
Density Functional Theory Study of Methanol Steam Reforming on Pt3Sn(111) and the Promotion Effect of a Surface Hydroxy Group
 , and
, and
Abstract
:
1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. Adsorption Structures and Energies
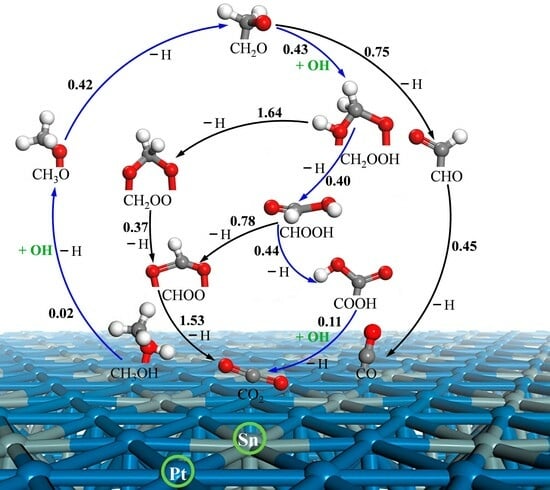
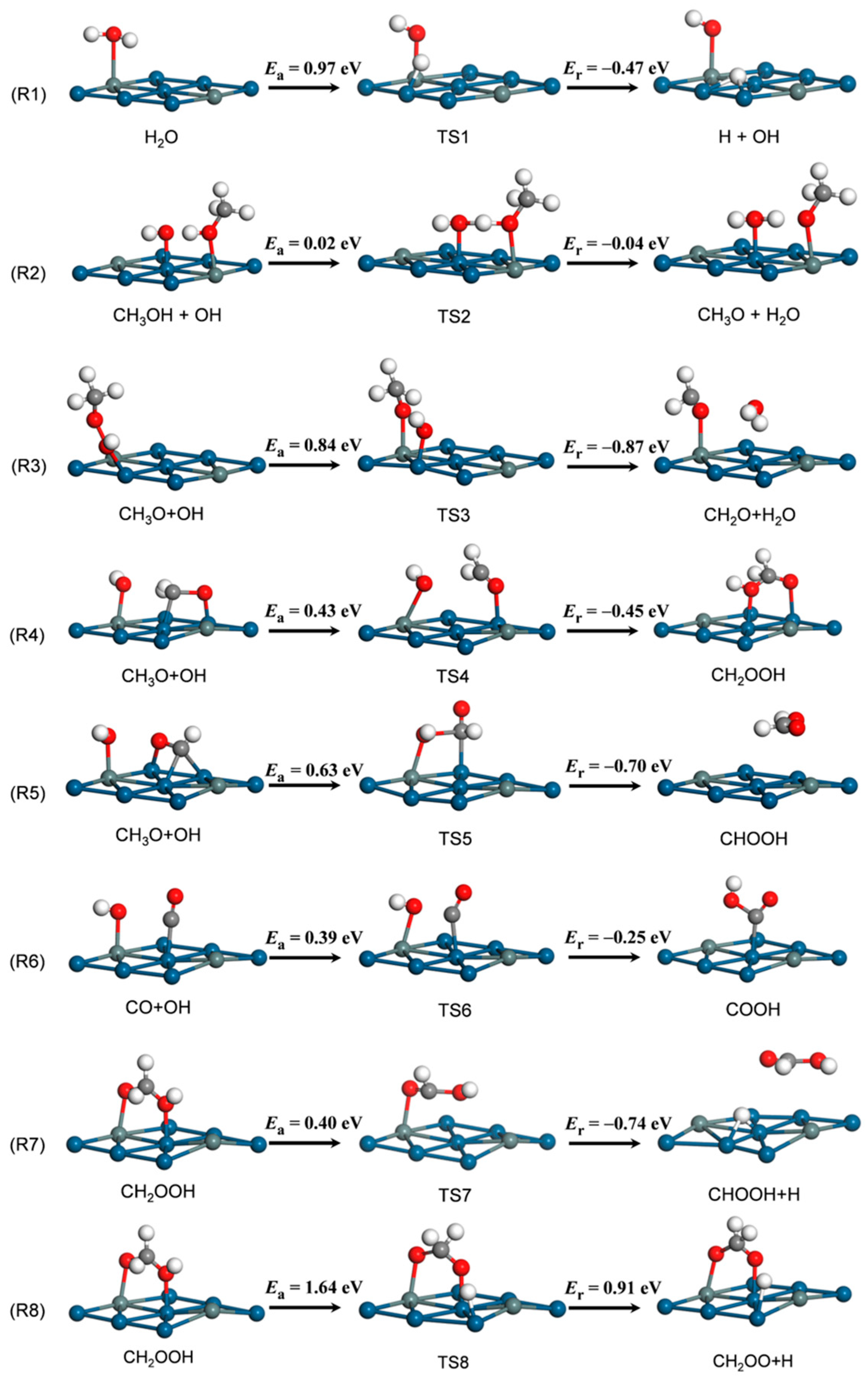
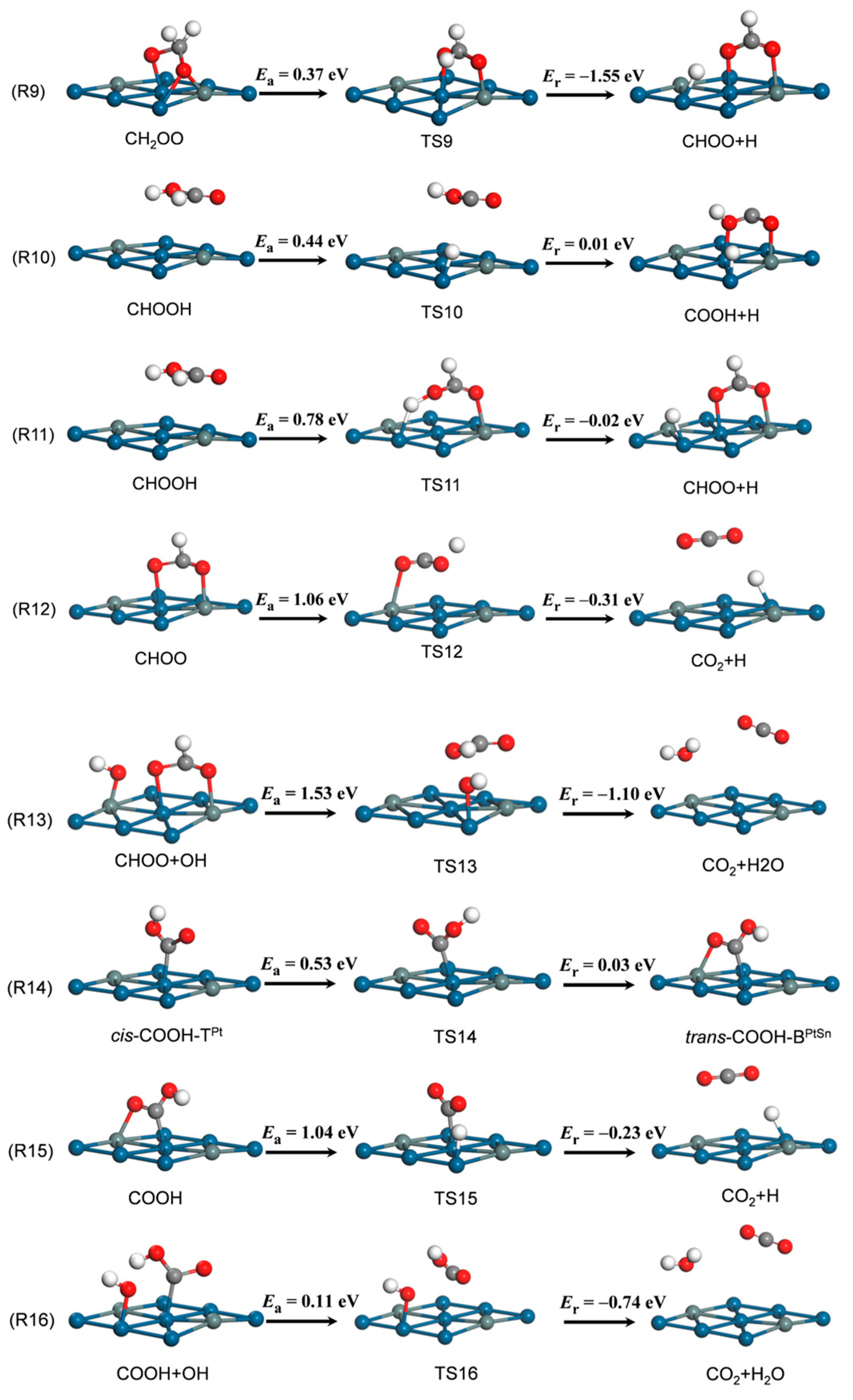
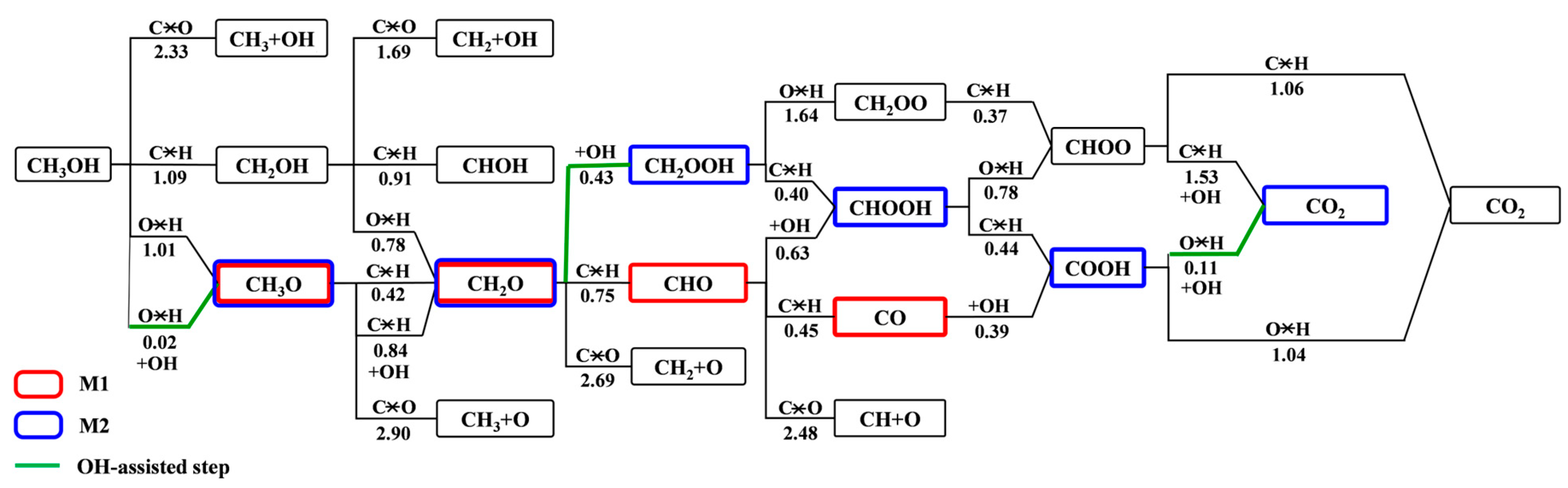
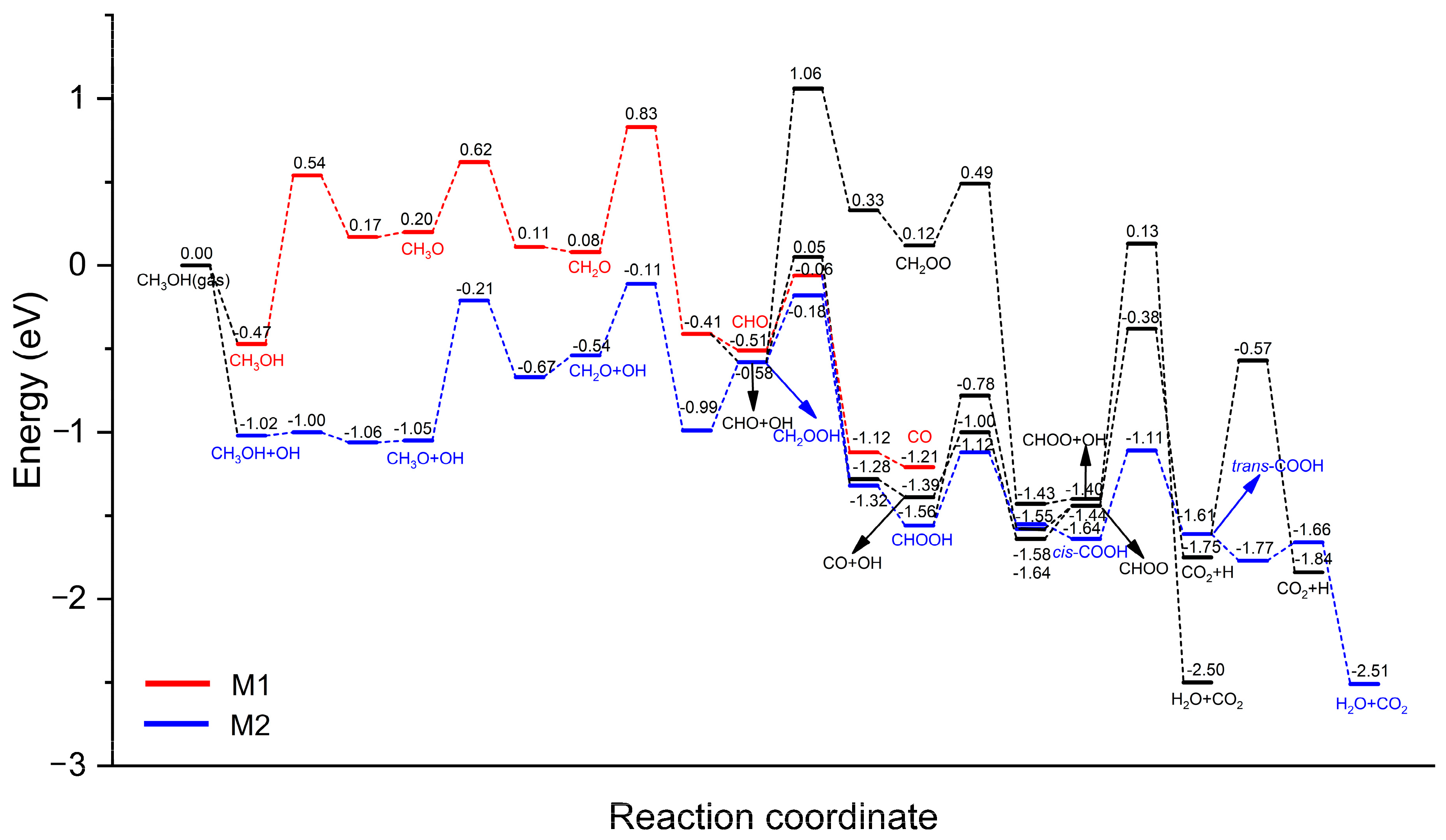
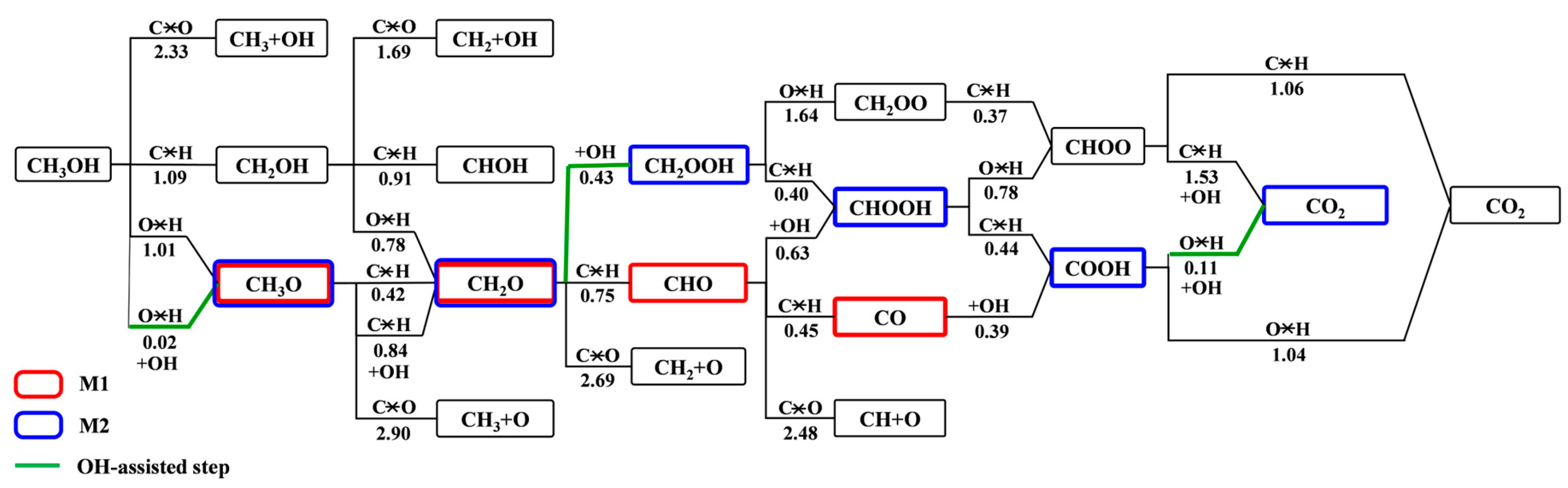
3.2. Elementary Reaction Steps
3.3. MSR Mechanisms
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Sá, S.; Silva, H.; Brandão, L.; Sousa, J.M.; Mendes, A. Catalysts for Methanol Steam Reforming—A Review. Appl. Catal. B Environ. 2010, 99, 43–57. [Google Scholar] [CrossRef]
- Song, S.Y.; Wang, X.; Li, S.L.; Wang, Z.; Zhu, Q.; Zhang, H.J. Pt Nanohelices with Highly Ordered Horizontal Pore Channels as Enhanced Photothermal Materials. Chem. Sci. 2015, 6, 6420–6424. [Google Scholar] [CrossRef] [PubMed]
- Antolini, E. Formation of Carbon-Supported PtM Alloys for Low Temperature Fuel Cells: A Review. Mater. Chem. Phys. 2003, 78, 563–573. [Google Scholar] [CrossRef]
- Zhao, L.M.; Wang, S.P.; Ding, Q.Y.; Xu, W.B.; Sang, P.P.; Chi, Y.H.; Lu, X.Q.; Guo, W.Y. The Oxidation of Methanol on PtRu(111): A Periodic Density Functional Theory Investigation. J. Phys. Chem. C 2015, 119, 20389–20400. