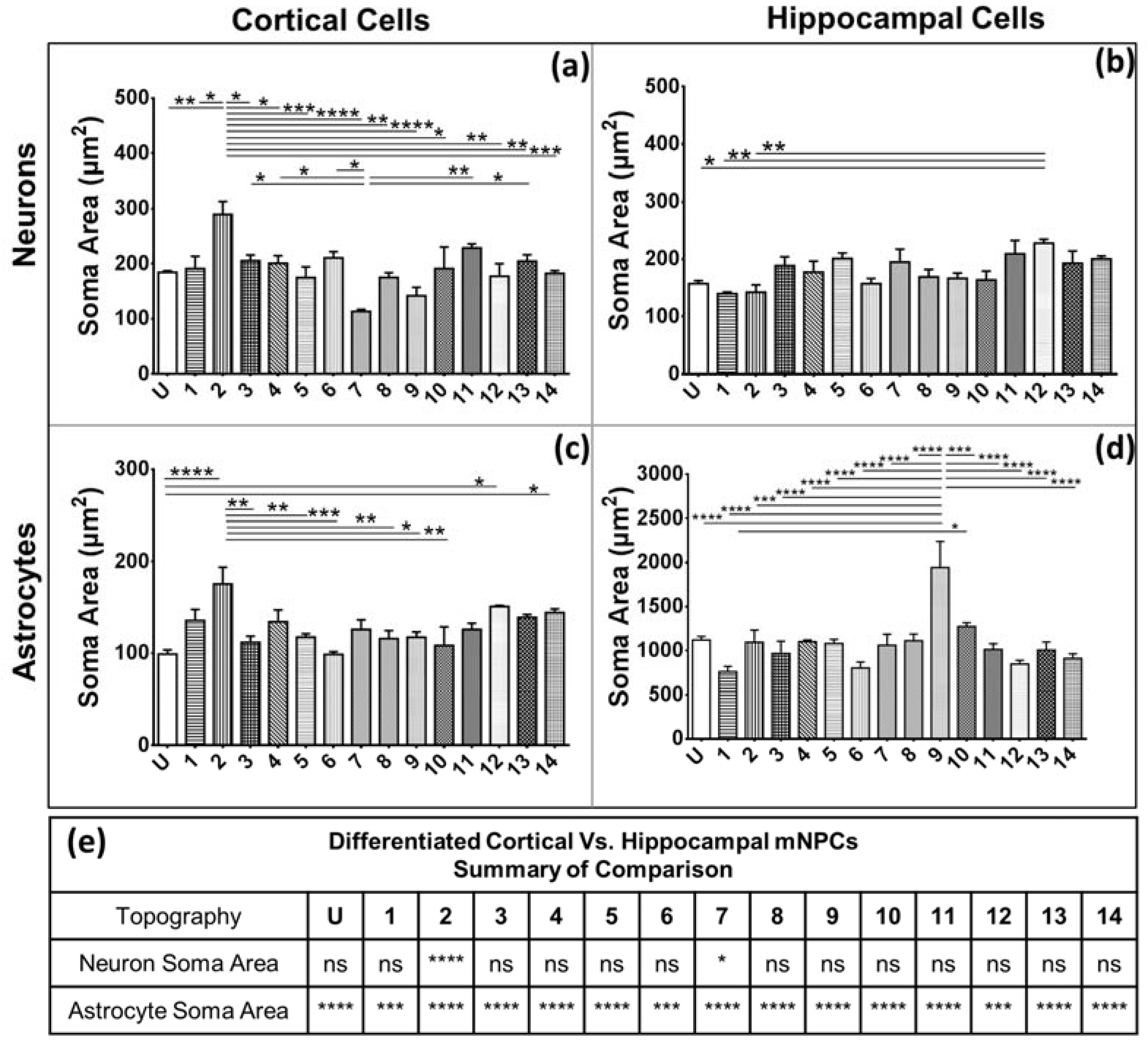
Secondly, soma areas of both cell types were measured on each of the topographies. Here, hippocampal mNPC-derived astrocytes were observed to possess much larger soma areas than other hippocampal NPC derived neurons, as well as neurons and astrocytes derived from cortical mNPCs. In addition, the area of these cells did not show much variation between topographies. These morphological attributes were also observed in immunofluorescence images (
Figure 1b) and may be an intrinsic trait of the cells as described earlier. The increase of soma size in association with topography would be an interesting tool to observe cells in disease states and developmental states. Soma sizes have been shown to drastically increase during human adolescence [
29]. Soma sizes also tend to be altered in diseases such as Costello Syndrome [
30], Rett Syndrome [
19], and depression [
31]. Although nanogrooved substrates have been reported to reduce soma sizes during the production of dopaminergic neurons [
32], the effect of topography has been largely unexplored in disease cells. It, would be interesting to observe patient iPSC derived neurons and astrocytes on topography because they have been shown to have significantly different soma sizes.
