First-Principles Study on the Structural and Electronic Properties of Monolayer MoS2 with S-Vacancy under Uniaxial Tensile Strain


Abstract
:1. Introduction
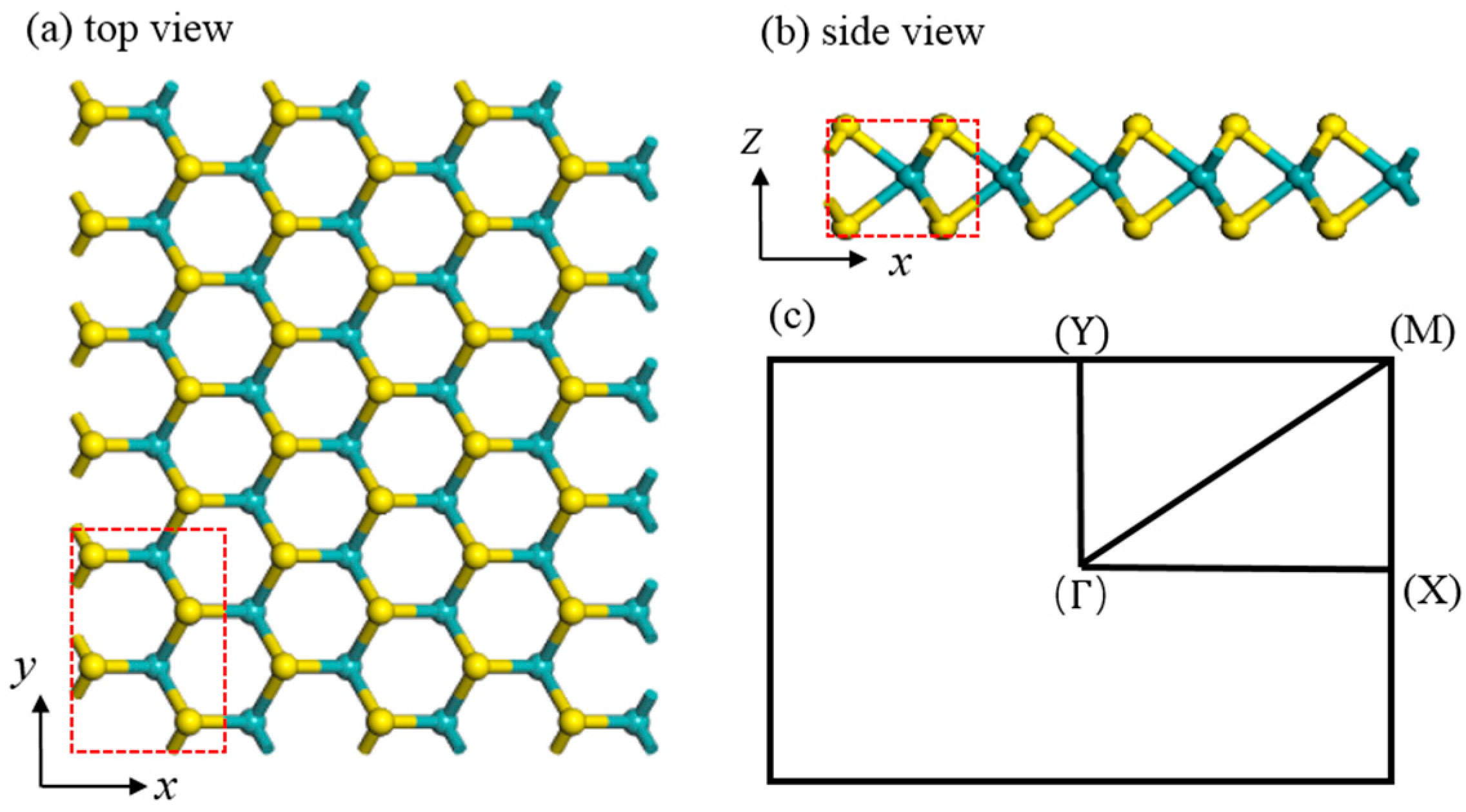
2. Method
3. Results and Discussion