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.B.; Song, S.Y.; Yang, X.G.; Wang, Z.; Jin, R.C.; Zhang, H.J. L-Arginine-Triggered Self-Assembly of CeO2 Nanosheaths on Palladium Nanoparticles in Water. Angew. Chem. Int. Ed. 2016, 55, 4542–4546. [Google Scholar] [CrossRef]
- Greeley, J.; Mavrikakis, M. A First-Principles Study of Methanol Decomposition on Pt(111). J. Am. Chem. Soc. 2002, 124, 7193–7201. [Google Scholar] [CrossRef]
- Hoseini, S.J.; Barzegar, Z.; Bahrami, M.; Roushani, M.; Rashidi, M. Organometallic Precursor Route for the Fabrication of PtSn Bimetallic Nanotubes and Pt3Sn/Reduced-graphene Oxide Nanohybrid Thin Films at Oilewater Interface and Study of Their Electrocatalytic Activity in Methanol Oxidation. J. Organomet. Chem. 2014, 769, 1–6. [Google Scholar] [CrossRef]
- Antolini, E.; Gonzalez, E.R. Effect of Synthesis Method and Structural Characteristics of Pt–Sn Fuel Cell Catalysts on the Electro-oxidation of CH3OH and CH3CH2OH in Acid Medium. Catal. Today 2011, 160, 28–38. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Liao, M.S.; Cabrera, C.R. Oxidation of Methanol on Platinum, Ruthenium and Mixed Pt–M Metals (M = Ru, Sn): A Theoretical Study. Surf. Sci. 2000, 463, 66–80. [Google Scholar] [CrossRef]
- Honma, I.; Toda, T. Temperature Dependence of Kinetics of Methanol Electro-oxidation on PtSn Alloys. J. Electrochem. Soc. 2003, 150, A1689–A1692. [Google Scholar] [CrossRef]
