
Microfluidic Vaterite Synthesis: Approaching the Nanoscale Particles
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
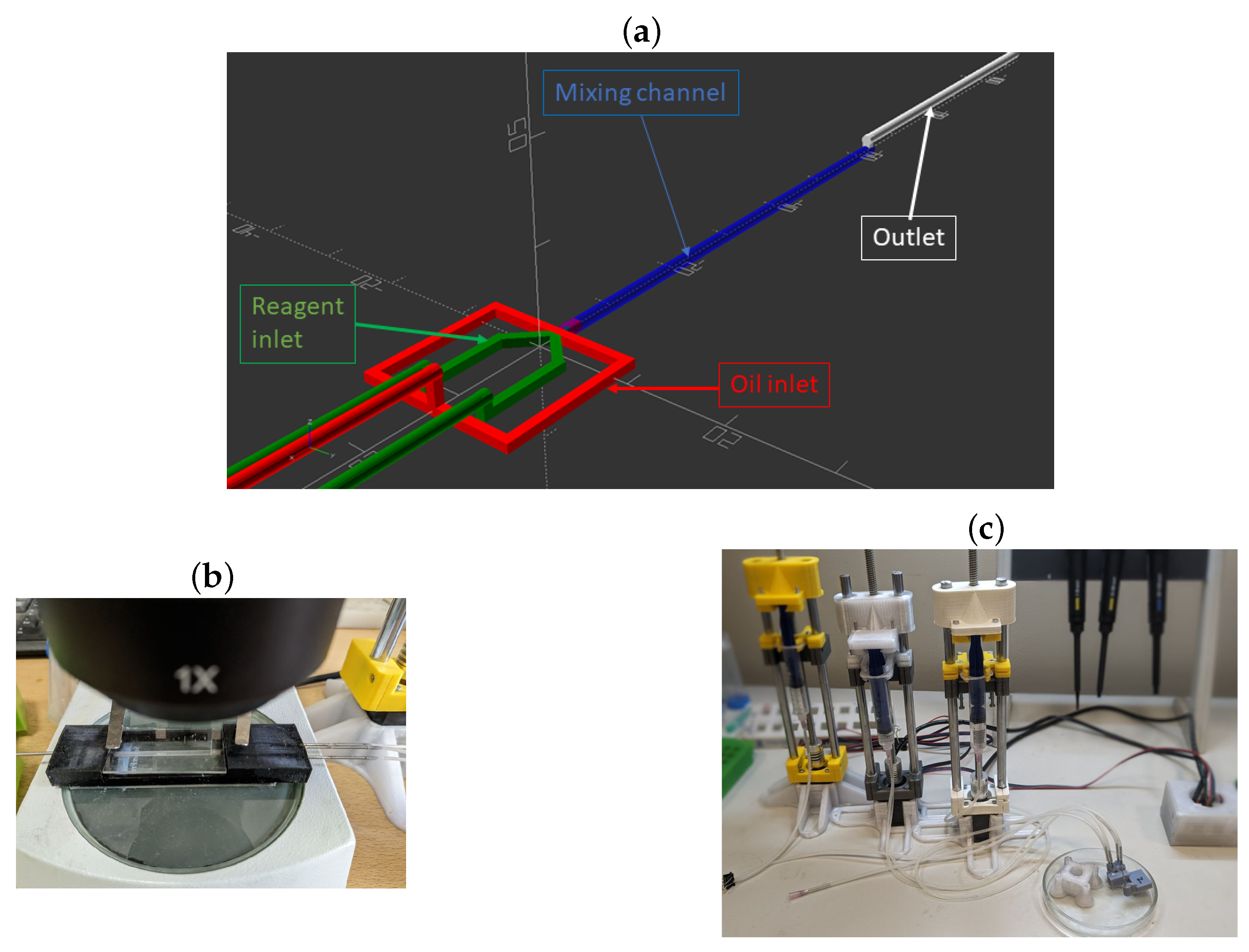
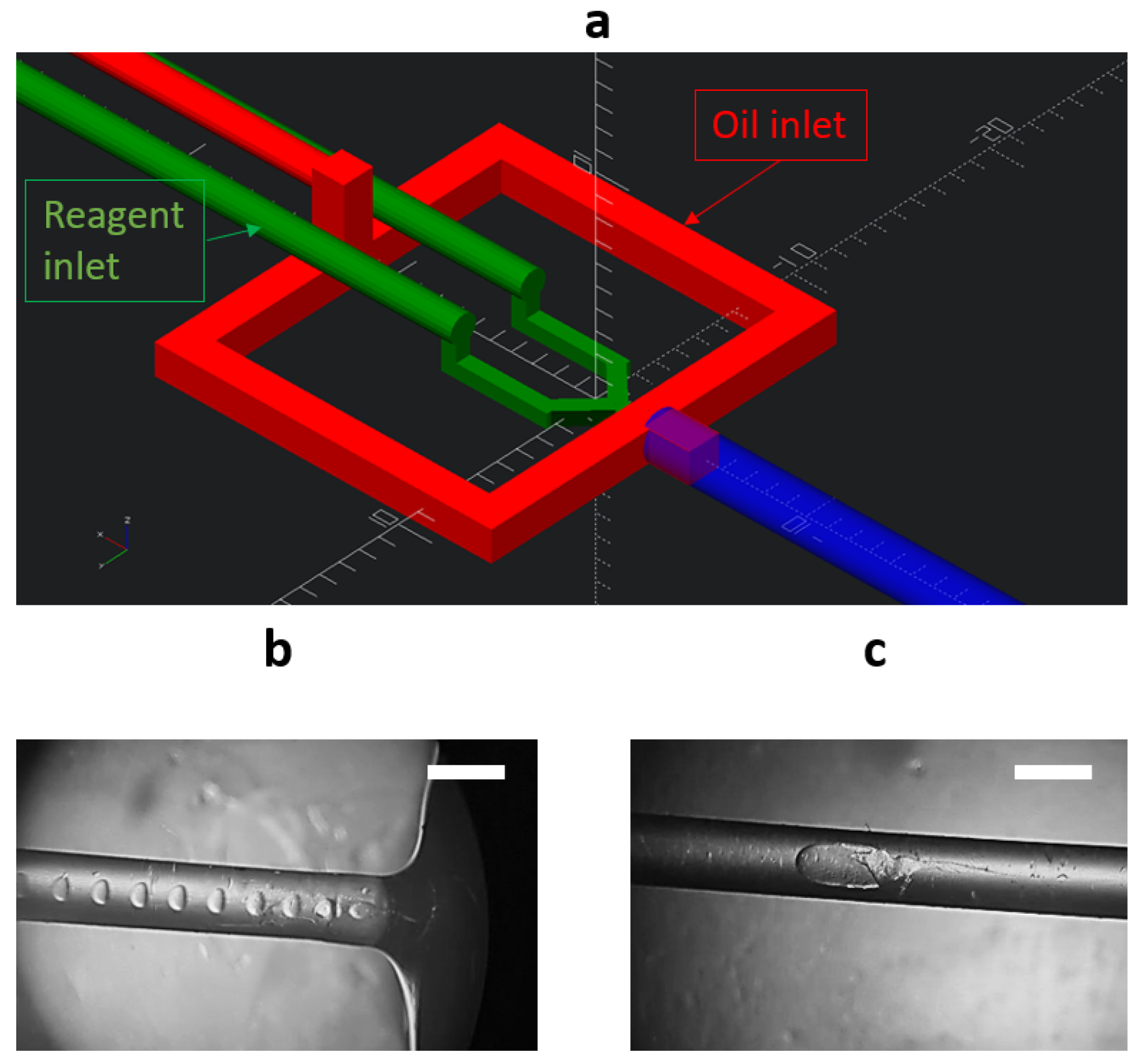
2.2. Microfluidic Chip Formation
2.3. Synthesis of CaCO3 Particles
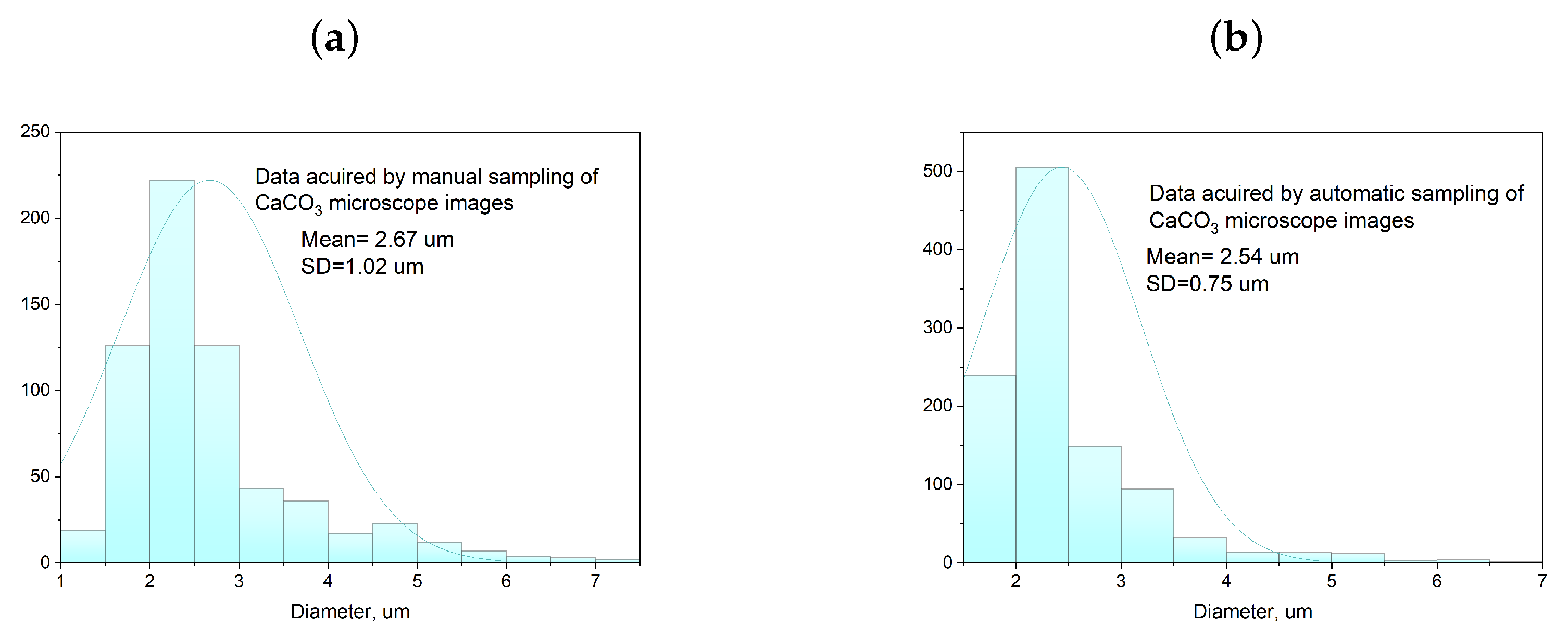
2.4. Characterization Methods
- (I)
- Direct search of particle size distribution was performed by the linear least squares method with Tikhonov regularization of the solution (program GNOM from the ATSAS package) [25,26]. In discrete form, the theoretical scattering intensity is written as followswhere is the desired volume size distribution function defined over size range ; is the square of the form factor, or the scattering intensity from a particle of a given shape with radius , and unit volume and density contrast; is the volume of the particle with effective radius ; and is the step on the radius grid. is found as the solution of a system of linear equations with regularization by the first derivative of the distribution.
- (II)