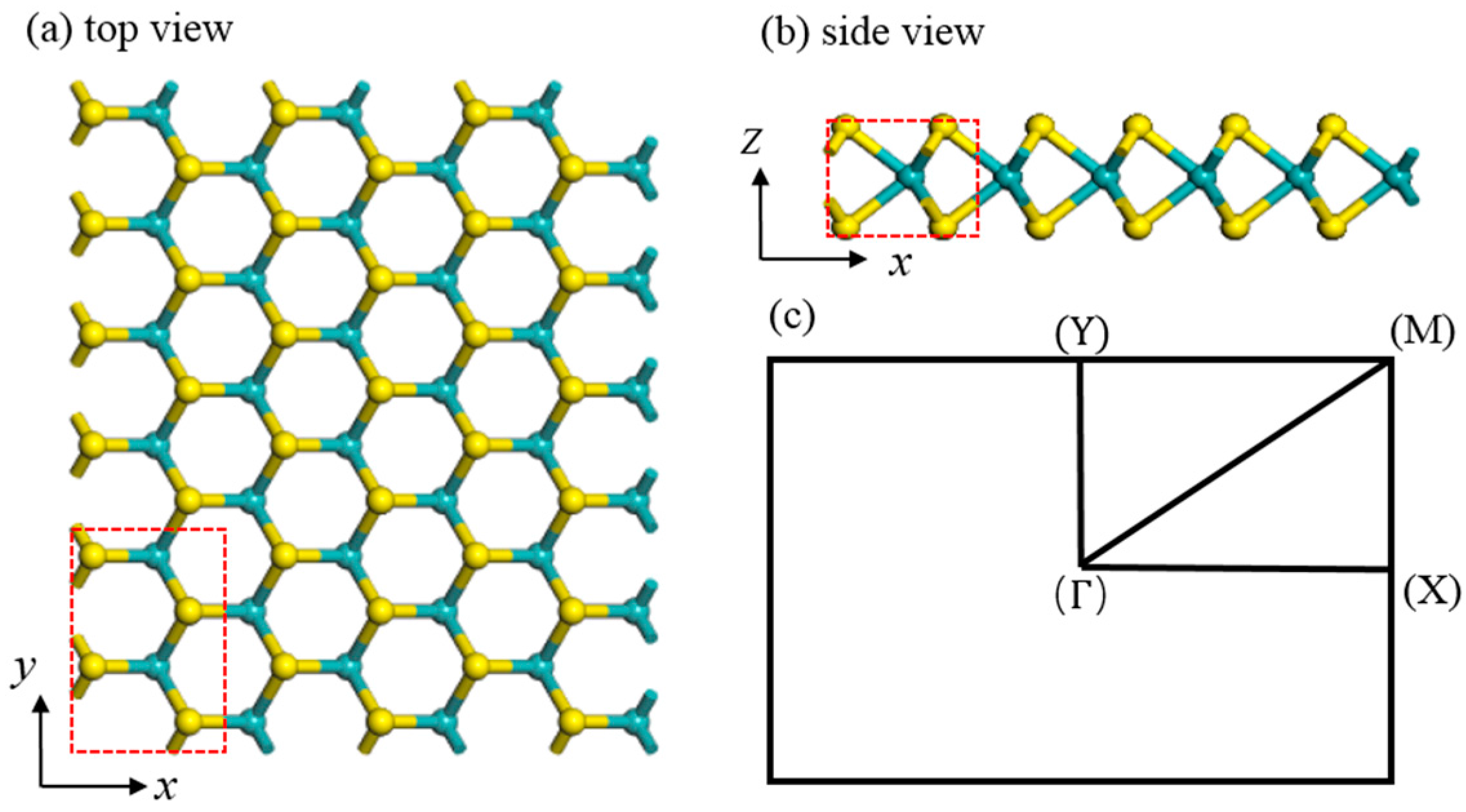
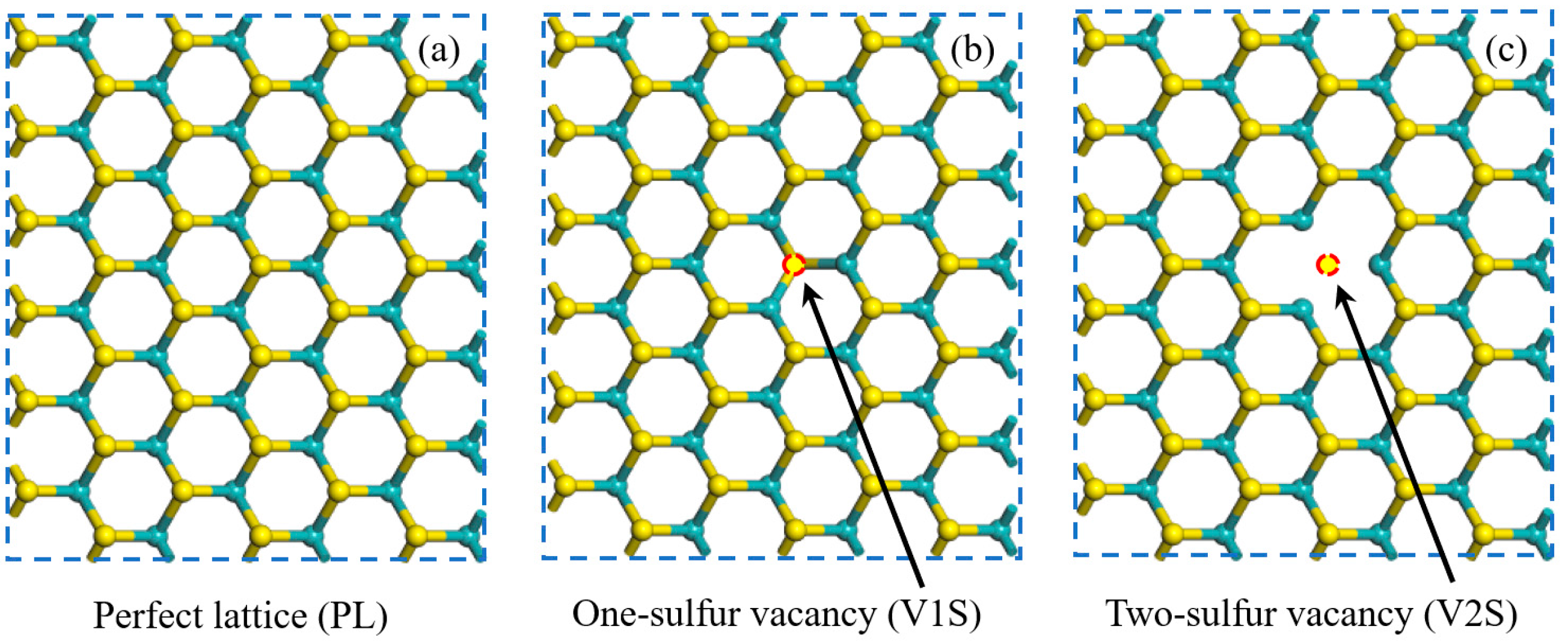