- Zhou, W.J.; Zhou, B.; Li, W.Z.; Zhou, Z.H.; Song, S.Q.; Sun, G.Q.; Xin, Q.; Douvartzides, S.; Goula, M.; Tsiakaras, P. Performance Comparison of Low-Temperature Direct Alcohol Fuel Cells with Different Anode Catalysts. J. Power Sources 2004, 126, 16–22. [Google Scholar] [CrossRef]
- Kim, M.; Pratt, S.J.; King, D.A. In Situ Characterization of the Surface Reaction between Chemisorbed Ammonia and Oxygen on Pt(100). J. Am. Chem. Soc. 2000, 122, 2409–2410. [Google Scholar] [CrossRef]
- Alcala, R.; Shabaker, J.W.; Huber, G.W.; Marco, A.; Sanchez-Castillo, M.A.; Dumesic, J.A. Experimental and DFT Studies of the Conversion of Ethanol and Acetic Acid on PtSn-Based Catalysts. J. Phys. Chem. B 2005, 109, 2074–2085. [Google Scholar] [CrossRef] [PubMed]
- Radael, G.N.; Oliveira, G.G.; Pontes, R.M. A DFT study of ethanol interaction with the bimetallic clusters of PtSn and its implications on reactivity. J. Mol. Graph. Model. 2023, 125, 108621. [Google Scholar] [CrossRef] [PubMed]
- García-Rodríguez, S.; Peña, M.A.; Fierro, J.L.G.; Rojas, S. Controlled Synthesis of Carbon-Supported Pt3Sn by Impregnation-reduction and Performance on the Electrooxidation of CO and Ethanol. J. Power Sources 2010, 195, 5564–5572. [Google Scholar] [CrossRef]
- Colmati, F.; Antolini, E.; Gonzalez, E.R. Pt–Sn/C Electrocatalysts for Methanol Oxidation Synthesized by Reduction with Formic Acid. Electrochim. Acta 2005, 50, 5496–5503. [Google Scholar] [CrossRef]
