Cycle Stability and Hydration Behavior of Magnesium Oxide and Its Dependence on the Precursor-Related Particle Morphology

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
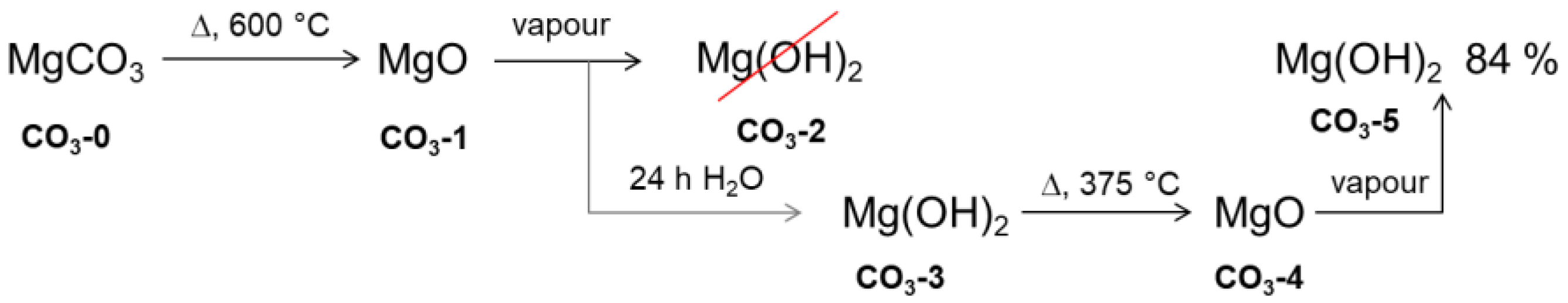
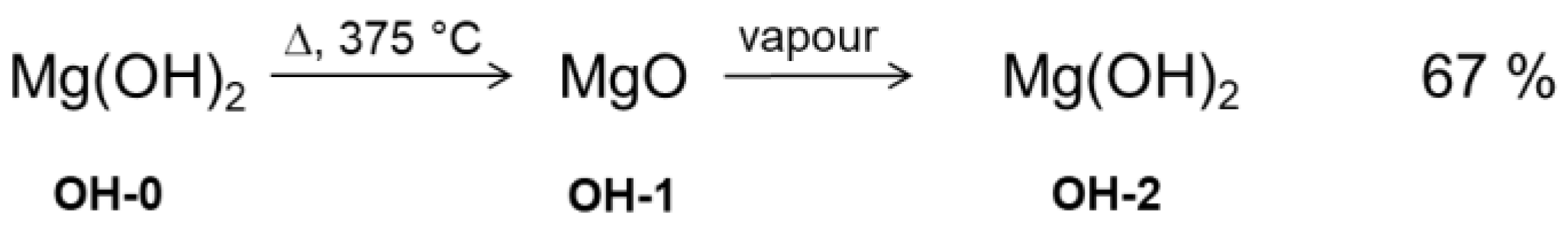
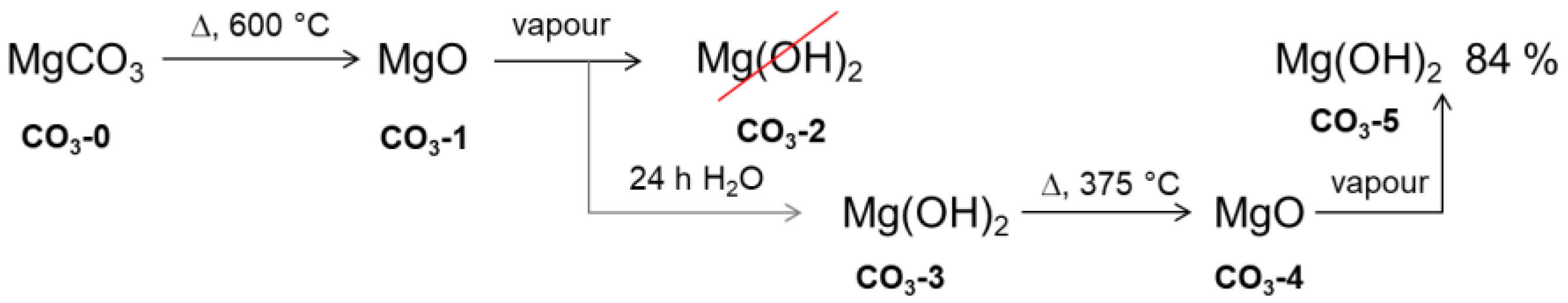
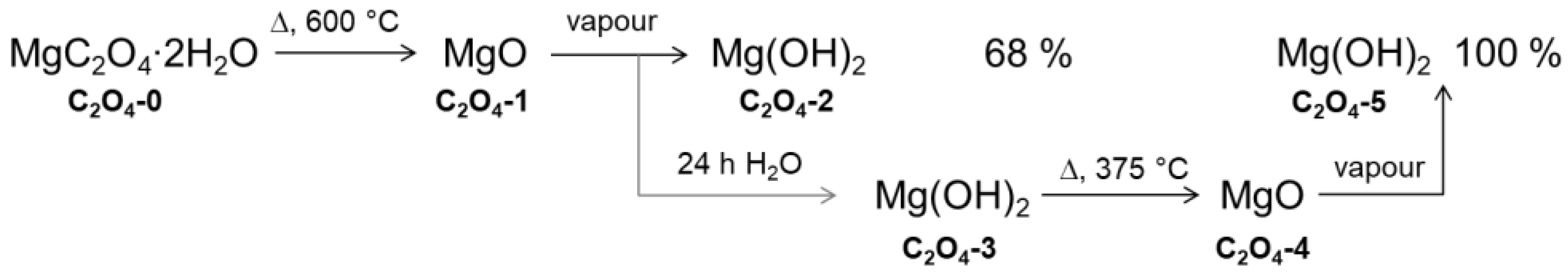
2.1. Material
2.2. BET Surface
2.3. Powder X-ray Diffraction with In-Situ Hydration (P-XRD)
2.4. Scanning Electron Microscopy (SEM)
2.5. Small-Angle X-ray Scattering (SAXS)
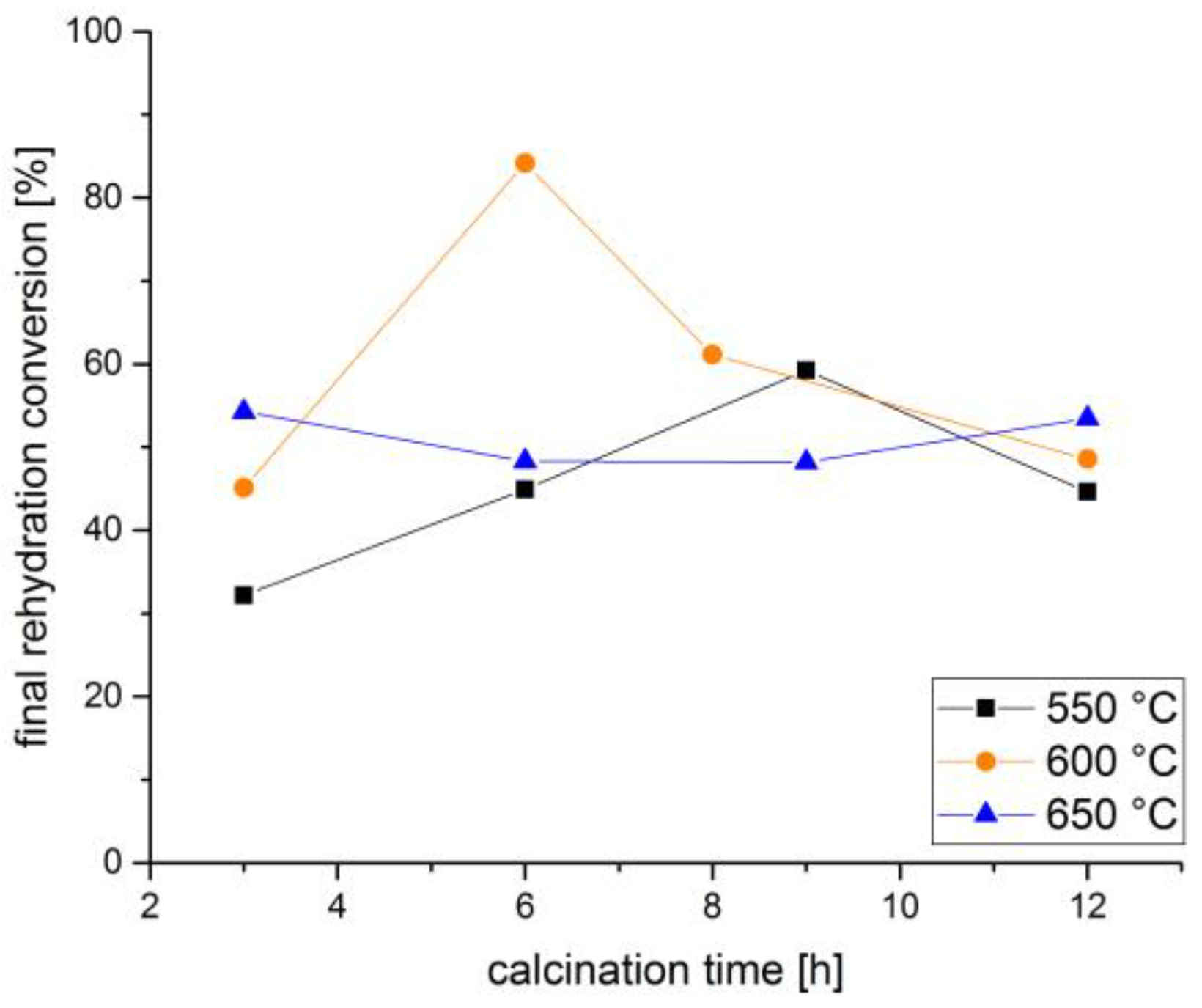
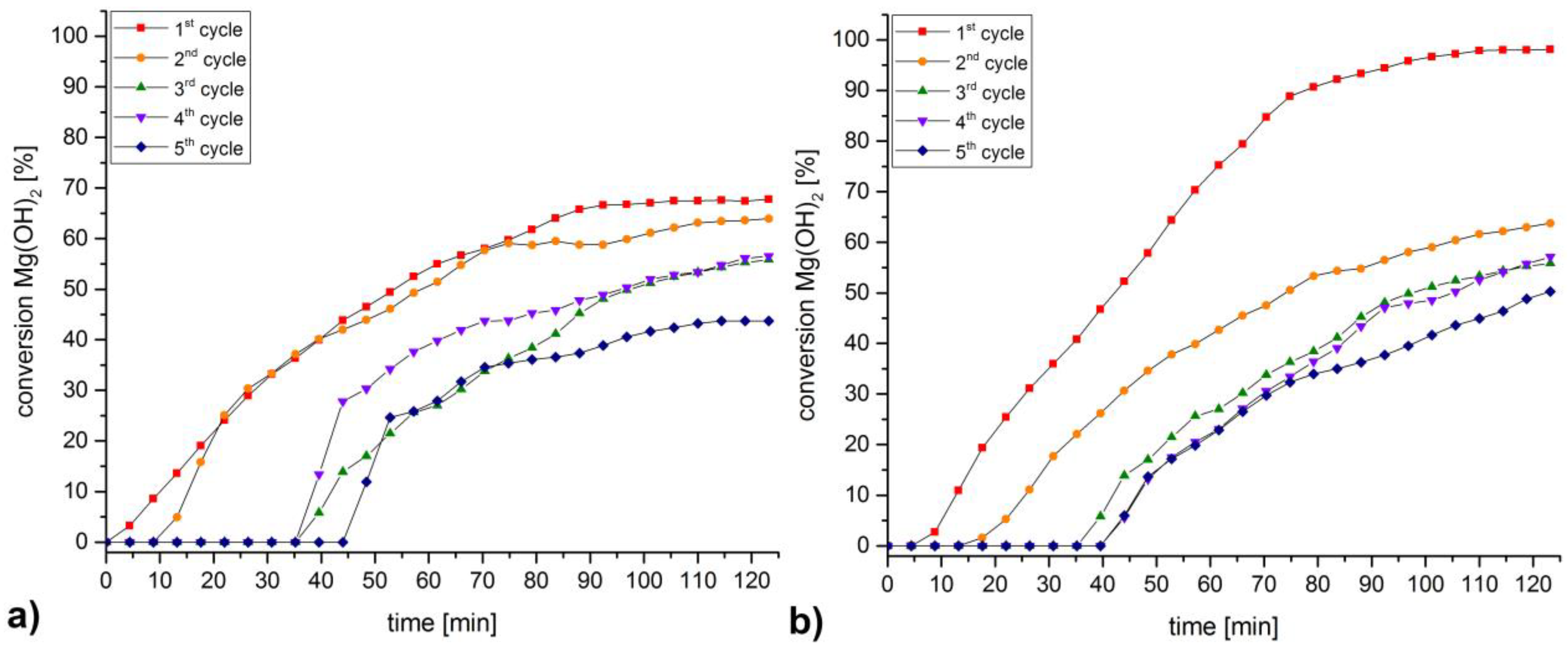
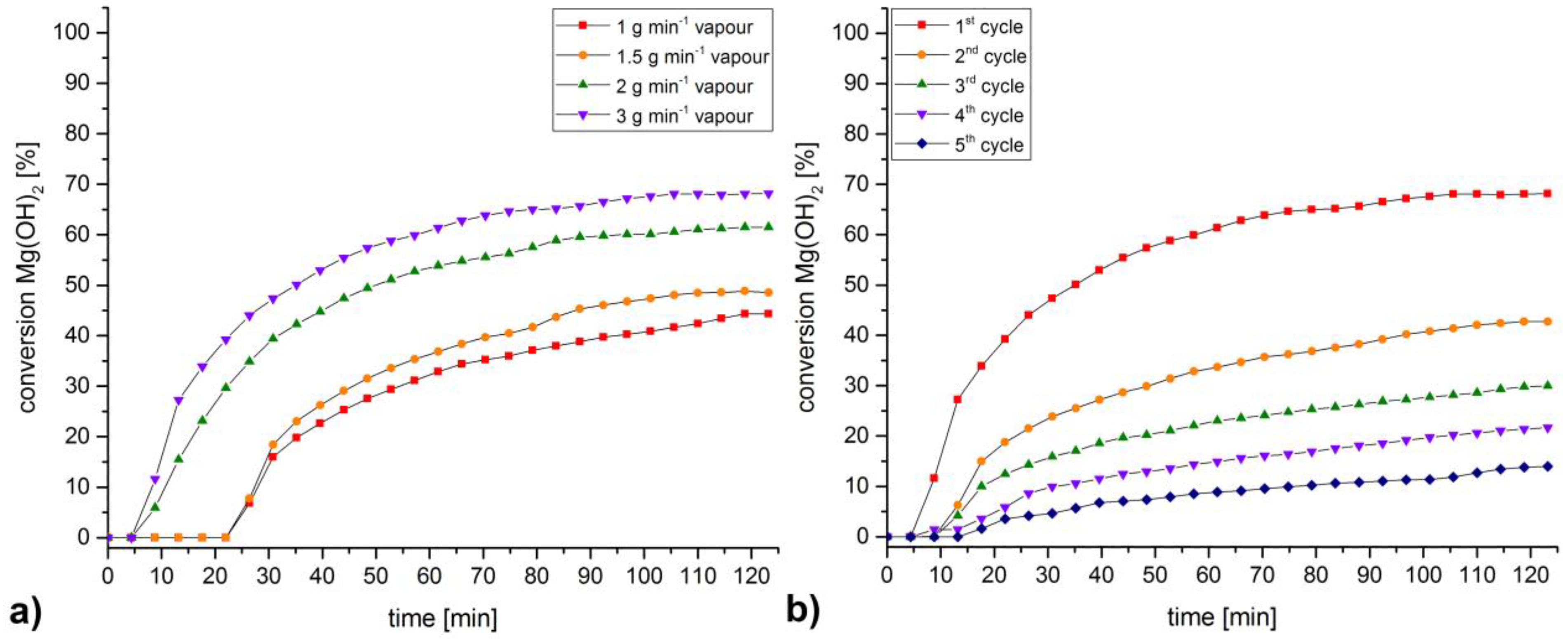
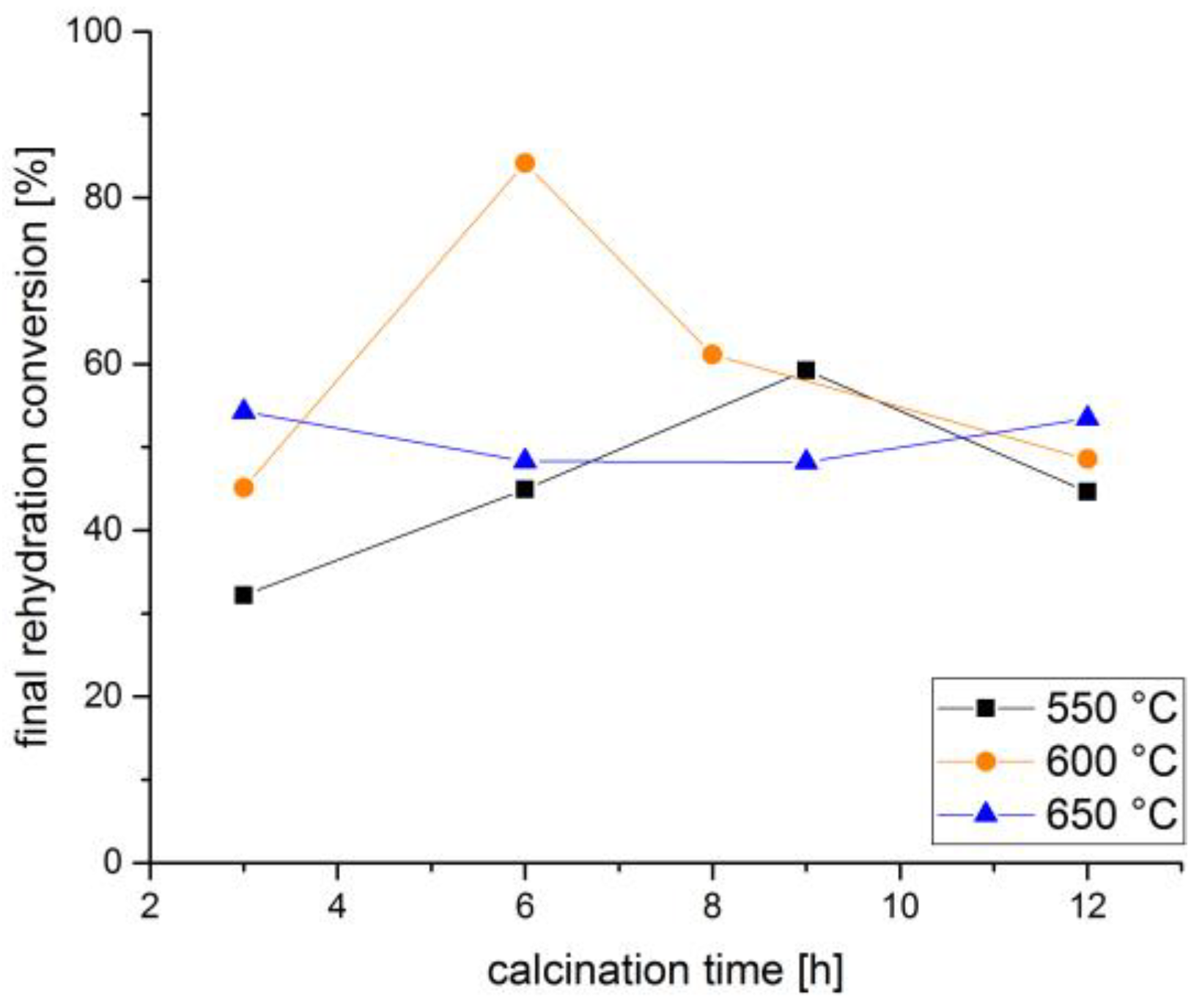
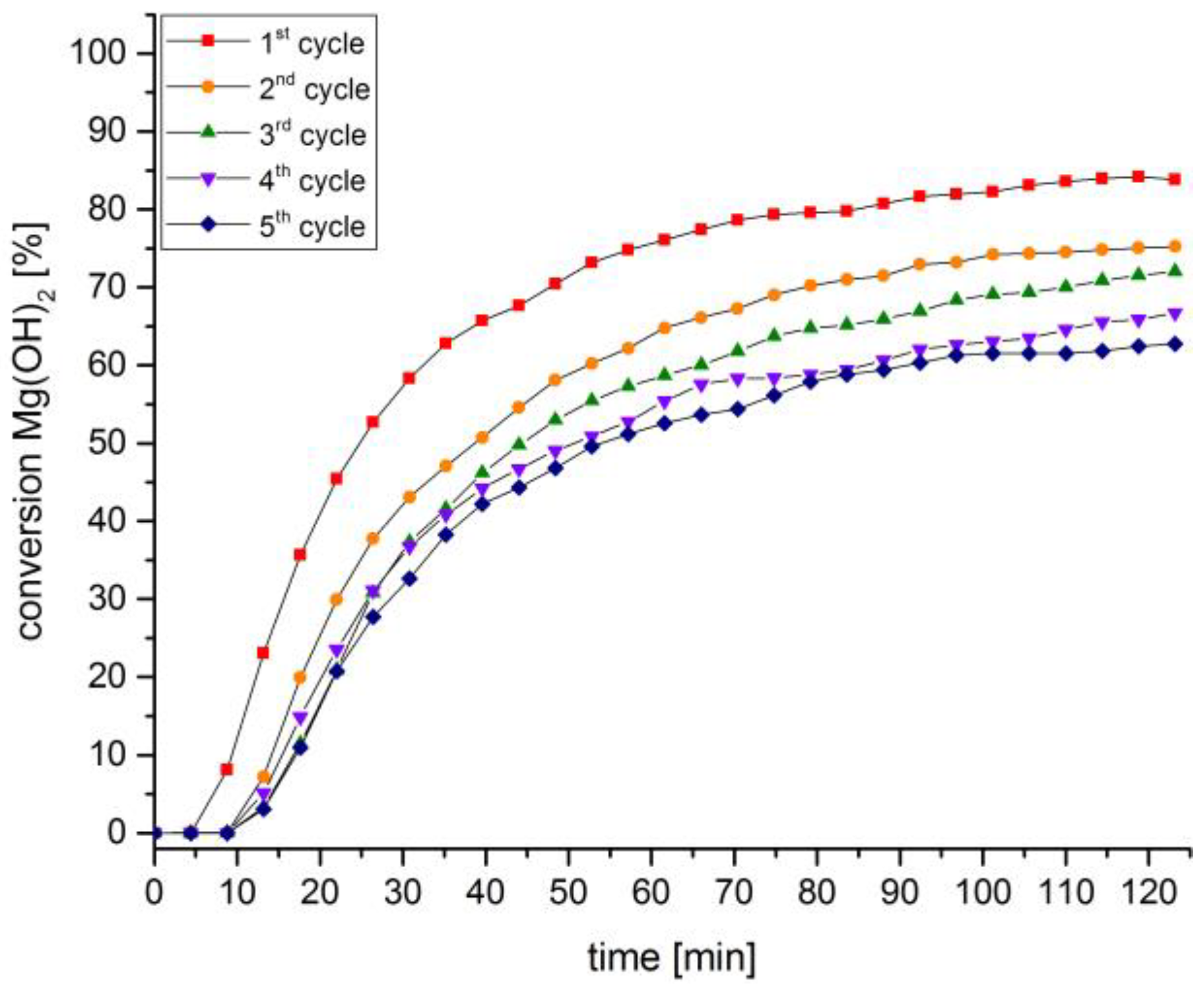
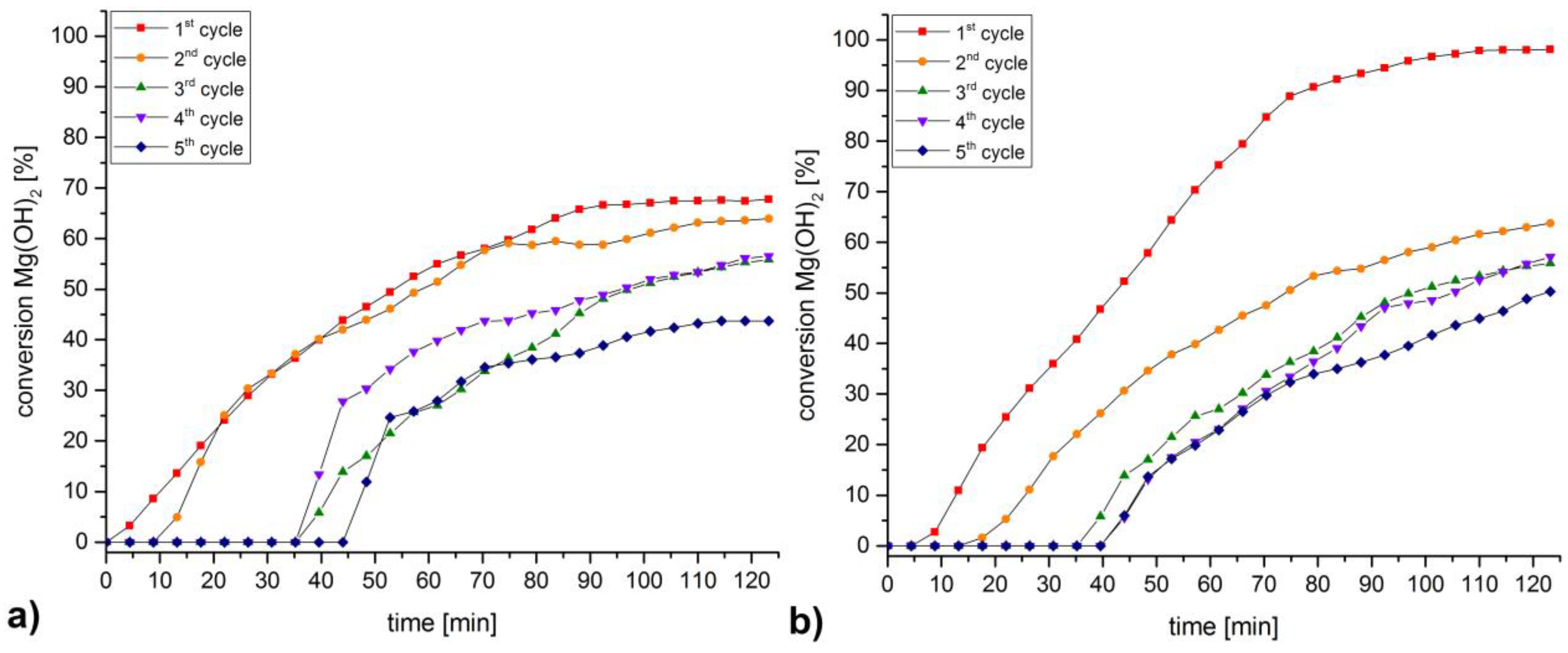
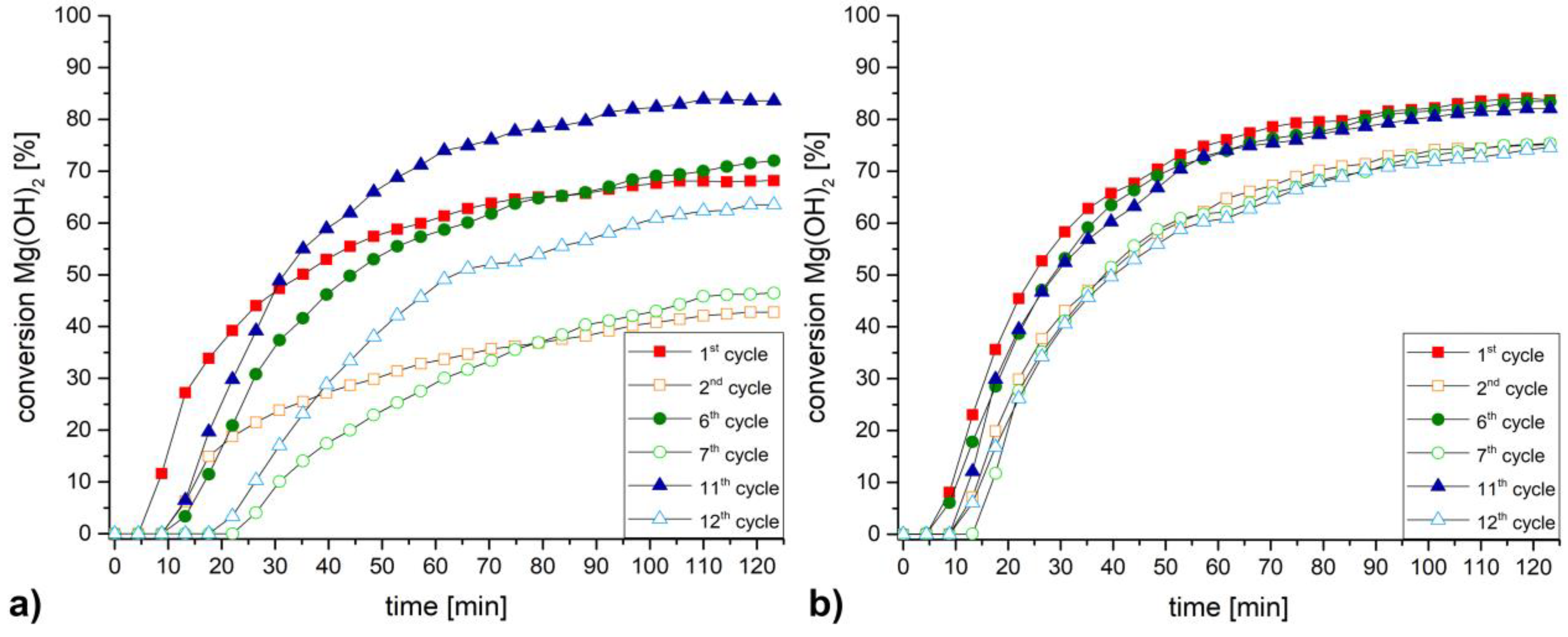
3. Discussion and Results
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Rahm, D. Sustainable Energy and the States, Essay on Politics Markets and Leadership, 1st ed.; McFarland: Jefferson, NC, USA, 2002. [Google Scholar]
- IEA. Heating without Global Warming: Market Developments and Policy Considerations for Renewable Heat. Available online: https://www.google.com.hk/url?sa=t&rct=j&q=&esrc=s&source=web&cd=1&cad=rja&uact=8&ved=2ahUKEwiWq76i5-ndAhULUd4KHYqlAxoQFjAAegQICRAC&url=https%3A%2F%2Fwww.iea.org%2Fpublications%2Ffreepublications%2Fpublication%2FFeaturedInsight_HeatingWithoutGlobalWarming_FINAL.pdf&usg=AOvVaw1PEQh4uZihy8UNUEKlI6EJ (accessed on 31 August 2018).