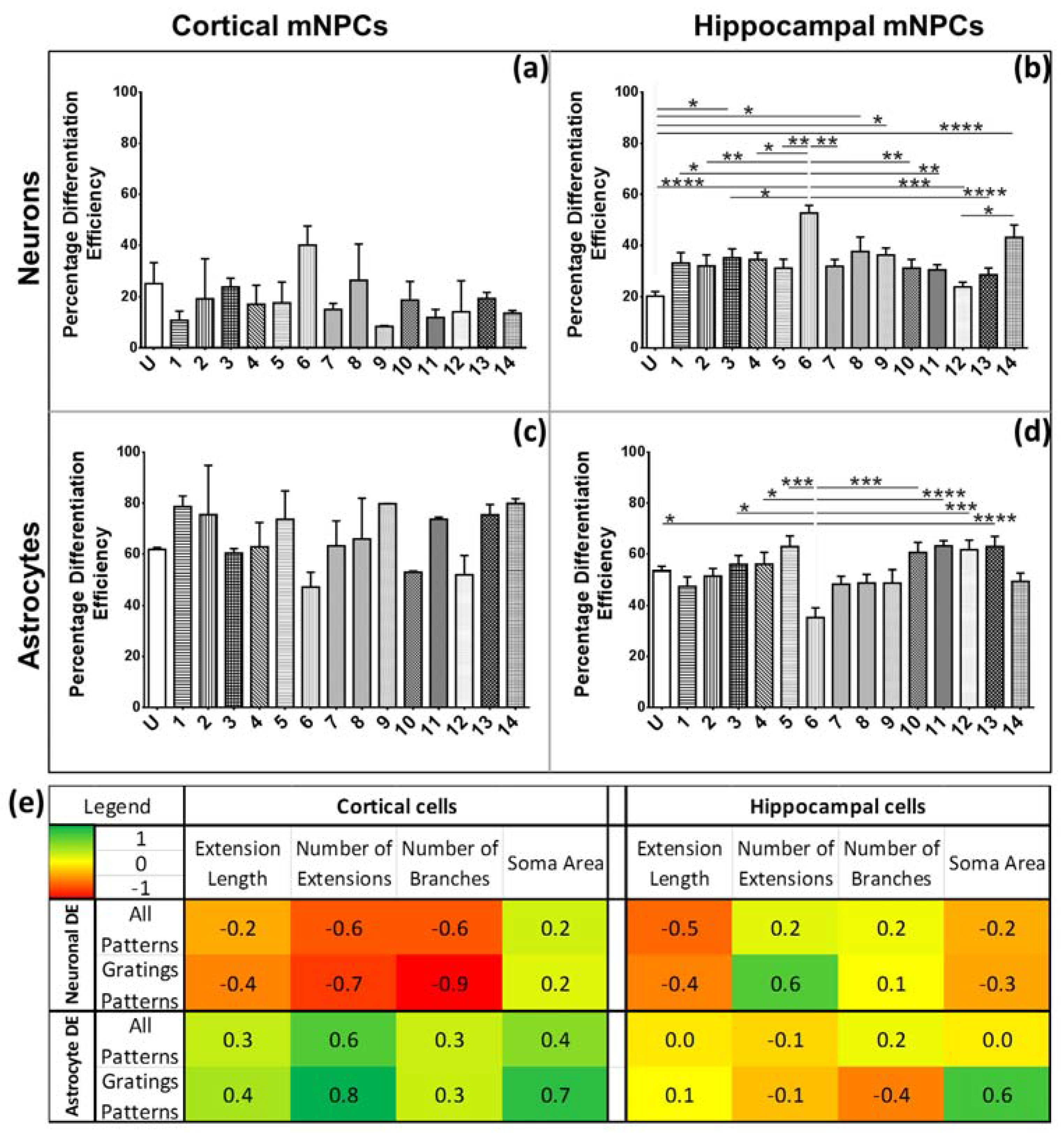
The characteristic morphological traits for cortical and hippocampal mNPC-derived neurons and astrocytes have been identified in the current study. These traits were expressed by cells regardless of topographical influence on cell morphology and have been summarized in
Figure 8. There is a range of morphological changes, as well as various differentiation efficiencies caused by the influence of an array of different topographies. This allowed for the study of the correlation between cell shape and cell fate. Different trends were observed for the two types of differentiated mNPCs. For example, while cortical neural progenitor derived neurons showed a negative correlation between differentiation efficiency and the number of extensions from the cell center, an opposite relation was observed for hippocampal neural progenitor derived neurons. However, astrocyte soma areas showed similar correlations with differentiation in both types of NPCs. Notably, gratings topography was shown to enhance the correlation between cell shape and differentiation in all cell types. The correlations have been summarized in
Figure 8. Thus, gratings that produce morphological effects, such as an increase in the number of extensions, were associated with an increased or decreased production of neurons, depending on the type of neural progenitor. The enhancement of neuronal differentiation by gratings substrates has been extensively reported [
4,
10,
18,
33]. Our findings suggest that this enhancement could be attributed to the morphological effects of gratings topography on neural progenitor cells. Therefore, gratings may promote the morphological characteristics necessary for enhanced production of neurons. However, the morphological characteristics associated with increased neuronal differentiation are largely dependent on the source of neural progenitor cells.
The differentiation of neural progenitors on topography has been reported in numerous studies. Similarly, neuronal morphology has also been established as a field. The difference between neural cells from different regions of the brain has been established in literature. The current study quantitatively compares the differentiation and morphology of neural progenitors from the hippocampus and cortex on an array of topographies. Moreover, a correlation between cell shape and differentiation was established. However, further studies are necessary to determine a causative relation between neuron production and cell shape. Grating substrates prove to be the ideal candidates for these studies.
