
Maximizing Roughness Factors in Oxide-Derived Copper Coatings through Electrodeposition Parameters for Enhanced Electrocatalytic Performance

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Electrodeposition
- For this experiment, 0.02 M Cu(OAc)2 was added to the acetate buffer solution with pH 4.8. The buffer solution was prepared by mixing 87.2 mL of 1 M CH3COOH and 50 mL of 1 M NaOH in 500 mL of water. After adding copper acetate, the pH of the solution was adjusted to the original buffer pH by adding a few drops of 1 M NaOH.
- For this experiment, 0.02 M Cu(OAc)2 was added to acetate buffer solution with pH 5.5. The buffer solution was prepared by mixing 57.4 mL of 1 M CH3COOH and 50 mL of 1 M NaOH in 500 mL of water. After adding copper acetate, the pH of the solution was adjusted to the original buffer pH by adding a few drops of 1 M NaOH.
- For this experiment, 0.4 M CuSO4, 3 M C3H6O3, aged for 48 h, pH 7.9. The pH of the solution was initially adjusted to 8.5, but upon ageing at 25 °C, the pH lowered to 7.9 and was not changed further. Ageing is mandatory because the dissolved species require no less than 24 h to reach equilibrium [36].
2.2. Characterization
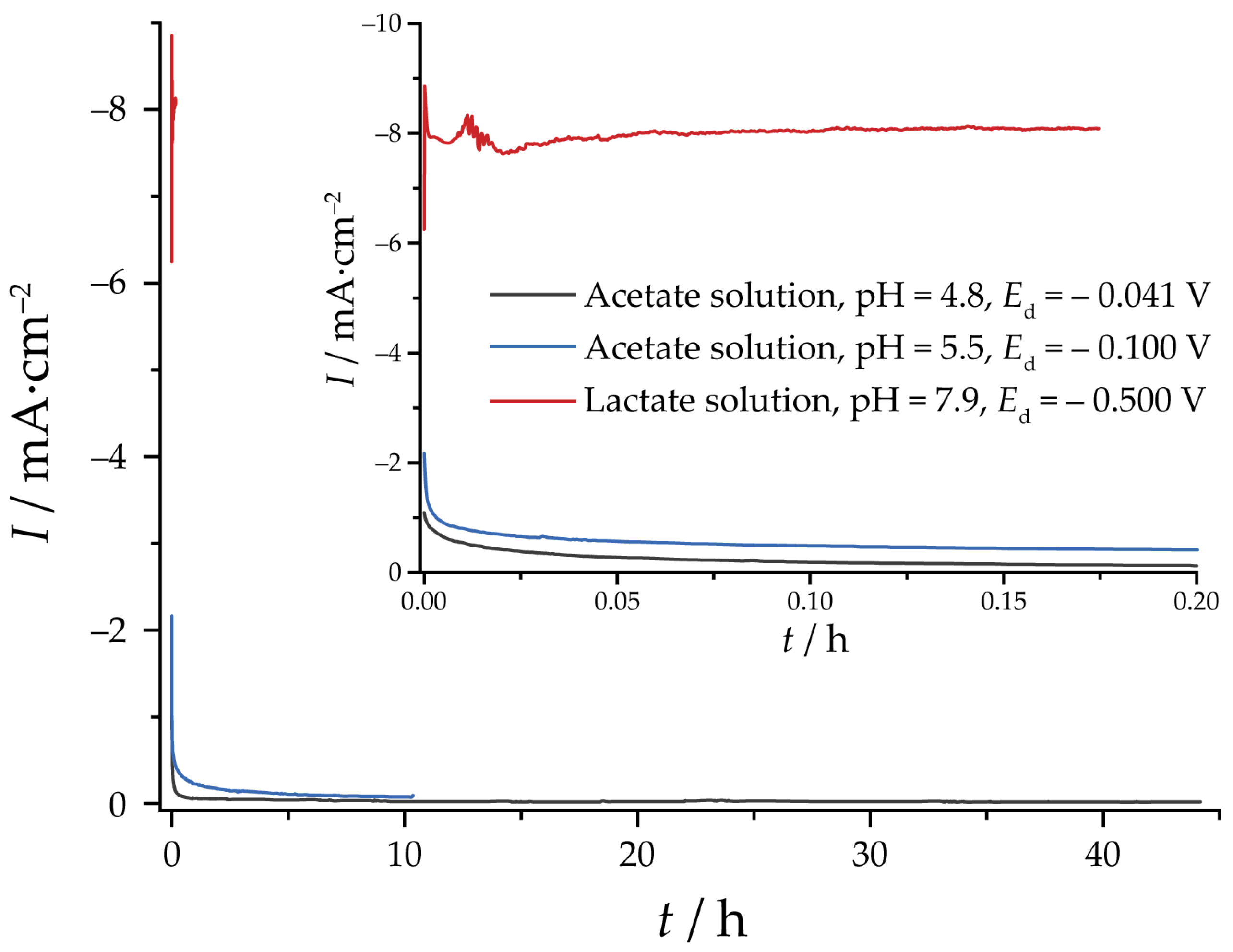
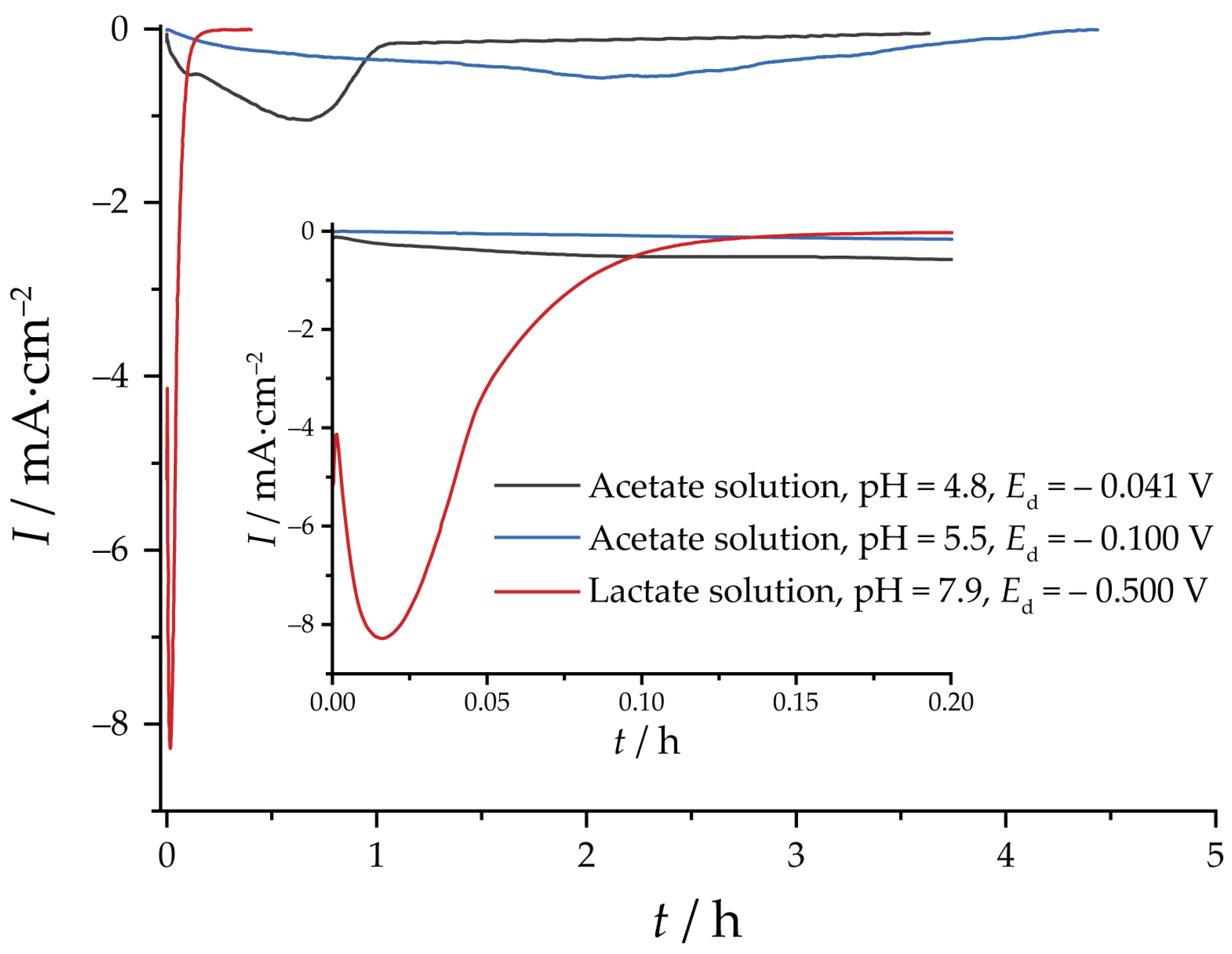
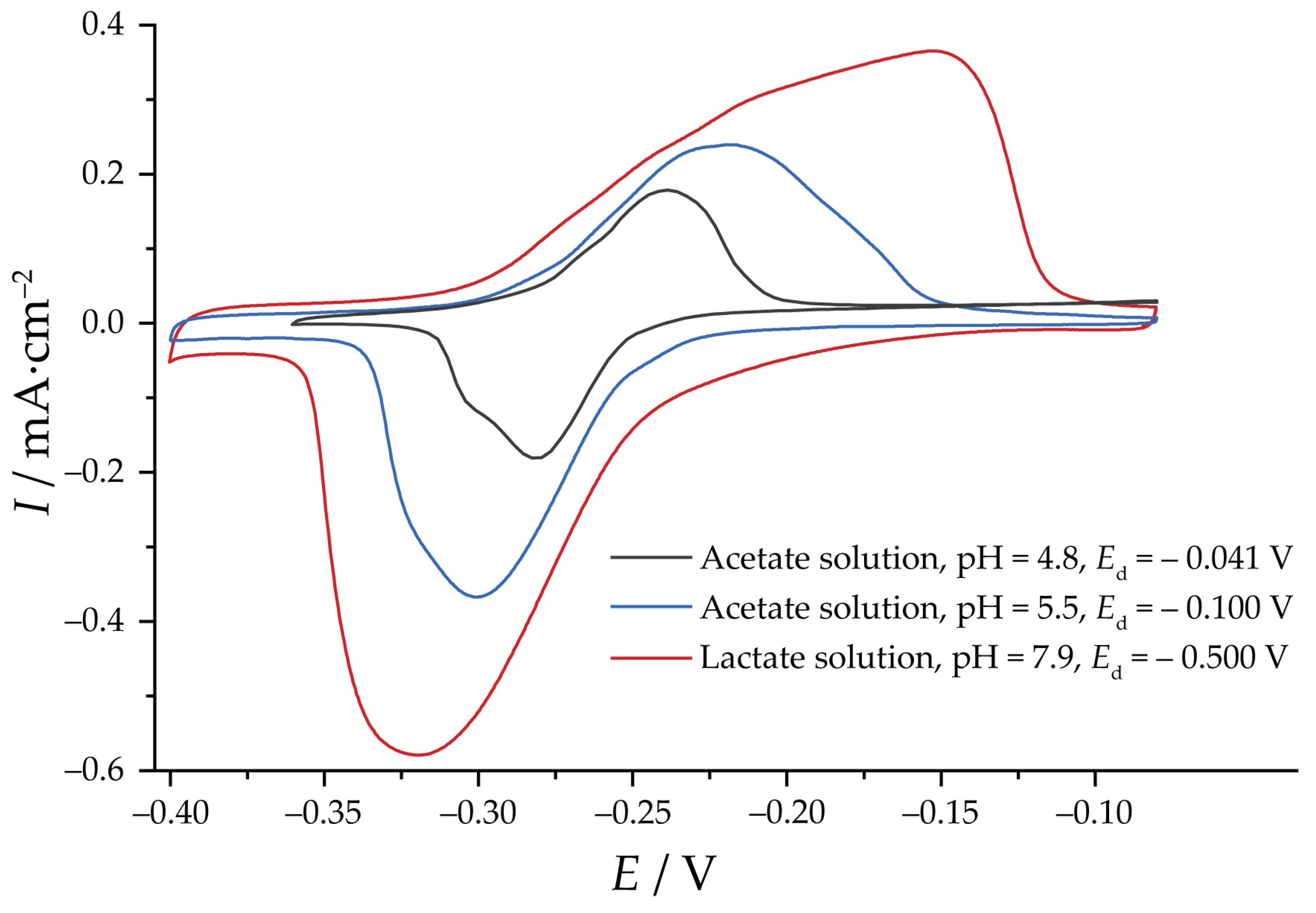
2.3. Electrochemical Reduction and EASA Measurements
3. Results and Discussion
3.1. Morphology and Composition of Electrodeposited Coatings
3.2. Reduction of Oxides and Determination of EASA
3.3. Cross-Section Examination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nitopi, S.; Bertheussen, E.; Scott, S.B.; Liu, X.; Engstfeld, A.K.; Horch, S.; Seger, B.; Stephens, I.E.L.; Chan, K.; Hahn, C.; et al. Progress and Perspectives of Electrochemical CO2 Reduction on Copper in Aqueous Electrolyte. Chem. Rev. 2019, 119, 7610–7672. [Google Scholar] [CrossRef]
- Hori, Y. Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Vayenas, C., Ed.; Springer: New York, NY, USA, 2008; Volume 42. [Google Scholar]