- IEA. Co-Generation and Renewables. Solutions for a Low-Carbon Energy Future. Available online: https://www.iea.org/publications/freepublications/publication/co-generation-and-renewables-solutions-for-a-low-carbon-energy-future.html (accessed on 31 August 2018).
- Bauer, T.; Steinmann, W.-D.; Laing, D.; Tamme, R. Thermal energy storage materials and systems. Annu. Rev. Heat Transf. 2012, 15, 131–177. [Google Scholar] [CrossRef]
- Hasnain, S.M. Review on sustainable thermal energy storage technologies, part i: Heat storage materials and techniques. Energy Convers. Manag. 1998, 39, 1127–1138. [Google Scholar] [CrossRef]
- Xu, J.; Wang, R.Z.; Li, Y. A review of available technologies for seasonal thermal energy storage. Sol. Energy 2014, 103, 610–638. [Google Scholar] [CrossRef]
- Zhang, H.; Baeyens, J.; Cáceres, G.; Degrève, J.; Lv, Y. Thermal energy storage: Recent developments and practical aspects. Prog. Energy Combust. Sci. 2016, 53, 1–40. [Google Scholar] [CrossRef]
- Cabeza, L.F.; Castell, A.; Barreneche, C.; de Gracia, A.; Fernández, A.I. Materials used as PCM in thermal energy storage in buildings: A review. Renew. Sustain. Energy Rev. 2011, 15, 1675–1695. [Google Scholar] [CrossRef]