- Lim, D.H.; Choi, D.H.; Lee, W.D.; Lee, H.I. A New Synthesis of a Highly Dispersed and CO Tolerant PtSn/C Electrocatalyst for Low-Temperature Fuel Cell; its Electrocatalytic Activity and Long-term Durability. Appl. Catal. B 2009, 89, 484–493. [Google Scholar] [CrossRef]
- Cai, F.F.; Jessica, J.I.; Fu, Y.; Kong, W.; Zhang, J.; Sun, Y. Low-temperature Hydrogen Production from Methanol Steam Reforming on Zn-modified Pt/MoC Catalysts. Appl. Catal. B Environ. 2020, 264, 118500. [Google Scholar] [CrossRef]
- Conant, T.; Karim, A.; Lebarbier, V.; Wang, Y.; Girgsdies, F.; Schlögl, R.; Datye, A. Stability of Bimetallic Pd–Zn Catalysts for the Steam Reforming of Methanol. J. Catal. 2008, 257, 64–70. [Google Scholar] [CrossRef]
- Liu, Z.; Guo, B. Microwave Heated Polyol Synthesis of Carbon-supported PtSn Nanoparticles for Methanol Electrooxidation. Electrochem. Commun. 2006, 8, 83–90. [Google Scholar] [CrossRef]
- Wang, K.; Gasteiger, H.A.; Markovic, N.M.; Ross, P.N., Jr. On the Reaction Pathway for Methanol and Carbon Monoxide Electrooxidation on Pt-Sn Alloy versus Pt-Ru Alloy Surfaces. Electrochim. Acta 1996, 41, 2587–2593. [Google Scholar] [CrossRef]
- Trimm, D.L.; Önsan, Z.I. On Board Fuel Conversion for Hydrogen-fuel-cell-driven Vehicles. Catal. Rev. 2001, 43, 31–84. [Google Scholar] [CrossRef]
- Takezawa, N.; Iwasa, N. Steam Reforming and Dehydrogenation of Methanol: Difference in the Catalytic Functions of Copper and Group VIII Metals. Catal. Today 1997, 36, 45–56. [Google Scholar] [CrossRef]