- Bushuyev, O.S.; De Luna, P.; Dinh, C.T.; Tao, L.; Saur, G.; van de Lagemaat, J.; Kelley, S.O.; Sargent, E.H. What Should We Make with CO2 and How Can We Make It? Joule 2018, 2, 825–832. [Google Scholar] [CrossRef]
- Gu, Z.; Shen, H.; Shang, L.; Lv, X.; Qian, L.; Zheng, G. Nanostructured Copper-Based Electrocatalysts for CO2 Reduction. Small Methods 2018, 2, 1800121. [Google Scholar] [CrossRef]
- Giziński, D.; Brudzisz, A.; Santos, J.S.; Trivinho-Strixino, F.; Stępniowski, W.J.; Czujko, T. Nanostructured Anodic Copper Oxides as Catalysts in Electrochemical and Photoelectrochemical Reactions. Catalysts 2020, 10, 1338. [Google Scholar] [CrossRef]
- Chen, C.; Yan, X.; Wu, Y.; Liu, S.; Sun, X.; Zhu, Q.; Feng, R.; Wu, T.; Qian, Q.; Liu, H.; et al. The in situ study of surface species and structures of oxide-derived copper catalysts for electrochemical CO2 reduction. Chem. Sci. 2021, 12, 5938–5943. [Google Scholar] [CrossRef]
- Löffler, M.; Khanipour, P.; Kulyk, N.; Mayrhofer, K.J.J.; Katsounaros, I. Insights into Liquid Product Formation during Carbon Dioxide Reduction on Copper and Oxide-Derived Copper from Quantitative Real-Time Measurements. ACS Catal. 2020, 10, 6735–6740. [Google Scholar] [CrossRef]
- Zhu, Q.; Sun, X.; Yang, D.; Ma, J.; Kang, X.; Zheng, L.; Zhang, J.; Wu, Z.; Han, B. Carbon dioxide electroreduction to C2 products over copper-cuprous oxide derived from electrosynthesized copper complex. Nat. Commun. 2019, 10, 3851. [Google Scholar] [CrossRef]
- Zahid, A.; Shah, A.; Shah, I. Oxide Derived Copper for Electrochemical Reduction of CO2 to C2+ Products. Nanomaterials 2022, 12, 1380. [Google Scholar] [CrossRef]
- Zhang, L.; Merino-Garcia, I.; Albo, J.; Sánchez-Sánchez, C.M. Electrochemical CO2 reduction reaction on cost-effective oxide-derived copper and transition metal–nitrogen–carbon catalysts. Curr. Opin. Electrochem. 2020, 23, 65–73. [Google Scholar] [CrossRef]
- Lei, Q.; Zhu, H.; Song, K.; Wei, N.; Liu, L.; Zhang, D.; Yin, J.; Dong, X.; Yao, K.; Wang, N.; et al. Investigating the Origin of Enhanced C2+ Selectivity in Oxide-/Hydroxide-Derived Copper Electrodes during CO2 Electroreduction. J. Am. Chem. Soc. 2020, 142, 4213–4222. [Google Scholar] [CrossRef]