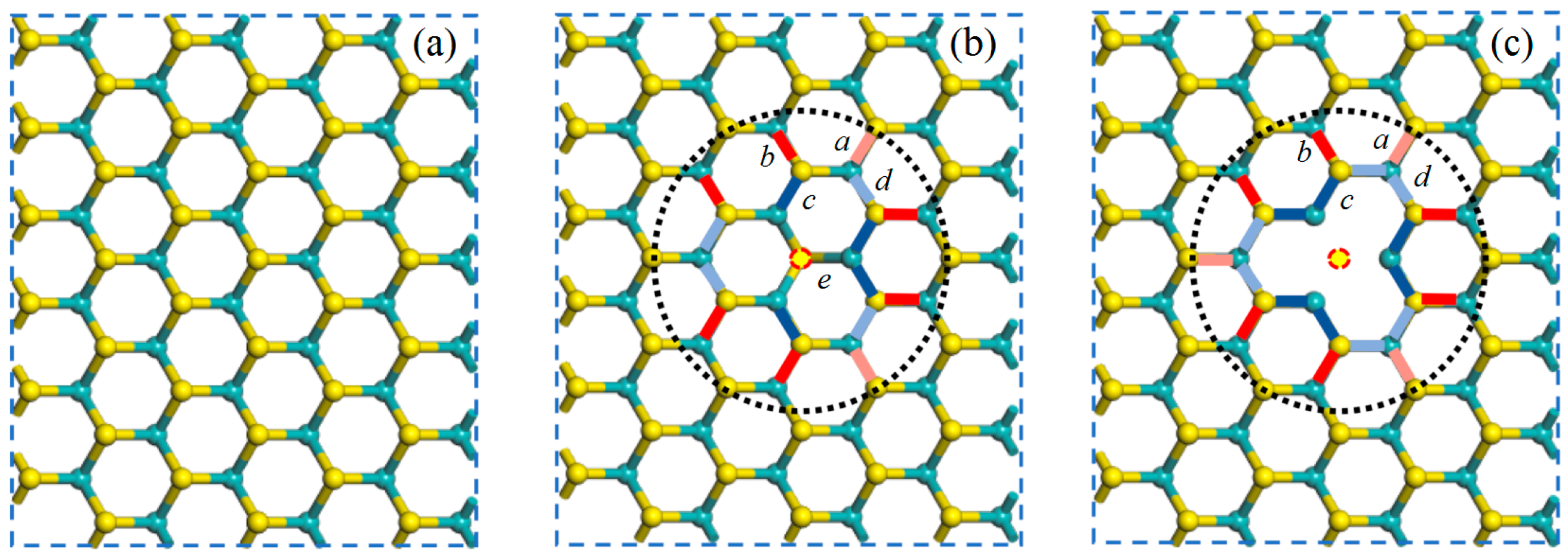
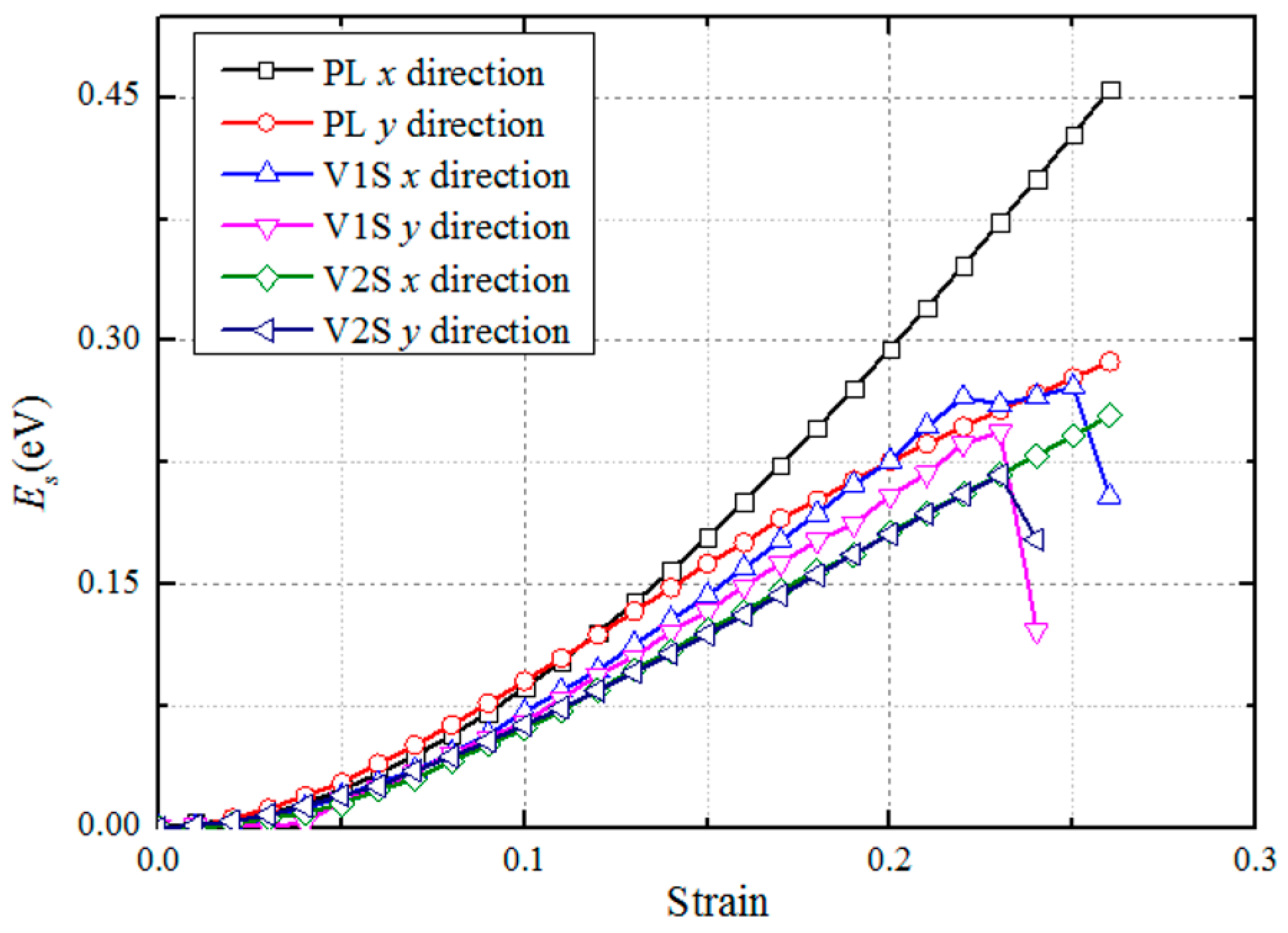