- Lin, S.; Johnson, R.S.; Smith, G.K.; Xie, D.; Guo, H. Pathways for Methanol Steam Reforming involving Adsorbed Formaldehyde and Hydroxyl Intermediates on Cu(111): Density Functional Theory Studies. Phys. Chem. Chem. Phys. 2011, 13, 9622–9631. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Xie, D.Q.; Guo, H. Pathways of Methanol Steam Reforming on PdZn and Comparison with Cu. J. Phys. Chem. C 2011, 115, 20583–20589. [Google Scholar] [CrossRef]
- Luo, W.J.; Asthagiri, A. Density Functional Theory Study of Methanol Steam Reforming on Co(0001) and Co(111) Surfaces. J. Phys. Chem. C 2014, 118, 15274–15285. [Google Scholar] [CrossRef]
- Fajín, J.L.C.; Cordeiro, M.N.D.S. Insights into the Mechanism of Methanol Steam Reforming for Hydrogen Production over Ni–Cu-Based Catalysts. ACS Catal. 2022, 12, 512–526. [Google Scholar] [CrossRef]
- Lin, S.; Xie, D.Q.; Guo, H. Methyl Formate Pathway in Methanol Steam Reforming on Copper: Density Functional Calculations. ACS Catal. 2011, 1, 1263–1271. [Google Scholar] [CrossRef]
- Lin, S.; Xie, D.Q.; Guo, H. First-principles Study of the Methyl Formate Pathway of Methanol Steam Reforming on PdZn(111) with Comparison to Cu(111). J. Mol. Catal. A Chem. 2012, 356, 165–170. [Google Scholar] [CrossRef]
- Li, J.; Wan, Q.; Lin, G.Z.; Lin, S. DFT Study on the Catalytic Role of α-MoC(100) in Methanol Steam Reforming. J. Chem. Phys. 2022, 35, 639–646. [Google Scholar] [CrossRef]