- Direct search of particle size distribution was performed in the form of a histogram (program VOLDIS) [27]. The elements of distribution vector are the search parameters for the minimum of the target functionwhere x is the vector of current values of the distribution , or parameter vector; is the theoretical intensity according to Formula (1); is a weighting function that compresses the range of scattering intensities to ratio ; and is an auxiliary least-squares multiplier that matches the scattering curves before calculating the difference.
- (III)
- A parametric approach based on the use of a superposition of several smooth distributions is given in analytical form (in this paper, Schulz distributions, program MIXTURE from the ATSAS package) [24,28]. The algorithm is also based on the minimization of the function of type (2), where vector x is calculated from the sum of several (usually 3–5) smooth analytic functions. The search parameters are the positions and half-widths of such functions and their relative contributions. In this study, we used scattering from a homogeneous sphere, the most commonly used in practice, at the unit contrast and volume as a formulator of inhomogeneities:Each of the above algorithms has its advantages and drawbacks, and applying only one of them is unlikely to provide artifact-free solutions. In particular, the distribution curves obtained with the GNOM and VOLDIS programs can show significant oscillations. These oscillations are due to the fact that during the search process, the increase in the scattering contribution from the particles of one radius is compensated by a decrease in the contribution from particles with nearby radii, without changing the shape of the theoretical scattering curve significantly. The MIXTURE algorithm is devoid of this disadvantage, as the resulting distribution curve is the sum of the smooth partial functions. However, the MIXTURE target function is multimodal, which requires setting starting values for the parameters of the partial distributions that are close enough to the global solution. These starting values can be determined through a visual analysis of the solutions obtained with the GNOM and VOLDIS programs, which was conducted in this paper.