- Dinker, A.; Agarwal, M.; Agarwal, G.D. Heat storage materials, geometry and applications: A review. J. Energy Inst. 2015, 90, 1–11. [Google Scholar] [CrossRef]
- Abedin, A.H.; Rosen, M.R. A critical review of thermochemical energy storage systems. Open Renew. Energy J. 2011, 4, 42–46. [Google Scholar] [CrossRef]
- Cot-Gores, J.; Castell, A.; Cabeza, L.F. Thermochemical energy storage and conversion: A-state-of-the-art review of the experimental research under practical conditions. Renew. Sustain. Energy Rev. 2012, 16, 5207–5224. [Google Scholar] [CrossRef]
- Solé, A.; Fontanet, X.; Barreneche, C.; Martorell, I.; Fernández, A.I.; Cabeza, L.F. Parameters to take into account when developing a new thermochemical energy storage system. Energy Procedia 2012, 30, 380–387. [Google Scholar] [CrossRef]
- Yan, T.; Wang, R.Z.; Li, T.X.; Wang, L.W.; Fred, I.T. A review of promising candidate reactions for chemical heat storage. Renew. Sustain. Energy Rev. 2015, 43, 13–31. [Google Scholar] [CrossRef]
- Pan, Z.; Zhao, C.Y. Dehydration/hydration of mgo/h2o chemical thermal storage system. Energy 2015, 82, 611–618. [Google Scholar] [CrossRef]
- Kuleci, H.; Schmidt, C.; Rybacki, E.; Petrishcheva, E.; Abart, R. Hydration of periclase at 350 °C to 620 °C and 200 mpa: Experimental calibration of reaction rate. Mineral. Petrol. 2015, 110, 1–10. [Google Scholar] [CrossRef]