- Wang, S.; Kou, T.; Baker, S.E.; Duoss, E.B.; Li, Y. Recent progress in electrochemical reduction of CO2 by oxide-derived copper catalysts. Mater. Today Nano 2020, 12, 100096. [Google Scholar] [CrossRef]
- Liang, Z.; Fu, J.; Vukmirovic, M.B.; Adzic, R.R. Electrochemical CO2 Reduction on Oxide-Derived Cu Surface with Various Oxide Thicknesses. ECS Trans. 2018, 85, 195–199. [Google Scholar] [CrossRef]
- Lee, S.H.; Sullivan, I.; Larson, D.M.; Liu, G.; Toma, F.M.; Xiang, C.; Drisdell, W.S. Correlating Oxidation State and Surface Area to Activity from Operando Studies of Copper CO Electroreduction Catalysts in a Gas-Fed Device. ACS Catal. 2020, 10, 8000–8011. [Google Scholar] [CrossRef]
- Ren, D.; Deng, Y.; Handoko, A.D.; Chen, C.S.; Malkhandi, S.; Yeo, B.S. Selective Electrochemical Reduction of Carbon Dioxide to Ethylene and Ethanol on Copper(I) Oxide Catalysts. ACS Catal. 2015, 5, 2814–2821. [Google Scholar] [CrossRef]
- Li, C.W.; Kanan, M.W. CO2 reduction at low overpotential on Cu electrodes resulting from the reduction of thick Cu2O films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef]
- Lum, Y.; Yue, B.; Lobaccaro, P.; Bell, A.T.; Ager, J.W. Optimizing C–C Coupling on Oxide-Derived Copper Catalysts for Electrochemical CO2 Reduction. J. Phys. Chem. C 2017, 121, 14191–14203. [Google Scholar] [CrossRef]
- Rudd, J.A.; Hernandez-Aldave, S.; Kazimierska, E.; Hamdy, L.B.; Bain, O.J.E.; Barron, A.R.; Andreoli, E. Investigation into the Re-Arrangement of Copper Foams Pre- and Post-CO2 Electrocatalysis. Chemistry 2021, 3, 687–703. [Google Scholar] [CrossRef]
- Reyter, D.; Odziemkowski, M.; Bélanger, D.; Roué, L. Electrochemically Activated Copper Electrodes. J. Electrochem. Soc. 2007, 154, K36. [Google Scholar] [CrossRef]
- Engstfeld, A.K.; Maagaard, T.; Horch, S.; Chorkendorff, I.; Stephens, I.E.L. Polycrystalline and Single-Crystal Cu Electrodes: Influence of Experimental Conditions on the Electrochemical Properties in Alkaline Media. Chem. Eur. J. 2018, 24, 17743–17755. [Google Scholar] [CrossRef]