3. Results
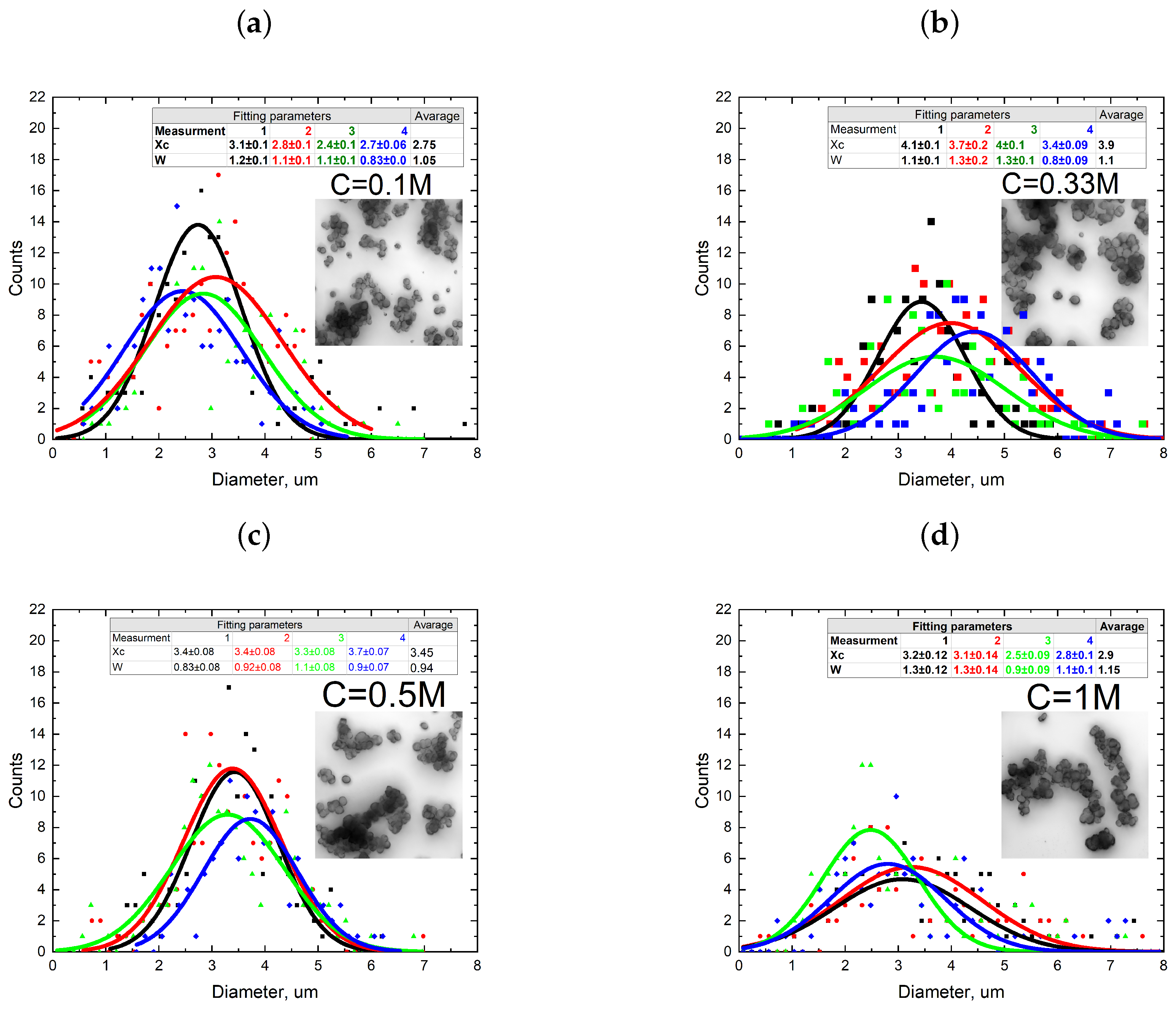
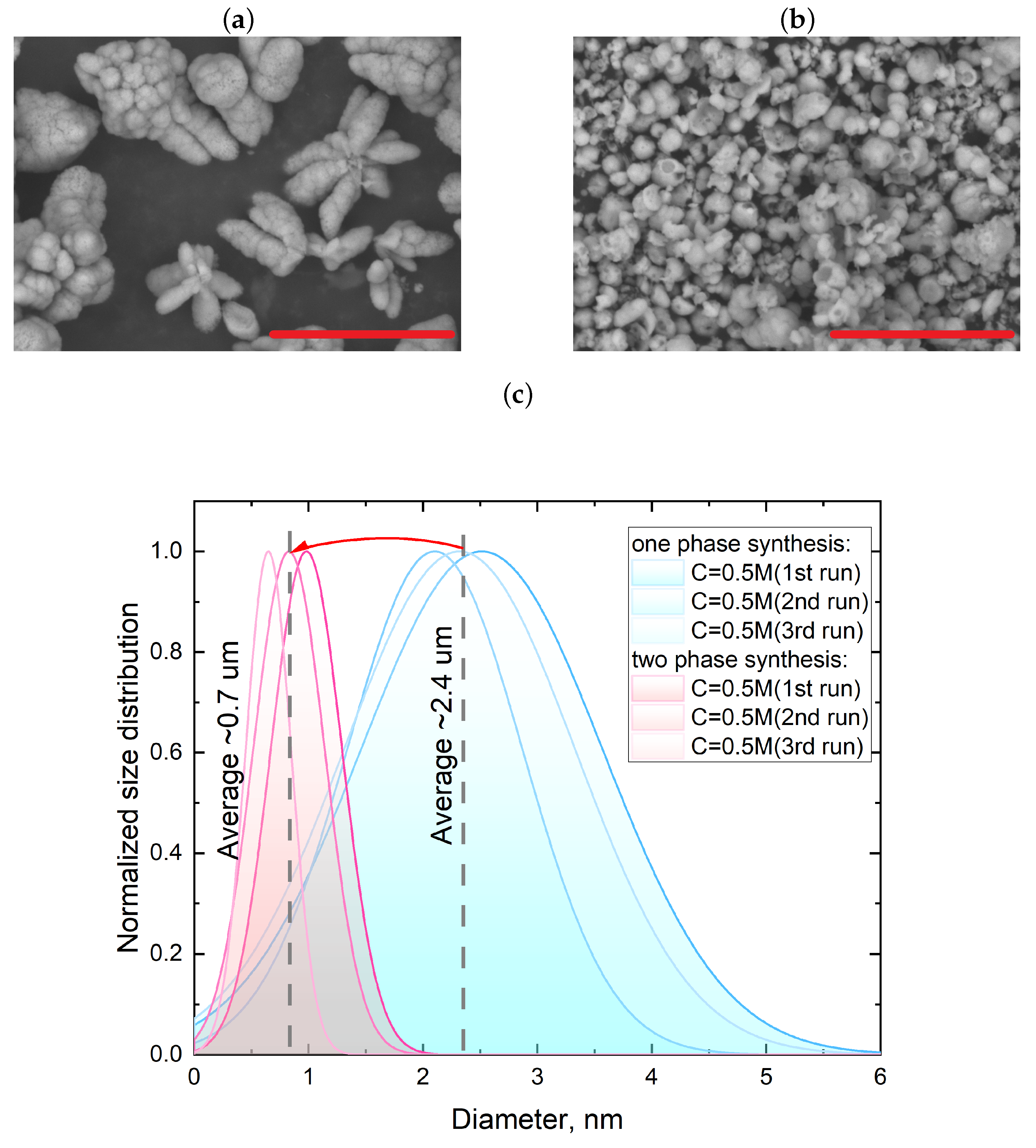
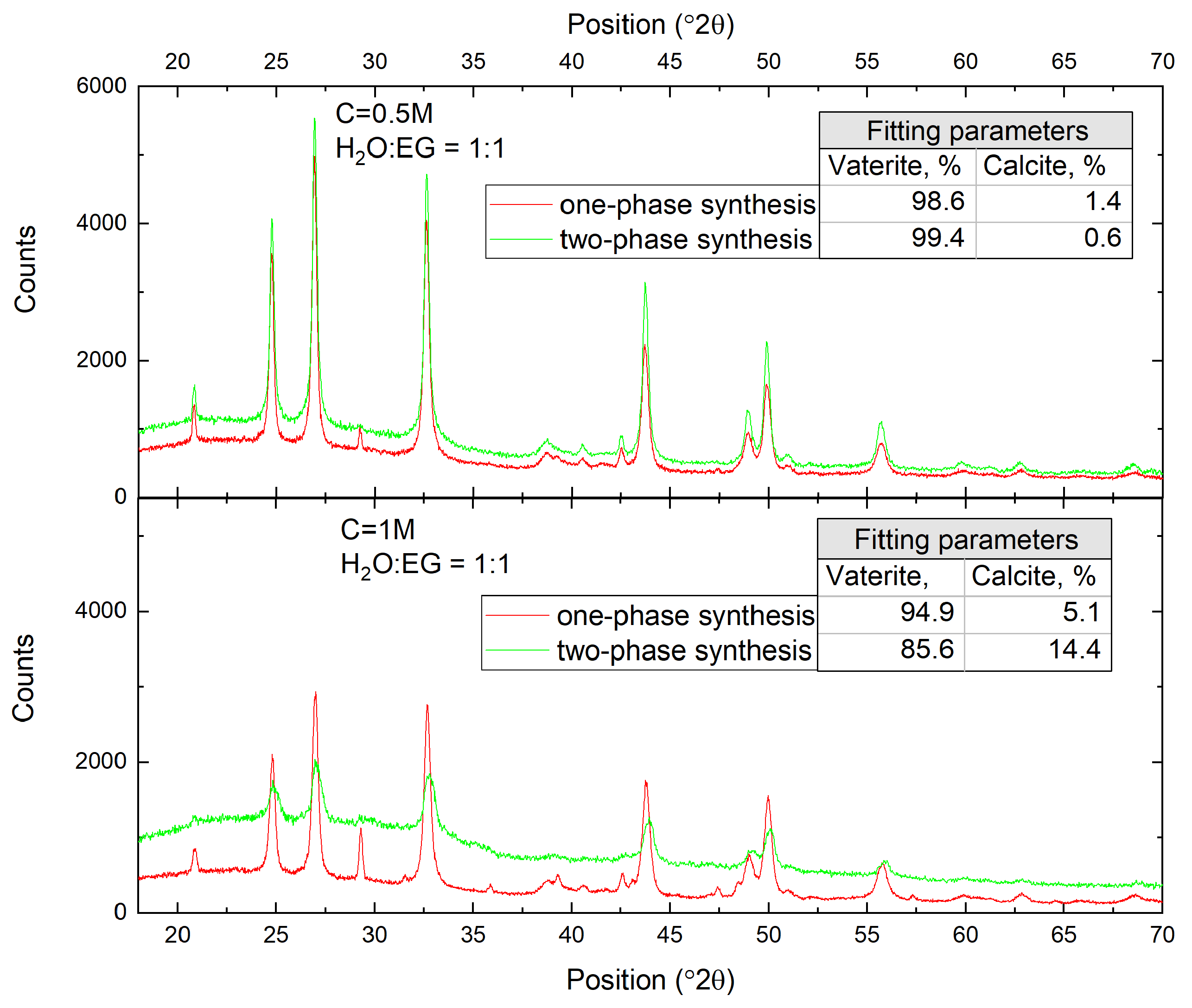
3.1. Single-Phase Synthesis in Water and Water-EG Conditions
3.2. Two-Phase Synthesis in Water and Water–EG Conditions
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| SAXS | Small-angle X-ray scattering |
| MSLA | Masked Stereolithography Apparatus |
| DLS | Dynamic light scattering |
| XRD | X-ray diffraction analysis |
Appendix A

References
- Volodkin, D. CaCO3 templated micro-beads and-capsules for bioapplications. Adv. Colloid Interface Sci. 2014, 207, 306–324. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Ma, G.H.; Hu, G.; Yu, D.; Mcleish, T.; Su, Z.G.; Shen, Z.Y. Preparation of hierarchical hollow CaCO3 particles and the application as anticancer drug carrier. J. Am. Chem. Soc. 2008, 130, 15808–15810. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ji, X.; Zhao, G.; Wang, X. Facile preparation of cubic calcium carbonate nanoparticles with hydrophobic properties via a carbonation route. Powder Technol. 2010, 200, 144–148. [Google Scholar] [CrossRef]
- Haložan, D.; Riebentanz, U.; Brumen, M.; Donath, E. Polyelectrolyte microcapsules and coated CaCO3 particles as fluorescence activated sensors in flowmetry. Colloids Surfaces A Physicochem. Eng. Asp. 2009, 342, 115–121. [Google Scholar] [CrossRef]
- Fujiwara, M.; Shiokawa, K.; Morigaki, K.; Zhu, Y.; Nakahara, Y. Calcium carbonate microcapsules encapsulating biomacromolecules. Chem. Eng. J. 2008, 137, 14–22. [Google Scholar] [CrossRef]
- Babou-Kammoe, R.; Hamoudi, S.; Larachi, F.; Belkacemi, K. Synthesis of CaCO3 nanoparticles by controlled precipitation of saturated carbonate and calcium nitrate aqueous solutions. Can. J. Chem. Eng. 2012, 90, 26–33. [Google Scholar] [CrossRef]
- Karimi, M.A.; Ranjbar, M. Hydrothermal synthesis and characterization of CaCO3 nanostructure. Synth. React. Inorg. Met.-Org. Nano-Met. Chem. 2016, 46, 635–637. [Google Scholar] [CrossRef]
- Nilsen, O.; Fjellvåg, H.; Kjekshus, A. Growth of calcium carbonate by the atomic layer chemical vapour deposition technique. Thin Solid Film. 2004, 450, 240–247. [Google Scholar] [CrossRef]