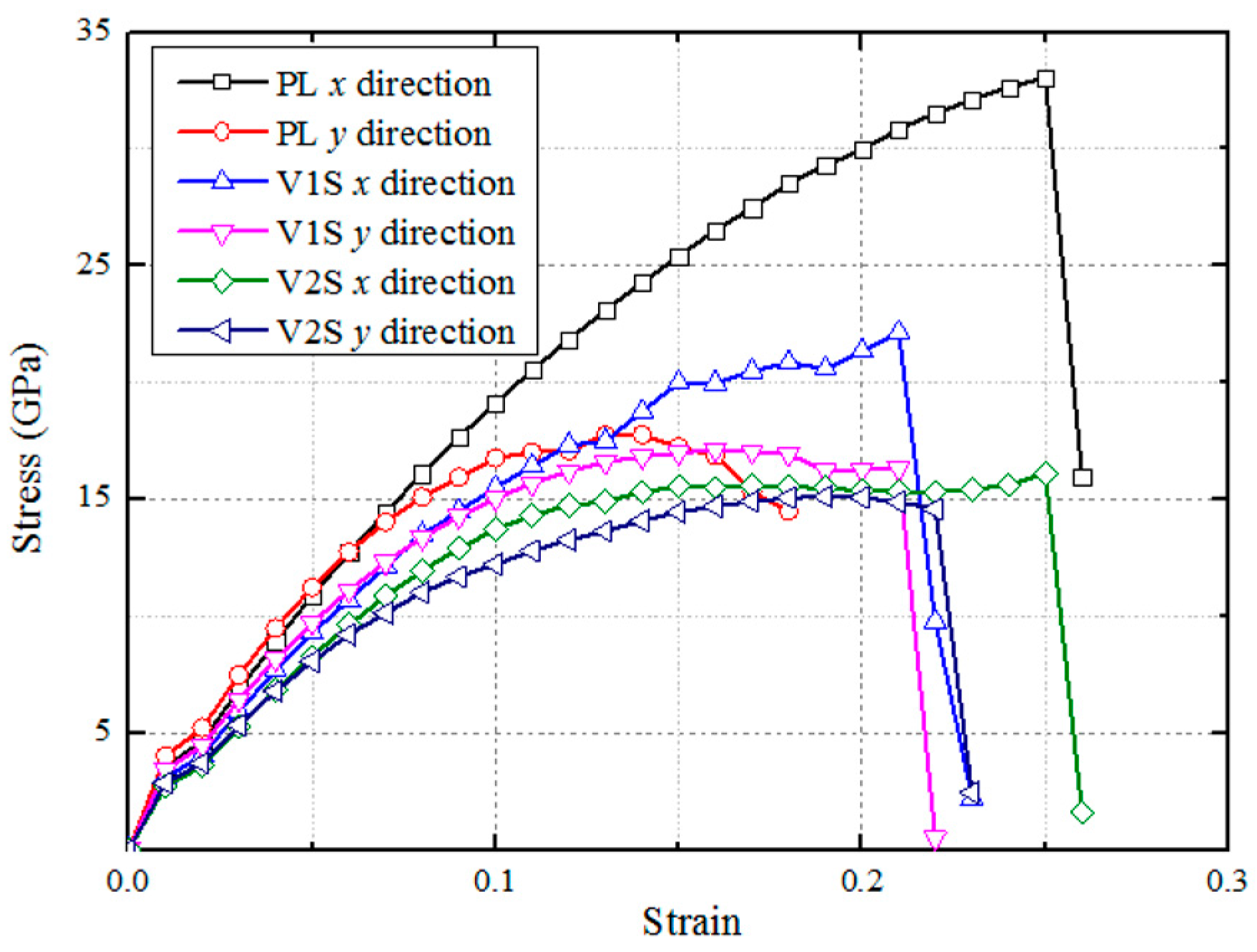
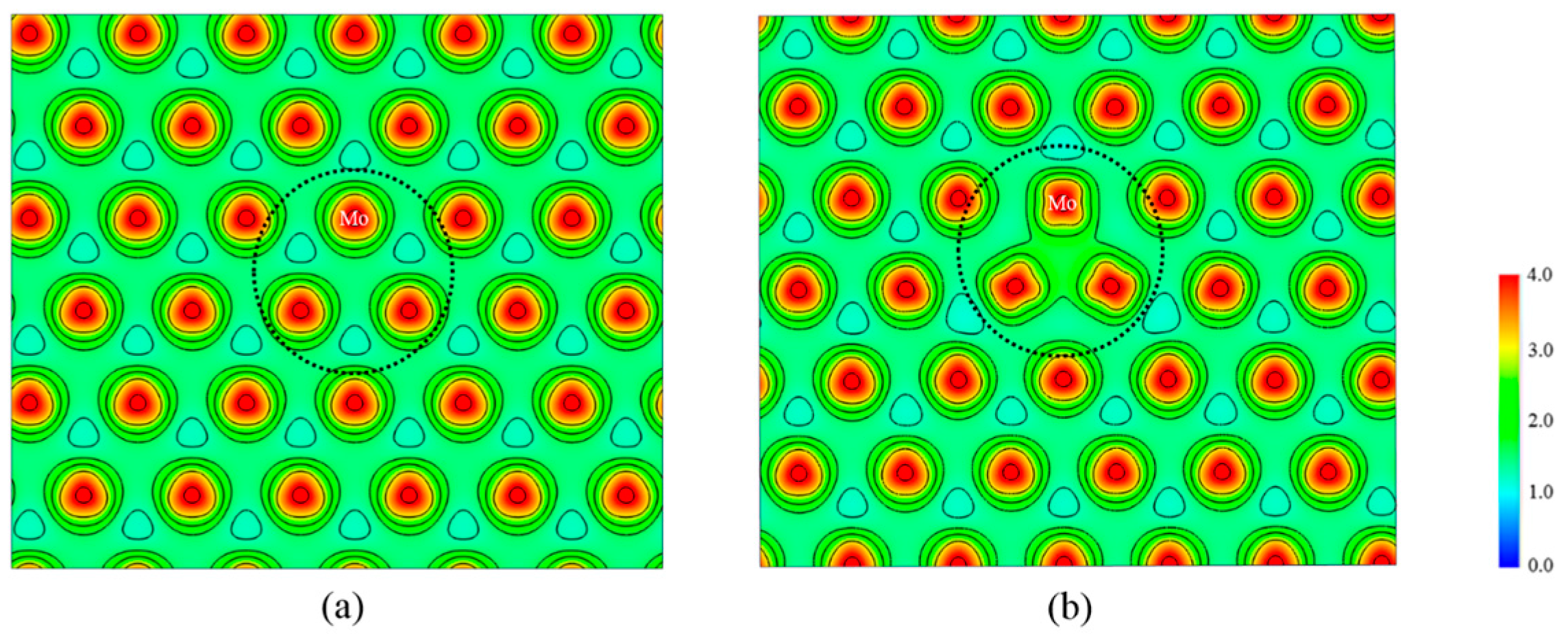