- Huang, Y.; He, X.; Chen, Z.X. Density Functional Study of Methanol Decomposition on Clean and O or OH Adsorbed PdZn(111). J. Chem. Phys. 2013, 138, 184701. [Google Scholar] [CrossRef]
- Delley, B. An All-electron Numerical Method for Solving the Local Density Functional for Polyatomic Molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. Fast Calculation of Electrostatics in Crystals and Large Molecules. J. Chem. Phys. 1996, 100, 6107–6110. [Google Scholar] [CrossRef]
- Delley, B. From Molecules to Solids with the DMol3 Approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Perdew, J.P. Density Functional Approximation for the Correlation Energy of the Inhomogeneous Electron Gas. Phys. Rev. B Condens. Matter Mater. Phys. 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Wang, Y. Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy. Phys. Rev. B Condens. Matter Mater. Phys. 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Delley, B. Hardness Conserving Semilocal Pseudopotentials. Phys. Rev. B Condens. Matter Mater. Phys. 2002, 66, 155125–155133. [Google Scholar] [CrossRef]
- Hoheisel, M.; Speller, S.; Kuntze, J.; Atrei, A.; Bardi, U.; Heiland, W. Structure of Pt3Sn(110) Studied by Scanning Tunneling Microscopy. Phys. Rev. B Condens. Matter Mater. Phys. 2001, 63, 245403. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Zhu, H.Y.; Guo, W.Y.; Jiang, R.B.; Zhao, L.M.; Lu, X.Q.; Li, M.; Fu, D.L.; Shan, H.H. Decomposition of Methanthiol on Pt(111): A Density Functional Investigation. Langmuir 2010, 26, 12017–12025. [Google Scholar] [CrossRef]