- Sommerdijk, N.A.; With, G.d. Biomimetic CaCO3 mineralization using designer molecules and interfaces. Chem. Rev. 2008, 108, 4499–4550. [Google Scholar] [CrossRef]
- Boyjoo, Y.; Pareek, V.K.; Liu, J. Synthesis of micro and nano-sized calcium carbonate particles and their applications. J. Mater. Chem. A 2014, 2, 14270–14288. [Google Scholar] [CrossRef]
- Konopacka-Łyskawa, D. Synthesis methods and favorable conditions for spherical vaterite precipitation: A review. Crystals 2019, 9, 223. [Google Scholar] [CrossRef]
- Besson, R.; Favergeon, L. Understanding the mechanisms of CaO carbonation: Role of point defects in CaCO3 by atomic-scale simulations. J. Phys. Chem. C 2014, 118, 22583–22591. [Google Scholar] [CrossRef]
- Remya, K.P.; Kim, S.; Kim, M.J. Surfactant-free hydrothermal fabrication of vaterite CaCO3 with hexagonal bipyramidal morphologies using seawater. Powder Technol. 2022, 410, 117865. [Google Scholar] [CrossRef]
- Weiss, I.M.; Tuross, N.; Addadi, L.; Weiner, S. Mollusc larval shell formation: Amorphous calcium carbonate is a precursor phase for aragonite. J. Exp. Zool. 2002, 293, 478–491. [Google Scholar] [CrossRef] [PubMed]
- Gal, A.; Habraken, W.; Gur, D.; Fratzl, P.; Weiner, S.; Addadi, L. Calcite crystal growth by a solid-state transformation of stabilized amorphous calcium carbonate nanospheres in a hydrogel. Angew. Chem. Int. Ed. 2013, 52, 4867–4870. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Wu, Y.; Yue, X.; Wang, C.; Zhang, J. Biomimetic synthesis of vaterite CaCO3 microspheres under threonine for preparation of pH-responsive antibacterial biofilm. J. Mater. Res. 2020, 35, 2427–2440. [Google Scholar] [CrossRef]
- Qu, F.; Meng, T.; Dong, Y.; Sun, H.; Tang, Q.; Liu, T.; Wang, Y. Aqueous two-phase droplet-templated colloidosomes composed of self-formed particles via spatial confined biomineralization. ACS Appl. Mater. Interfaces 2019, 11, 35613–35621. [Google Scholar] [CrossRef]
- Baranov, K.; Reznik, I.; Karamysheva, S.; Swart, J.W.; Moshkalev, S.; Orlova, A. Optical Properties of AgInS2 Quantum Dots Synthesized in a 3D-Printed Microfluidic Chip. Technologies 2023, 11, 93. [Google Scholar] [CrossRef]
- Wijnen, B.; Hunt, E.J.; Anzalone, G.C.; Pearce, J.M. Open-source syringe pump library. PLoS ONE 2014, 9, e107216. [Google Scholar] [CrossRef]