- Myagmarjav, O.; Ryu, J.; Kato, Y. Dehydration kinetic study of a chemical heat storage material with lithium bromide for a magnesium oxide/water chemical heat pump. Prog. Nucl. Energy 2015, 82, 153–158. [Google Scholar] [CrossRef]
- Birchal, V.S.; Rocha, S.D.F.; Mansur, M.B.; Ciminelli, V.S.T. A simplified mechanistic analysis of the hydration of magnesia. Can. J. Chem. Eng. 2001, 79, 507–511. [Google Scholar] [CrossRef]
- Zhou, S. Hydration Mechanisms of Magnesia-Based Refractory Bricks. Ph.D. Thesis, The University of Brithish Columbia, Vancouver, BC, Canada, December 2004. [Google Scholar]
- Ishitobi, H.; Hirao, N.; Ryu, J.; Kato, Y. Evaluation of heat output densities of lithium chloride-modified magnesium hydroxide for thermochemical energy storage. Ind. Eng. Chem. Res. 2013, 52, 5321–5325. [Google Scholar] [CrossRef]
- Myagmarjav, O.; Ryu, J.; Kato, Y. Lithium bromide-mediated reaction performance enhancement of a chemical heat-storage material for magnesium oxide/water chemical heat pumps. Appl. Therm. Eng. 2014, 63, 170–176. [Google Scholar] [CrossRef]
- Shkatulov, A.; Ryu, J.; Kato, Y.; Aristov, Y. Composite material “Mg(OH)2/vermiculite”: A promising new candidate for storage of middle temperature heat. Energy 2012, 44, 1028–1034. [Google Scholar] [CrossRef]