- Lu, X.Q.; Deng, Z.G.; Guo, C.; Wang, W.L.; Wei, S.X.; Ng, S.P.; Chen, X.F.; Ding, N.; Guo, W.Y.; Wu, C.M.L. Methanol Oxidation on Pt3Sn (111) for Direct Methanol Fuel Cells: Methanol Decomposition. ACS Appl. Mater. Interfaces 2016, 8, 12194–12204. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Lipscomb, W.N. The Synchronous-transit Method for Determining Reaction Pathways and Locating Molecular Transition States. Chem. Phys. Lett. 1977, 49, 225–232. [Google Scholar] [CrossRef]
- Ma, Y.; Hernández, L.; Guadarrama-Pérez, C.; Balbuena, P.B. Ethanol Reforming on Co(0001) Surfaces: A Density Functional Theory Study. J. Phys. Chem. A 2012, 116, 1409–1416. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.X.; Wang, Y.F.; Zhang, R.X. Theoretical Insight into Hydrogen Production from Methanol Steam Reforming on Pt(111). Mol. Catal. 2022, 532, 112745. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
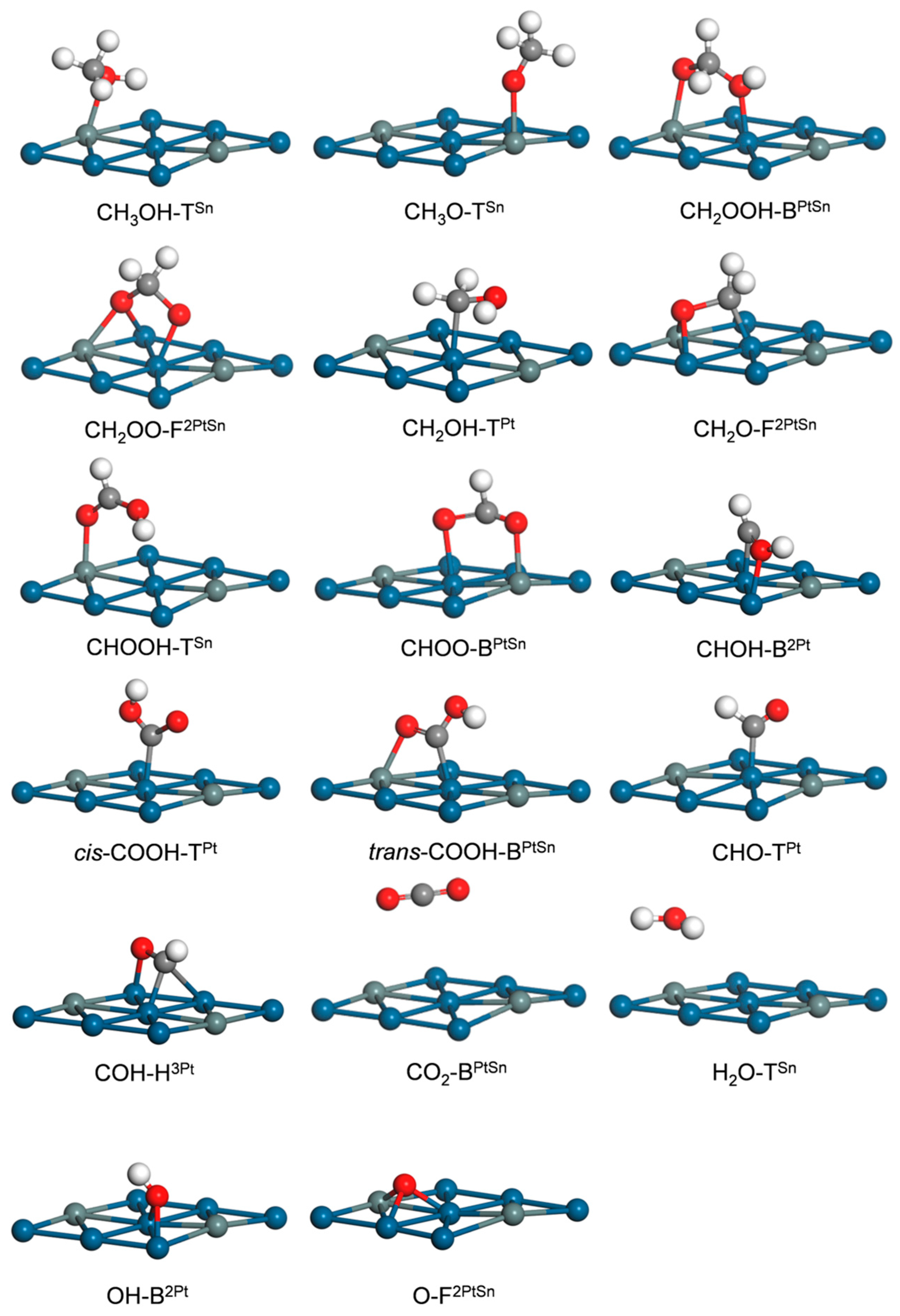
| Species | Site | Mode | dC/O-Pt/Sn | Eads |
|---|---|---|---|---|
| CH3OH | TSn | η1(O) | 2.64 | 0.47 |
| CH3O | TSn | η1(O) | 2.05 | 1.71 |
| CH2OOH | BPtSn | η1(O)-η1(O) | 2.15, 2.31 | 1.89 |
| CH2OO | F2PtSn | η2(O)-η1(O) | 2.09, 2.26, 2.27 | 3.24 |
| CH2OH | TPt | η1(C) | 2.14 | 1.94 |
| CH2O | F2PtSn | η1(C)-η2(O) | 2.13, 2.28, 2.43 | 0.38 |