- Wartchow, R. Datensammlung nach der ‘‘learnt profile”-methode (LP) für calcit und vergleich mit der ‘‘background peak background”-methode (BPB). Z. Krist. 1989, 186, 300–302. [Google Scholar]
- Kamhi, S.R. On the structure of vaterite CaCO3. Acta Crystallogr. 1963, 16, 770–772. [Google Scholar] [CrossRef]
- Mogilevskiy, L.Y.; Dembo, A.; Svergun, D.; Feygin, L. Small-angle X-ray diffractometer with single coordinate detector. Crystallogr. Rep. 1984, 29, 587–591. [Google Scholar]
- Feigin, L.A.; Svergun, D.I. Structure Analysis by Small-Angle X-ray and Neutron Scattering; Plenum Press: New York, NY, USA, 1987; pp. 280–281. [Google Scholar]
- Konarev, P.V.; Volkov, V.V.; Sokolova, A.V.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Feigin, L.A.; Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar]
- Manalastas-Cantos, K.; Konarev, P.V.; Hajizadeh, N.R.; Kikhney, A.G.; Petoukhov, M.V.; Molodenskiy, D.S.; Panjkovich, A.; Mertens, H.D.T.; Gruzinov, A.; Borges, C.; et al. ATSAS 3.0: Expanded functionality and new tools for small-angle scattering data analysis. J. Appl. Crystallogr. 2021, 54, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Volkov, V.V. VOLDIS: A Direct Search for the Size Distribution of Nanoparticles from Small-Angle Scattering Data and Stability of the Solution. Crystals 2022, 12, 1659. [Google Scholar] [CrossRef]
- Volkov, V.V.; Konarev, P.V.; Kryukova, A.E. Combined Scheme of Reconstruction of the Particle Size Distribution Function Using Small-Angle Scattering Data. JETP Lett. 2020, 112, 591–595. [Google Scholar] [CrossRef]
- Pallaeva, T.; Mikheev, A.; Eurov, D.; Kurdyukov, D.; Popova, V.; Dmitrienko, E.; Akasov, R.; Trushina, D. High-Capacity CaCO3 Containers: The Effect of Size on Drug Loading and Interaction with Cells. Med. Sci. Forum. 2022, 14, 87. [Google Scholar]
- Cubaud, T.; Mason, T.G. Interacting viscous instabilities in microfluidic systems. Soft Matter 2012, 8, 10573–10582. [Google Scholar] [CrossRef]
- Patiño-Nariño, E.A.; Galvis, A.F.; Pavanello, R.; Moshkalev, S.A. Numerical study of single bubble rising dynamics for the variability of moderate Reynolds and sidewalls influence: A bi-phase SPH approach. Eng. Anal. Bound. Elem. 2021, 129, 1–26. [Google Scholar] [CrossRef]