- Zamengo, M.; Ryu, J.; Kato, Y. Composite block of magnesium hydroxide – expanded graphite for chemical heat storage and heat pump. Appl. Therm. Eng. 2014, 69, 29–38. [Google Scholar] [CrossRef]
- Müller, D.; Knoll, C.; Ruh, T.; Artner, W.; Welch, J.M.; Peterlik, H.; Eitenberger, E.; Friedbacher, G.; Harasek, M.; Blaha, P.; et al. Calcium doping facilitates water dissociation in magnesium oxide. Adv. Sustain. Syst. 2018, 2, 1700096. [Google Scholar] [CrossRef]
- Kato, Y.; Sasaki, Y.; Yoshizawa, Y. Magnesium oxide/water chemical heat pump to enhance energy utilization of a cogeneration system. Energy 2005, 30, 2144–2155. [Google Scholar] [CrossRef]
- Morozov, S.A. Synthesis of porous magnesium oxide by thermal decomposition of basic magnesium carbonate. Russ. J. Gen. Chem. 2003, 73, 37–42. [Google Scholar] [CrossRef]
- Shand, M.A. The Chemistry and Technology of Magnesia; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2006; Available online: https://www.wiley.com/en-us/The+Chemistry+and+Technology+of+Magnesia-p-9780471656036 (accessed on 31 August 2018).
- Criado, Y.A.; Alonso, M.; Abanades, J.C. Kinetics of the CaO/Ca(OH)2 hydration/dehydration reaction for thermochemical energy storage applications. Ind. Eng. Chem. Res. 2014, 53, 12594–12601. [Google Scholar] [CrossRef]