- Tiwari, A.; Maagaard, T.; Chorkendorff, I.; Horch, S. Effect of Dissolved Glassware on the Structure-Sensitive Part of the Cu(111) Voltammogram in KOH. ACS Energy Lett. 2019, 4, 1645–1649. [Google Scholar] [CrossRef]
- Li, D.; Batchelor-McAuley, C.; Compton, R.G. Some thoughts about reporting the electrocatalytic performance of nanomaterials. Appl. Mater. Today 2020, 18, 100404. [Google Scholar] [CrossRef]
- Brisard, G.M.; Zenati, E.; Gasteiger, H.A.; Marković, N.M.; Ross, P.N. Underpotential Deposition of Lead on Cu(100) in the Presence of Chloride: Ex-Situ Low-Energy Electron Diffraction, Auger Electron Spectroscopy, and Electrochemical Studies. Langmuir 1997, 13, 2390–2397. [Google Scholar] [CrossRef]
- Brisard, G.M.; Zenati, E.; Gasteiger, H.A.; Markovic, N.; Ross, P.N. Underpotential Deposition of Lead on Copper(111): A Study Using a Single-Crystal Rotating Ring Disk Electrode and ex Situ Low-Energy Electron Diffraction and Scanning tunneling Microscopy. Langmuir 1995, 11, 2221–2230. [Google Scholar] [CrossRef]
- Siegenthaler, H.; Jüttner, K. Voltammetric investigation of lead adsorption on Cu(111) single crystal substrates. J. Electroanal. Chem. 1984, 163, 327–343. [Google Scholar] [CrossRef]
- Giri, S.D.; Mahajani, S.M.; Suresh, A.K.; Sarkar, A. Electrochemical reduction of CO2 on activated copper: Influence of surface area. Mater. Res. Bull. 2020, 123, 110702. [Google Scholar] [CrossRef]
- Tang, Y.; Chen, Z.; Jia, Z.; Zhang, L.; Li, J. Electrodeposition and characterization of nanocrystalline cuprous oxide thin films on TiO2 films. Mater. Lett. 2005, 59, 434–438. [Google Scholar] [CrossRef]
- Li, G.; Huang, Y.; Fan, Q.; Zhang, M.; Lan, Q.; Fan, X.; Zhou, Z.; Zhang, C. Effects of bath pH on structural and electrochemical performance of Cu2O. Ionics 2016, 22, 2213–2223. [Google Scholar] [CrossRef]
- McShane, C.M.; Choi, K.S. Photocurrent enhancement of n-type Cu2O electrodes achieved by controlling dendritic branching growth. J. Am. Chem. Soc. 2009, 131, 2561–2569. [Google Scholar] [CrossRef]