- Guignon, B.; Aparicio, C.; Sanz, P.D. Volumetric properties of pressure-transmitting fluids up to 350 MPa: Water, ethanol, ethylene glycol, propylene glycol, castor oil, silicon oil, and some of their binary mixture. J. Chem. Eng. Data 2010, 55, 3017–3023. [Google Scholar] [CrossRef]
- Gibaud, A.; Younas, D.; Matthews, L.; Narayanan, T.; Longkaew, K.; Hageberg, I.; Chushkin, Y.; Breiby, D.; Chattopadhyay, B. Insights into the precipitation kinetics of CaCO3 particles in the presence of polystyrene sulfonate using in situ small-angle X-ray scattering. J. Appl. Crystallogr. 2023, 56, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pancera, S.; Boyko, V.; Shukla, A.; Narayanan, T.; Huber, K. Evaluation of the particle growth of amorphous calcium carbonate in water by means of the porod invariant from SAXS. Langmuir 2010, 26, 17405–17412. [Google Scholar] [CrossRef] [PubMed]
- Bolze, J.; Peng, B.; Dingenouts, N.; Panine, P.; Narayanan, T.; Ballauff, M. Formation and growth of amorphous colloidal CaCO3 precursor particles as detected by time-resolved SAXS. Langmuir 2002, 18, 8364–8369. [Google Scholar] [CrossRef]
- Strumendo, M.; Segre, C.; Ilavsky, J.; Kuzmenko, I. A novel SAXS model for multi-texture systems: Application to CaCO3 calcination using in-situ USAXS-SAXS-WAXS. Appl. Mater. Today 2022, 29, 101568. [Google Scholar] [CrossRef]
- Natsi, P.; Rokidi, S.; Koutsoukos, P. Precipitation of Calcium Carbonate (CaCO3) in Water–Monoethylene Glycol Solutions. Ind. Eng. Chem. Res. 2019, 58, 4732–4743. [Google Scholar] [CrossRef]
- Lin, Y.H.; Singuru, M.M.R.; Marpaung, D.S.S.; Liao, W.C.; Chuang, M.C. Ethylene Glycol-Manipulated Syntheses of Calcium Carbonate Particles and DNA Capsules toward Efficient ATP-Responsive Cargo Release. ACS Appl. Bio Mater. 2023, 6, 3351–3360. [Google Scholar] [CrossRef]
- Parakhonskiy, B.V.; Haase, A.; Antolini, R. Sub-micrometer vaterite containers: Synthesis, substance loading, and release. Angew. Chem. 2012, 124, 1221–1223. [Google Scholar] [CrossRef]
- Parakhonskiy, B.; Svenskaya, Y.I.; Yashchenok, A.; Fattah, H.; Inozemtseva, O.; Tessarolo, F.; Antolini, R.; Gorin, D. Size controlled hydroxyapatite and calcium carbonate particles: Synthesis and their application as templates for SERS platform. Colloids Surfaces Biointerfaces 2014, 118, 243–248. [Google Scholar] [CrossRef]