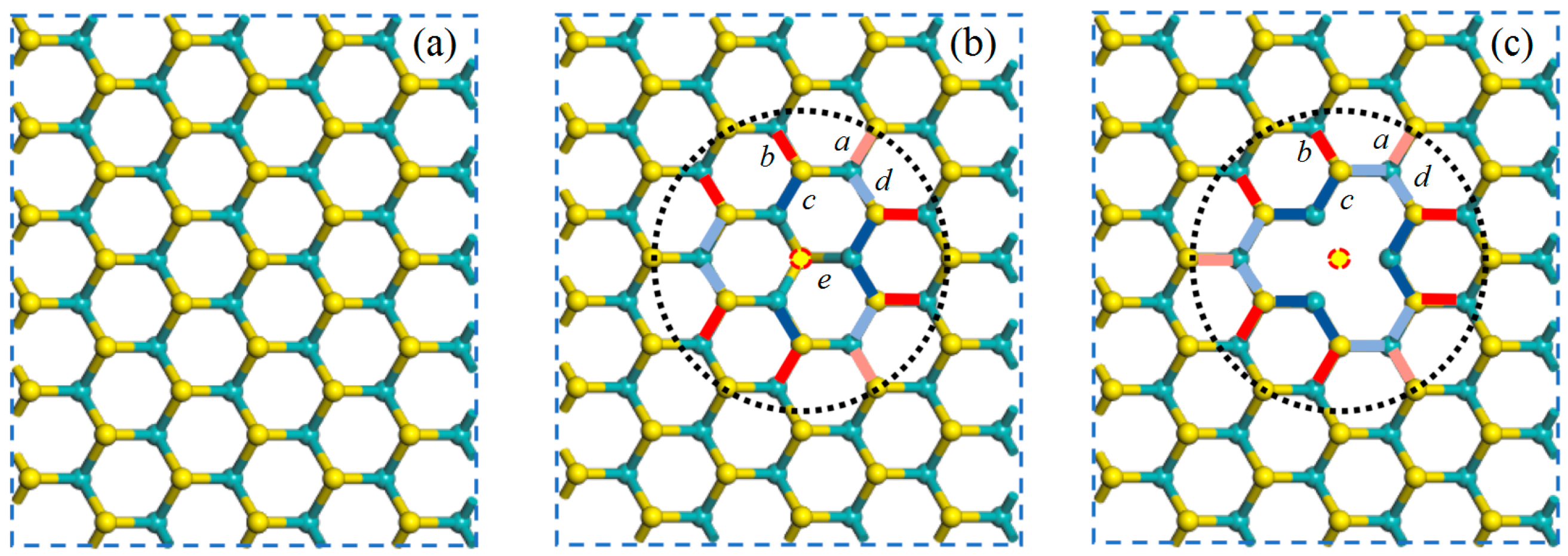
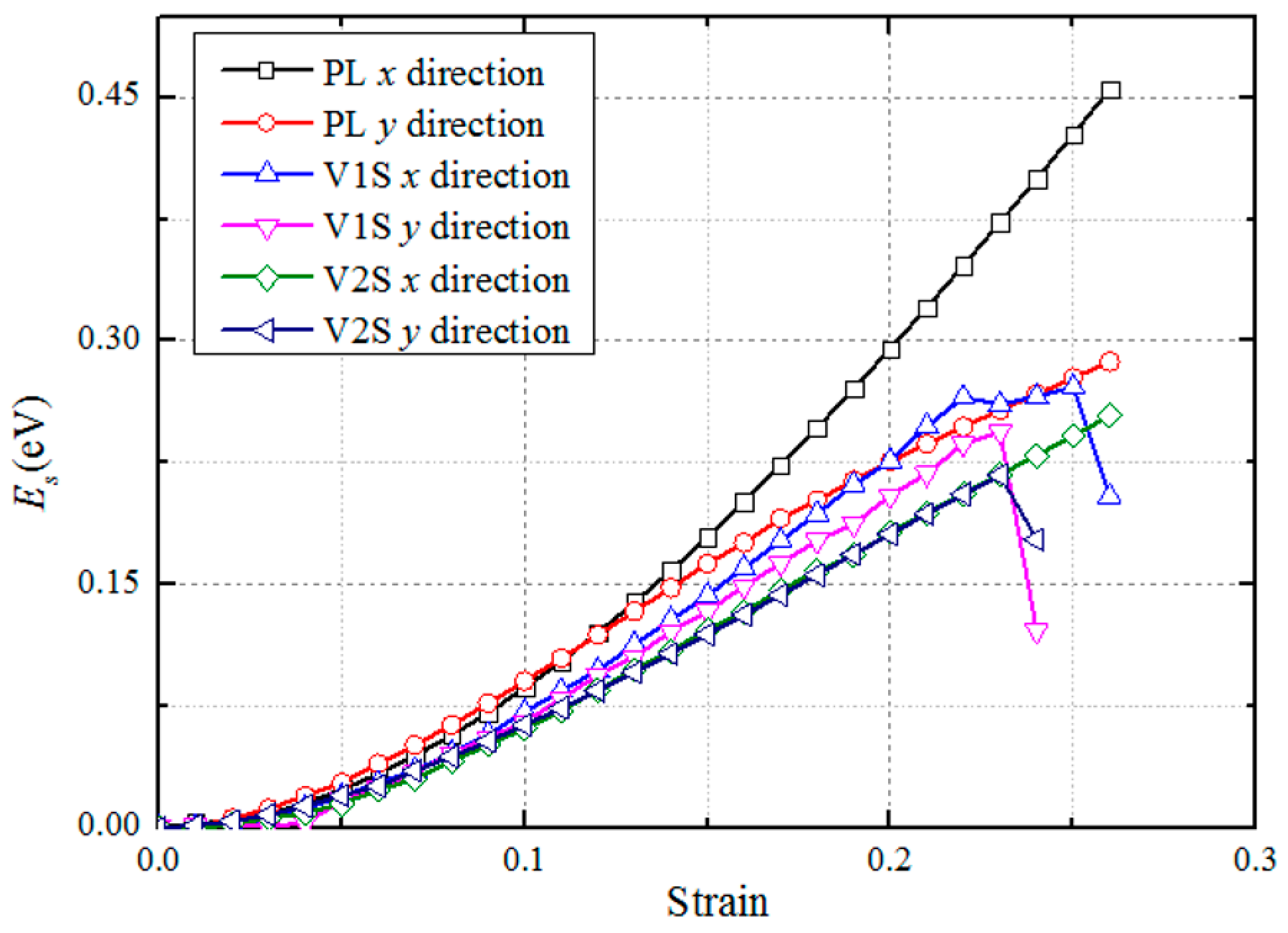