| HCOOH | TSn | η1(O) | 2.58 | 0.49 |
| CHOO | BPtSn | η1(O)-η1(O) | 2.17, 2.29 | 2.52 |
| CHOH | B2Pt | η2(C) | 2.09, 2.12 | 3.14 |
| cis-COOH | TPt | η1(C) | 2.03 | 2.48 |
| trans-COOH | BPtSn | η1(C)-η1(O) | 2.04, 2.55 | 2.41 |
| CHO | TPt | η1(C) | 2.01 | 2.28 |
| COH | H3Pt | η3(C) | 2.04, 2.06, 2.11 | 4.05 |
| CO2 | BPtSn | - | - | 0.11 |
| H2O | TSn | - | - | 0.01 |
| OH | B2Pt | η2(O) | 2.23, 2.24 | 2.51 |
| O | F2PtSn | η3(O) | 2.13, 2.14, 2.14 | 4.12 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, P.; Zhu, H.; Sun, Q.; Li, M.; Liu, D.; Li, R.; Lu, X.; Zhao, W.; Chi, Y.; Ren, H.; et al. Density Functional Theory Study of Methanol Steam Reforming on Pt3Sn(111) and the Promotion Effect of a Surface Hydroxy Group. Nanomaterials 2024, 14, 318. https://doi.org/10.3390/nano14030318
He P, Zhu H, Sun Q, Li M, Liu D, Li R, Lu X, Zhao W, Chi Y, Ren H, et al. Density Functional Theory Study of Methanol Steam Reforming on Pt3Sn(111) and the Promotion Effect of a Surface Hydroxy Group. Nanomaterials. 2024; 14(3):318. https://doi.org/10.3390/nano14030318
Chicago/Turabian StyleHe, Ping, Houyu Zhu, Qianyao Sun, Ming Li, Dongyuan Liu, Rui Li, Xiaoqing Lu, Wen Zhao, Yuhua Chi, Hao Ren, and et al. 2024. "Density Functional Theory Study of Methanol Steam Reforming on Pt3Sn(111) and the Promotion Effect of a Surface Hydroxy Group" Nanomaterials 14, no. 3: 318. https://doi.org/10.3390/nano14030318