4. Materials and Methods
4.1. Fabrication and Replication of PDMS Substrates
The multi-architecture (MARC) chip is an array of multiple nano- and micro-topographical patterns. To assemble the MARC chip, the fabrication of individual patterns of 2 mm × 2 mm in size (large enough to allow for statistical analysis of cells) via nano-imprinting lithography was completed. Subsequently, these individual patterns were arranged onto a single silicon chip pre-coated with poly-dimethylsiloxane (PDMS, Sylgard 184 polymer, Dow Corning, Auburn, MI, USA) as a bonding material. Based on how the individual patterns are cut, they may either be circular [
10] or square-shaped. The versatility of the MARC chip lies in the choice of patterns one can arrange onto each chip, making it possible to combine different or repeating patterns. The MARC chip can also be customized to be of a different shape and size to accommodate different applications, such as microfluidic systems [
25]. Replicas of the MARC chip were generated by soft lithography using PDMS and replication was verified by scanning electron microscopy (JEOL JSM 6010 LV, JEOL, Tokyo, Japan) by sputter coating the samples with platinum at 30 mA for 30 s and observing them at various magnifications at high vacuum (
Figure S1a–h). Silicon molds of 44-pattern and 18-pattern MARC chips were used for testing multiple topographies at once, along with an unpatterned substrate as a control.
Table 1 shows the topographical patterns that were studied.
For the generation of replicas, PDMS was mixed in a 10:1 ratio with the curing agent and placed under vacuum for removal of air bubbles. The mixture was then poured onto the MARC chip molds. After additional removal of air under vacuum for 30 min, the PDMS was cured at 70 °C for 2 h. Replicas were generated upon demolding. The PDMS replicas were cut according to the wells of the culture plates. To remove hydrophobicity, replicas were treated with oxygen-plasma (Harrick Plasma, Ithaca, NY, USA) for 1 min at 30 W. For sterilization, samples were washed with 70% ethanol and placed under ultraviolet light for 20 min. To allow attachment of laminin, replicas were coated overnight with poly-L-ornithine solution (33 μg/mL) in sterile deionized water (Sigma-Aldrich, St. Louis, MO, USA). After two washes with sterile deionized water, they were coated overnight with 20 μg/mL mouse laminin in Dulbecco’s modified Eagle medium (DMEM)/nutrient mixture F12 (Biological Industries, Cromwell, CT, USA) in a 1:1 ratio (1:1 DMEM/F12) (Life Technologies, Waltham, MA, USA) to carry out differentiation of mNPCs.