- Golden, T.D.; Shumsky, M.G.; Zhou, Y.; VanderWerf, R.A.; Van Leeuwen, R.A.; Switzer, J.A. Electrochemical Deposition of Copper(I) Oxide Films. Chem. Mater. 1996, 8, 2499–2504. [Google Scholar] [CrossRef]
- Zhou, Y.C.; Switzer, J.A. Galvanostatic electrodeposition and microstructure of copper (I) oxide film. Mater. Res. Innov. 1998, 2, 22–27. [Google Scholar] [CrossRef]
- Rakhshani, A.E.; Al-Jassar, A.A.; Varghese, J. Electrodeposition and characterization of cuprous oxide. Thin Solid Films 1987, 148, 191–201. [Google Scholar] [CrossRef]
- Jayasingha, J.L.K.; Kaumal, M.N.; Jayathilaka, K.M.D.C.; Gunewardene, M.S.; Dissanayake, D.P.; Jayanetti, J.K.D.S. Surfactant free template assisted electrodeposited n-type nano-cubic Cu2O thin films for nonenzymatic glucose sensing. Phys. Status Solidi A 2017, 214, 1700135. [Google Scholar] [CrossRef]
- de Jongh, P.E.; Vanmaekelbergh, D.; Kelly, J.J. Cu2O: Electrodeposition and Characterization. Chem. Mater. 1999, 11, 3512–3517. [Google Scholar] [CrossRef]
- Zheng, J.Y.; Jadhav, A.P.; Song, G.; Kim, C.W.; Kang, Y.S. Cu and Cu2O films with semi-spherical particles grown by electrochemical deposition. Thin Solid Films 2012, 524, 50–56. [Google Scholar] [CrossRef]
- Chen, T.; Kitada, A.; Seki, Y.; Fukami, K.; Usmanov, D.T.; Chen, L.C.; Hiraoka, K.; Murase, K. Identification of Copper(II)–Lactate Complexes in Cu2O Electrodeposition Baths: Deprotonation of the α-Hydroxyl Group in Highly Concentrated Alkaline Solution. J. Electrochem. Soc. 2018, 165, D444–D451. [Google Scholar] [CrossRef]
- Zhou, Y.; Switzer, J.A. Electrochemical Deposition and Microstructure of Copper (I) Oxide Films. Scripta Mater. 1998, 38, 1731–1738. [Google Scholar] [CrossRef]
- Rietveld, H.M. A profile refinement method for nuclear and magnetic structures. J. Appl. Crystallogr. 1969, 2, 65–71. [Google Scholar] [CrossRef]
- Hill, R.J.; Howard, C.J. Quantitative phase analysis from neutron powder diffraction data using the Rietveld method. J. Appl. Crystallogr. 1987, 20, 467–474. [Google Scholar] [CrossRef]
- Solovyov, L.A. Full-profile refinement by derivative difference minimization. J. Appl. Crystallogr. 2004, 37, 743–749. [Google Scholar] [CrossRef]