- Müller, D.; Knoll, C.; Artner, W.; Welch, J.M.; Freiberger, N.; Nilica, R.; Eitenberger, E.; Friedbacher, G.; Harasek, M.; Hradil, K.; et al. Influence of the particle morphology on cycle stability and hydration behavior of magnesium oxide. Appl. Energy 2017. submitted. [Google Scholar]
- Hu, X.L.; Carrasco, J.; Klimeš, J.; Michaelides, A. Trends in water monomer adsorption and dissociation on flat insulating surfaces. Phys. Chem. Chem. Phys. 2011, 13, 12447–12453. [Google Scholar] [CrossRef] [PubMed]
- Stankic, S.; Bernardi, J.; Diwald, O.; Knözinger, E. Optical surface properties and morphology of Mgo and Cao nanocrystals. J. Phys. Chem. B 2006, 110, 13866–13871. [Google Scholar] [CrossRef] [PubMed]
- Kazimirov, V.Y.; Smirnov, M.B.; Bourgeois, L.; Guerlou-Demourgues, L.; Servant, L.; Balagurov, A.M.; Natkaniec, I.; Khasanova, N.R.; Antipov, E.V. Atomic structure and lattice dynamics of Ni and Mg hydroxides. Solid State Ion. 2010, 181, 1764–1770. [Google Scholar] [CrossRef]
- Effenberger, H.; Mereiter, K.; Zemann, J. Crystal structure refinements of magnesite, calcite, rhodochrosite, siderite, smithonite, and dolomite, with discussion of some aspects of the stereochemistry of calcite type carbonates. Zeitschrift für Kristallographie–Cryst. Mater. 1981, 156. [Google Scholar] [CrossRef]
- Chen, X.-A.; Song, F.-P.; Chang, X.-A.; Zang, H.-G.; Xiao, W.-Q. A new polymorph of magnesium oxalate dihydrate. Acta Crystallogr. Sect. E Struct. Rep. Online 2008, 64, m863. [Google Scholar] [CrossRef] [PubMed]