4.2. Culture of Murine Neural Progenitor Cells (mNPCs)
Hippocampal neural progenitor cells were isolated from the hippocampus of five day old mice [
10]. Cortical NPCs were isolated from the whole cortex of 13 day old mouse embryos (E13). Each NPC was established as a monolayer culture, as previously described for hippocampal NPCs [
34] Both mNPCs were a gift from Dr. Eyleen Goh’s group (Duke-NUS Graduate Medical School, Singapore). The cells were cultured in neural progenitor expansion medium, which contains 1:1 DMEM/F12Dulbecco’s modified Eagle medium (DMEM)/nutrient mixture F12 (Biological Industries, Cromwell, CT, USA) in a 1:1 ratio supplemented with 1× N2 supplement (Life Technologies, Waltham, MA, USA), and 1× penicillin-streptomycin (Caisson Biotech, Austin, TX, USA). This medium was supplemented daily with 20 ng/mL basic fibroblast growth factor (bFGF) (Life Technologies, Waltham, MA, USA) and 20 ng/mL epidermal growth factor (EGF) (R&D Systems, Minneapolis, MN, USA). It was changed every two days. Cells were passaged at 80% confluence with Accutase (Innovative Cell Technologies Inc., San Diego, CA, USA). After centrifugation of the cell suspension at 1000 rpm for 5 min, cells were split into a 1:4 ratio and transferred to plates coated with laminin. To maintain phenotypic integrity, cells were cultured to a maximum passage number of 25.
4.3. Differentiation of Cortical mNPCs
For cortical mNPCs, cells were seeded at a density of 5 × 10
4 cells per cm
2 on PDMS replicas with neuronal differentiation medium consisting of DMEM/F12 with 1× N2 supplement, 1× B27 medium, and 1× penicillin-streptomycin solution, without any growth factors. Every two days, half of the medium volume was refreshed. Samples were fixed on day 5 for further analysis (
Figure S2).
4.4. Differentiation of Hippocampal mNPCs
Differentiation of hippocampal mNPCs was carried out at a seeding density of 7.5 × 10
3 cells per cm
2 on PDMS replicas as described in Moe et al. [
10]. Briefly, the differentiation period was divided into two phases: the induction phase and the maturation phase (
Figure S2). The neural induction medium induces neurogenesis by withdrawal of the growth factors necessary to maintain the cells as progenitors. The induction medium consists of DMEM/F12 with 1× N2 supplement, 1× B27 medium, 5 ng/mL FGF2 (Life Technologies, Waltham, MA, USA), laminin (1 μg/mL), and 1× penicillin-streptomycin solution. Every two days, half of the medium volume was refreshed. On day 7, the induction medium was replaced with the maturation medium. The maturation medium consisted of a 1:1 ratio of Neurobasal (Life Technologies, Waltham, MA, USA) and DMEM/F12 with 1× B27 supplement (Life Technologies, Waltham, MA, USA), 0.25× N2 supplement, and 1× penicillin-streptomycin. Every two days, half of the medium volume was refreshed. On day 14 of culture, samples were fixed for further analysis. Cells cultured in this manner from the dataset described in Moe et al. [
10] were analysed in this study.
4.5. Immunofluorescence Staining of Class III β-Tubulin and Glial Fibrillary Acidic Protein
Cells were fixed for 20 min with 4% paraformaldehyde, and then blocked for 60 min with 10% goat serum (Life Technologies, Waltham, MA, USA) and 0.1% Triton X-100 in PBS. Primary antibodies against Class III β-Tubulin (TUJ1) (1:600 rabbit polyclonal AB_262133, Sigma, St. Louis, MO, USA) were used to mark neuronal cells. Antibodies against and glial fibrillary acidic protein (GFAP) (1:600 mouse monoclonal AB_2109815, Millipore, Billerica, MA, USA) were used to mark astrocytes. The antibodies were diluted in 1% goat serum and incubated overnight at 4 °C. This was followed by 1 h of incubation with the appropriate secondary antibodies at room temperature. The secondary antibodies used were goat anti-rabbit Alexa Fluor 488 (polyclonal AB_2576217, Molecular Probes, Eugene, OR, USA), and goat anti-mouse Alexa Fluor 546 (polyclonal AB_144695, Molecular Probes, Eugene, OR, USA) at the concentration of 1:500. Cells were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, St. Louis, MO, USA) for 20 min before mounting with Prolong Gold antifade mounting media (Life Technologies, Waltham, MA, USA). The images were taken with an inverted fluorescence DMIRM microscope (Leica, Wetzlar, Germany), confocal microscope SP5 (Leica, Wetzlar, Germany), and confocal microscope Nikon A1r. For comparison with differentiated hippocampal neural progenitor cells, images from Moe et al. [
10] were analysed. Uniform brightness and contrast (histogram stretch) adjustments were made for each panel of immunofluorescence images represented in
Figure 1 using ImageJ.