- Flaten, E.M.; Seiersten, M.; Andreassen, J.P. Polymorphism and morphology of calcium carbonate precipitated in mixed solvents of ethylene glycol and water. J. Cryst. Growth 2009, 311, 3533–3538. [Google Scholar] [CrossRef]
- Rong, W.; Li, Z.; Zhang, W.; Sun, L. An improved CANNY edge detection algorithm. In Proceedings of the 2014 IEEE International Conference on Mechatronics and Automation, Tianjin, China, 3–6 August 2014; IEEE: Piscataway, NJ, USA, 2014; pp. 577–582. [Google Scholar]
- Hramm, O.; Bilous, N.; Ahekian, I. Configurable Cell Segmentation Solution Using Hough Circles Transform and Watershed Algorithm. In Proceedings of the 2019 IEEE 8th International Conference on Advanced Optoelectronics and Lasers (CAOL), Sozopol, Bulgaria, 6–8 September 2019; IEEE: Piscataway, NJ, USA, 2019; pp. 602–605. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthesis Type | Parameters | Results |
|---|---|---|
| One-phase with use of pristine reagent mixture | Concentration of 0.1, 0.33, 0.5, and 1 M | Diameter ∼ 3 m, FWHM ∼ 1 m |
| One-phase with use of EG and H2O reagent mixture | Concentration of 0.5 and 1 M | Diameter ∼ 2 m, FWHM ∼ 0.7 m |
| Two-phase with use of pristine reagent mixture reagent mixture | Concentration of 0.5 and 1 M | Diameter ∼ 0.7 m, FWHM ∼ 0.3 m |
| Two-phase with use of EG and H2O reagent mixture | Concentration of 0.5 and 1 M | Diameter ∼ 0.05 m, FWHM ∼ 0.01 m |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reznik, I.; Baranov, M.A.; Cherevkov, S.A.; Konarev, P.V.; Volkov, V.V.; Moshkalev, S.; Trushina, D.B. Microfluidic Vaterite Synthesis: Approaching the Nanoscale Particles. Nanomaterials 2023, 13, 3075. https://doi.org/10.3390/nano13233075
Reznik I, Baranov MA, Cherevkov SA, Konarev PV, Volkov VV, Moshkalev S, Trushina DB. Microfluidic Vaterite Synthesis: Approaching the Nanoscale Particles. Nanomaterials. 2023; 13(23):3075. https://doi.org/10.3390/nano13233075
Chicago/Turabian StyleReznik, Ivan, Mikhail A. Baranov, Sergei A. Cherevkov, Petr V. Konarev, Vladimir V. Volkov, Stanislav Moshkalev, and Daria B. Trushina. 2023. "Microfluidic Vaterite Synthesis: Approaching the Nanoscale Particles" Nanomaterials 13, no. 23: 3075. https://doi.org/10.3390/nano13233075