- Boiocchi, M.; Caucia, F.; Merli, M.; Prella, D.; Ungaretti, L. Crystal-chemical reasons for the immiscibility of periclase and wüstite under lithospheric p,t conditions. Eur. J. Mineral. 2001, 13, 871–881. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Degen, T.; Sadki, M.; Bron, E.; König, U.; Nénert, G. The highscore suite. Powder Diffr. 2014, 29, S13–S18. [Google Scholar] [CrossRef]
- Beaucage, G. Approximations leading to a unified exponential/power-law approach to small-angle scattering. J. Appl. Crystallogr. 1995, 28, 717–728. [Google Scholar] [CrossRef]
- Kinning, D.J.; Thomas, E.L. Hard-sphere interactions between spherical domains in diblock copolymers. Macromolecules 1984, 17, 1712–1718. [Google Scholar] [CrossRef]
- Pabisch, S.; Feichtenschlager, B.; Kickelbick, G.; Peterlik, H. Effect of interparticle interactions on size determination of zirconia and silica based systems—A comparison of SAXS, DLS, BET, XRD and TEM. Chem. Phys. Lett. 2012, 521, 91–97. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
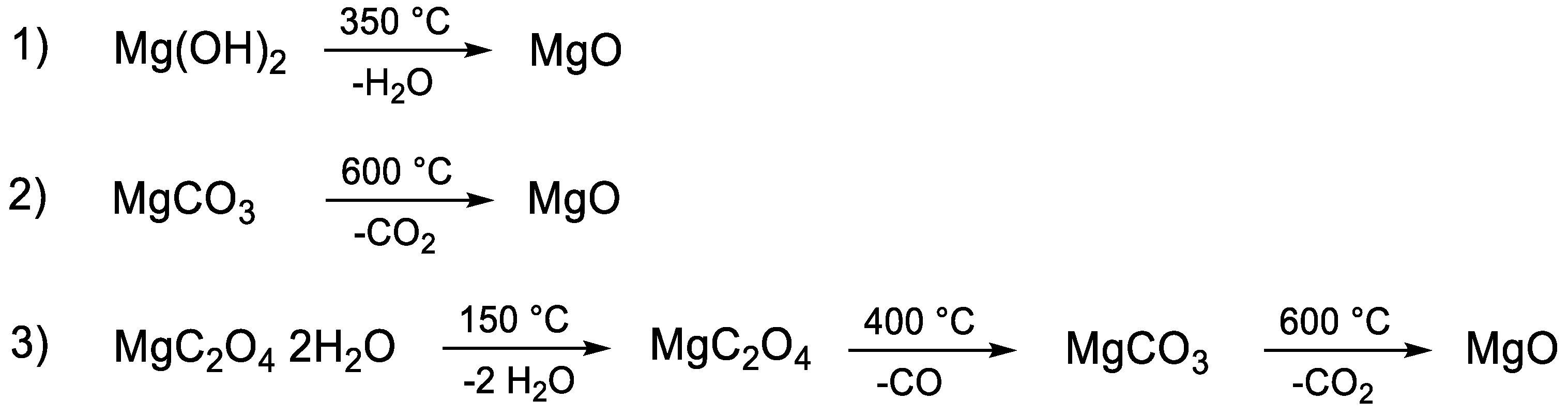{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mg(OH)2 [31] | MgCO3 [32] | MgC2O4·2H2O [33] | MgO [34] | |
|---|---|---|---|---|
| Space group | Pm1 (164) | Rc (167) | Fddd (70) | Fmm (225) |