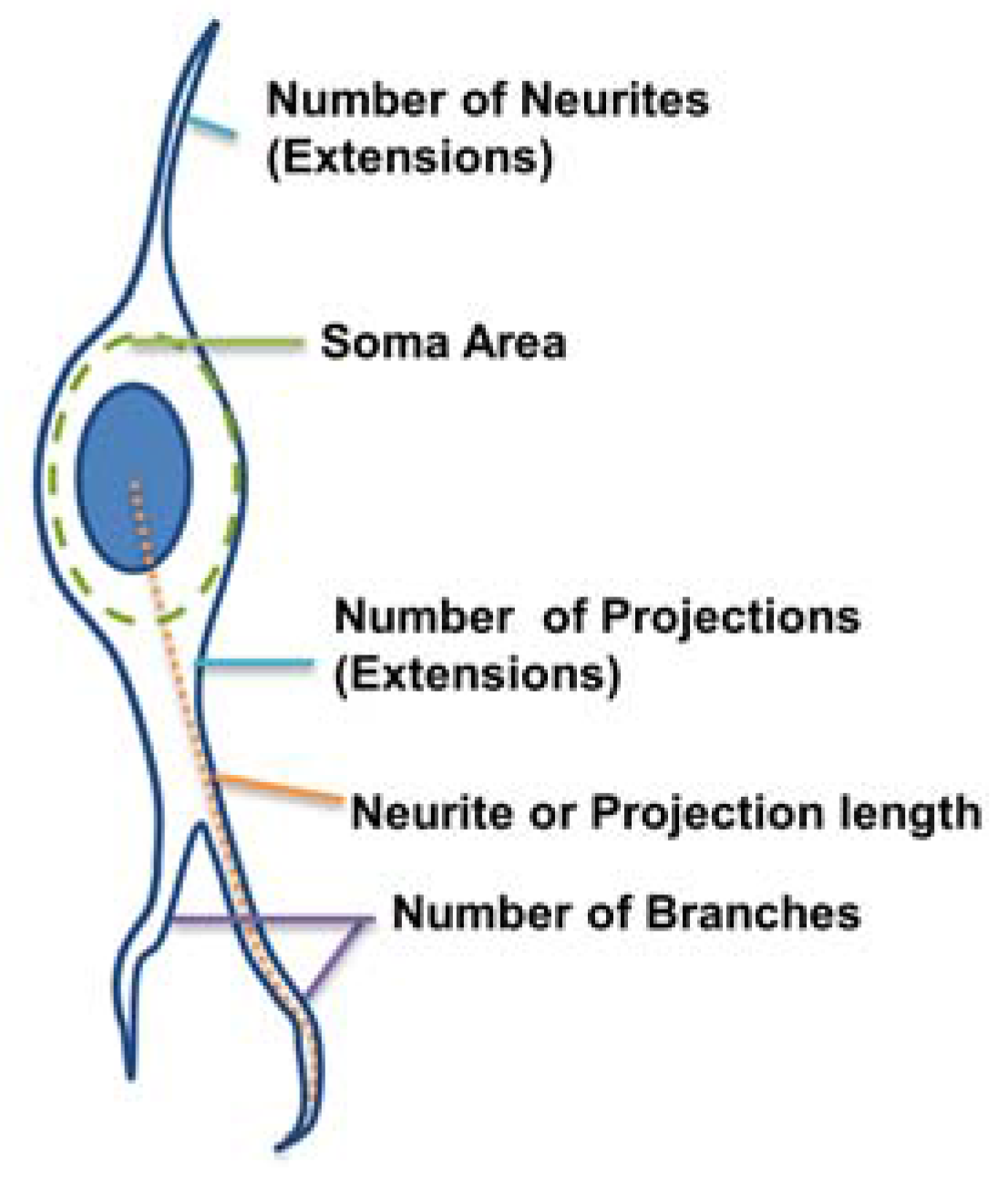
4.6. Morphological Parameter Measurements
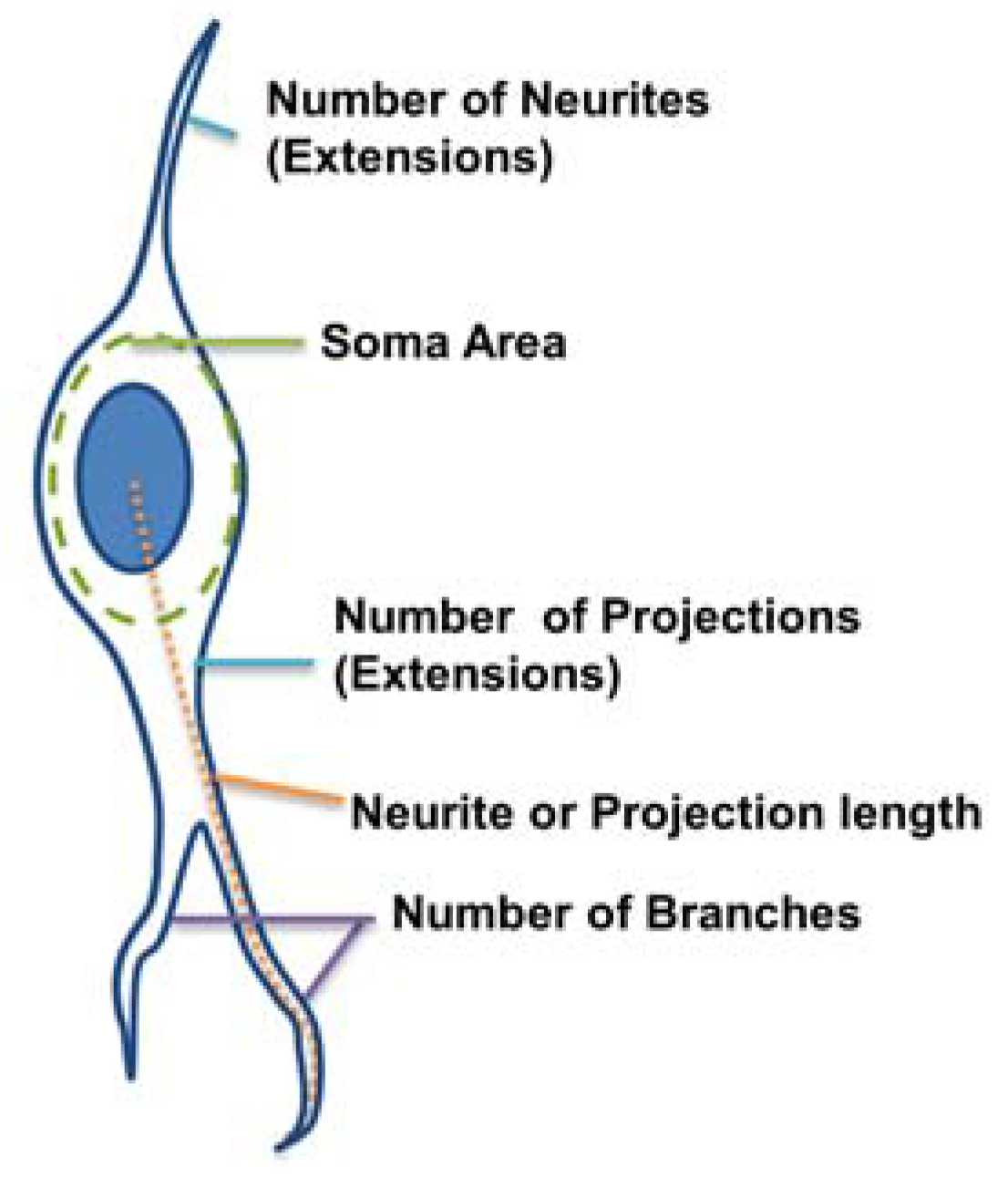
The morphological parameters of the astrocytes and neurons were measured from the GFAP and TUJ1 populations. respectively. For image analysis, ImageJ (version 1.46r) and a custom-made program in MATLAB (version R2013a, MathWorks, Natick, MA, USA) were used. The MATLAB program had a graphical user interface for the tracing of neurite extensions (
Figure S3) that was designed to rapidly process large data sets. The algorithms were semi-automated and relied on initial user input regarding the rough localization of cell features. The tracing was then iteratively refined using active contour algorithms [
35]. For each cell processed, the program automatically created and saved a dataset which described the topology and geometry of the neuron, using the adjacency matrix and other means from graph theory [
36]. From this dataset, the following morphological parameters were measured (
Figure 2):
Number of extensions: Number of extensions from the cell center (i.e., the number of neurites per neuron and number of projections per astrocyte).