- Bewick, A.; Jovićević, J.; Thomas, B. Phase formation in the underpotential deposition of metals. Faraday Symp. Chem. Soc. 1977, 12, 24–35. [Google Scholar] [CrossRef]
- Mezine, Z.; Kadri, A.; Hamadou, L.; Benbrahim, N.; Chaouchi, A. Electrodeposition of copper oxides (CuxOy) from acetate bath. J. Electroanal. Chem. 2018, 817, 36–47. [Google Scholar] [CrossRef]
- Min, C.; Li, S.; Shi, Z.; Xie, J.; Ma, R. Effect of pH on the electrodeposition nucleation and growth mechanism of cuprous oxide. J. Solid State Electrochem. 2023, 27, 1085–1093. [Google Scholar] [CrossRef]
- Bijani, S.; Schrebler, R.; Dalchiele, E.A.; Gabás, M.; Martínez, L.; Ramos-Barrado, J.R. Study of the Nucleation and Growth Mechanisms in the Electrodeposition of Micro- and Nanostructured Cu2O Thin Films. J. Phys. Chem. C 2011, 115, 21373–21382. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Soultions; National Association of Corrosion: Houston, TX, USA, 1974; p. 644. [Google Scholar]
- Swanson, H.E.; Tatge, E. Circular of the Bureau of Standards No. 539 Volume 1: Standard X-ray Diffraction Powder Patterns; National Bureau of Standards: Washington, DC, USA, 1953; Volume 1, pp. 15–16. [Google Scholar] [CrossRef]
- Swanson, H.E.; Fuyat, R.K. Circular of the Bureau of Standards No. 539 Volume 2: Standard X-ray Diffraction Powder Patterns; National Bureau of Standards: Washington, DC, USA, 1953; Volume 2, pp. 23–25. [Google Scholar] [CrossRef]
- Bijani, S.; Martínez, L.; Gabás, M.; Dalchiele, E.A.; Ramos-Barrado, J.R. Low-Temperature Electrodeposition of Cu2O Thin Films: Modulation of Micro-Nanostructure by Modifying the Applied Potential and Electrolytic Bath pH. J. Phys. Chem. C 2009, 113, 19482–19487. [Google Scholar] [CrossRef]
- Switzer, J.A.; Hung, C.-J.; Huang, L.-Y.; Miller, F.S.; Zhou, Y.; Raub, E.R.; Shumsky, M.G.; Bohannan, E.W. Potential oscillations during the electrochemical self-assembly of copper/cuprous oxide layered nanostructures. J. Mater. Res. 2011, 13, 909–916. [Google Scholar] [CrossRef]