| a [Å] | 3.1486(1) | 4.637(1) | 5.3940(11) | 4.2113(5) |
| b [Å] | 3.1486(1 | 4.637(1) | 12.691(3) | 4.2113(5) |
| c [Å] | 4.7713(1) | 15.023(3) | 15.399(3) | 4.2113(5) |
| α [°] | 90 | 90 | 90 | 90 |
| γ [°] | 120 | 120 | 90 | 90 |
| V [Å3] | 40.96 | 279.74 | 1054.14 | 74.69 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gravogl, G.; Knoll, C.; Welch, J.M.; Artner, W.; Freiberger, N.; Nilica, R.; Eitenberger, E.; Friedbacher, G.; Harasek, M.; Werner, A.; et al. Cycle Stability and Hydration Behavior of Magnesium Oxide and Its Dependence on the Precursor-Related Particle Morphology. Nanomaterials 2018, 8, 795. https://doi.org/10.3390/nano8100795
Gravogl G, Knoll C, Welch JM, Artner W, Freiberger N, Nilica R, Eitenberger E, Friedbacher G, Harasek M, Werner A, et al. Cycle Stability and Hydration Behavior of Magnesium Oxide and Its Dependence on the Precursor-Related Particle Morphology. Nanomaterials. 2018; 8(10):795. https://doi.org/10.3390/nano8100795
Chicago/Turabian StyleGravogl, Georg, Christian Knoll, Jan M. Welch, Werner Artner, Norbert Freiberger, Roland Nilica, Elisabeth Eitenberger, Gernot Friedbacher, Michael Harasek, Andreas Werner, and et al. 2018. "Cycle Stability and Hydration Behavior of Magnesium Oxide and Its Dependence on the Precursor-Related Particle Morphology" Nanomaterials 8, no. 10: 795. https://doi.org/10.3390/nano8100795