Number of branches per extension: Total number of branches divided by total number of extensions per cell. A branch was defined as a secondary protrusion from an extension originating from the cell center. Thus, each extension could possess multiple branches. When an extension from the cell center displayed no secondary protrusion, the neurite was counted as possessing zero branches.
Extension Length: The length of the longest extension from the cell body.
Soma area: Area of the cell body excluding extensions. This was identified by ellipse approximation using a function of ImageJ.
All images used for quantification were TIFF images. For quantification of the morphology of differentiated cortical mNPCs, maximum intensity projections of confocal image stacks were taken. For the measurement of the number of extensions, extension length, and the number of branches of both, images were adjusted in brightness and contrast individually for maximization of signal recognition by the MATLAB active contour algorithms and to ensure visibility of as many cells as possible. The algorithm was semi-automated such that the cell center and specific points along the extensions were manually defined to limit the program to the cell space from which individual cell skeletons were created (
Figure S3). The accuracy of the active contour algorithm was manually adjusted as necessary. This was achieved by adjusting parameters, such as point spacing, smoothing, fitting edge, and adjusting image and mechanical forces, such as contribution, tension, and bending. The iteration time was also set to maximize signal recognition by the software. Taken together, the presence of noise did not influence the parameter measurement.
For the measurement of the soma area, ImageJ software was used. Images were individually adjusted for brightness and contrast to ensure the visibility of cell somas; then they were ”despecked”, threshold-adjusted for noise reduction, and converted to binary. The “watershed” function was used for the preparation of images for calculation of the soma area through the “fit ellipse” function of the “analyse particles” macro.
Prism 6.0 (Graphpad Software Inc., La Jolla, CA, USA) was used for data analysis. Data were presented as means and the standard error of means. For the comparison of data on different topographies, one-way analysis of variance (ANOVA) was performed, followed by a Tukey’s multiple comparisons test. For the comparison of data between cortical and hippocampal mNPCs, two-way ANOVA was performed, followed by a Sidak’s multiple comparisons test. p values less than 0.05 were considered statistically significant. The number of cells examined for each replicate ranged from 30–150 cells. The total number of cells (differentiated neurons, astrocytes and undifferentiated cells) measured for each of the topographies was as follows: Cortical Cells- U: 583, 1:333, 2:342, 3:363, 4:269, 5:378, 6:131, 7:149, 8:167, 9:237, 10:149, 11:270, 12:158, 13:782, 14:716; Hippocampal cells- U: 1144, 1:911, 2:754, 3:738, 4:1035, 5:1109, 6:579, 7:1046, 8:436, 9:728, 10:889, 11:920, 12:812, 13:1075, 14:689.
Differentiation efficiency (DE) was calculated as the percentage of cells expressing TUJ1 (neuronal differentiation efficiency) or GFAP (astrocyte differentiation efficiency). Hippocampal mNPC differentiation efficiency was calculated from images belonging to the dataset described in Moe et al. [
10], which was performed in a similar manner, with permission of John Wiley and Sons.
For further analysis, correlation coefficients were calculated by comparing measured values of each morphological parameter on all topographies with the differentiation efficiencies of all topographies. For the gratings pattern subset, DE values and morphological measurements were selected from cells grown on grating topographies. Neuronal DE was compared exclusively with neuronal morphological parameters. Similarly, astrocyte correlations were calculated separately. In addition, correlation coefficients were not calculated between types of neural progenitor cells but rather within the same subset.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}