- Switzer, J.A.; Hung, C.-J.; Huang, L.-Y.; Switzer, E.R.; Kammler, D.R.; Golden, T.D.; Bohannan, E.W. Electrochemical Self-Assembly of Copper/Cuprous Oxide Layered Nanostructures. J. Am. Chem. Soc. 1998, 120, 3530–3531. [Google Scholar] [CrossRef]
- Eskhult, J.; Herranen, M.; Nyholm, L. On the origin of the spontaneous potential oscillations observed during galvanostatic deposition of layers of Cu and Cu2O in alkaline citrate solutions. J. Electroanal. Chem. 2006, 594, 35–49. [Google Scholar] [CrossRef]
- Leopold, S.; Herranen, M.; Carlsson, J.O.; Nyholm, L. In situ pH measurement of the self-oscillating Cu(II)–lactate system using an electropolymerised polyaniline film as a micro pH sensor. J. Electroanal. Chem. 2003, 547, 45–52. [Google Scholar] [CrossRef]
- Mech, K.; Bisztyga-Szklarz, M.; Szaciłowski, K. Brief Insights into Cu2O Electrodeposition: Detailed Progressive Voltammetric and Electrogravimetric Analysis of a Copper Lactate System. J. Electrochem. Soc. 2020, 167, 042504. [Google Scholar] [CrossRef]
- Levi-Setti, R.; Fox, T.R.; Lam, K. Ion channeling effects in scanning ion microscopy with a 60 keV Ga+ probe. Nucl. Instrum. Methods Phys. Res. 1983, 205, 299–309. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Levin, E.E.; Kokin, A.A.; Morozov, D.A.; Nikitina, V.A. Maximizing Roughness Factors in Oxide-Derived Copper Coatings through Electrodeposition Parameters for Enhanced Electrocatalytic Performance. Nanomaterials 2023, 13, 3064. https://doi.org/10.3390/nano13233064
Levin EE, Kokin AA, Morozov DA, Nikitina VA. Maximizing Roughness Factors in Oxide-Derived Copper Coatings through Electrodeposition Parameters for Enhanced Electrocatalytic Performance. Nanomaterials. 2023; 13(23):3064. https://doi.org/10.3390/nano13233064
Chicago/Turabian StyleLevin, Eduard E., Alexander A. Kokin, Dmitriy A. Morozov, and Victoria A. Nikitina. 2023. "Maximizing Roughness Factors in Oxide-Derived Copper Coatings through Electrodeposition Parameters for Enhanced Electrocatalytic Performance" Nanomaterials 13, no. 23: 3064. https://doi.org/10.3390/nano13233064