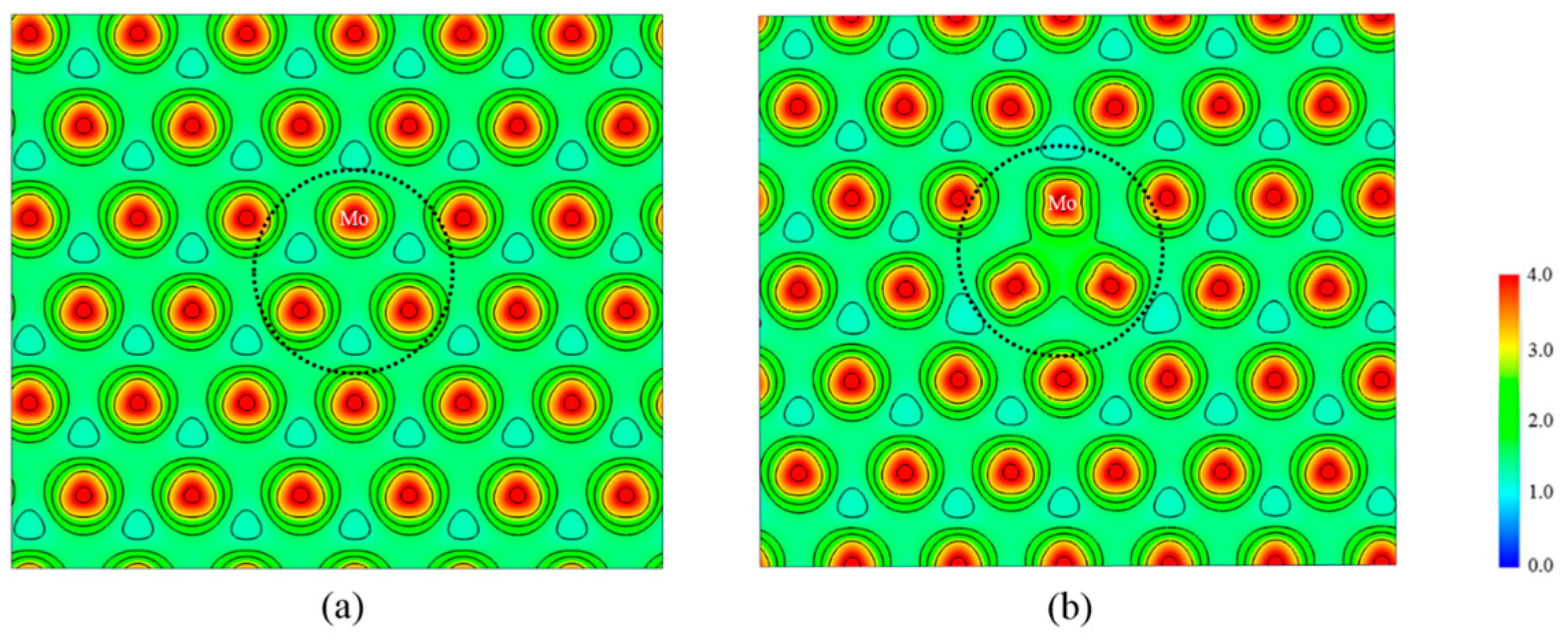
3.1. Structural Properties
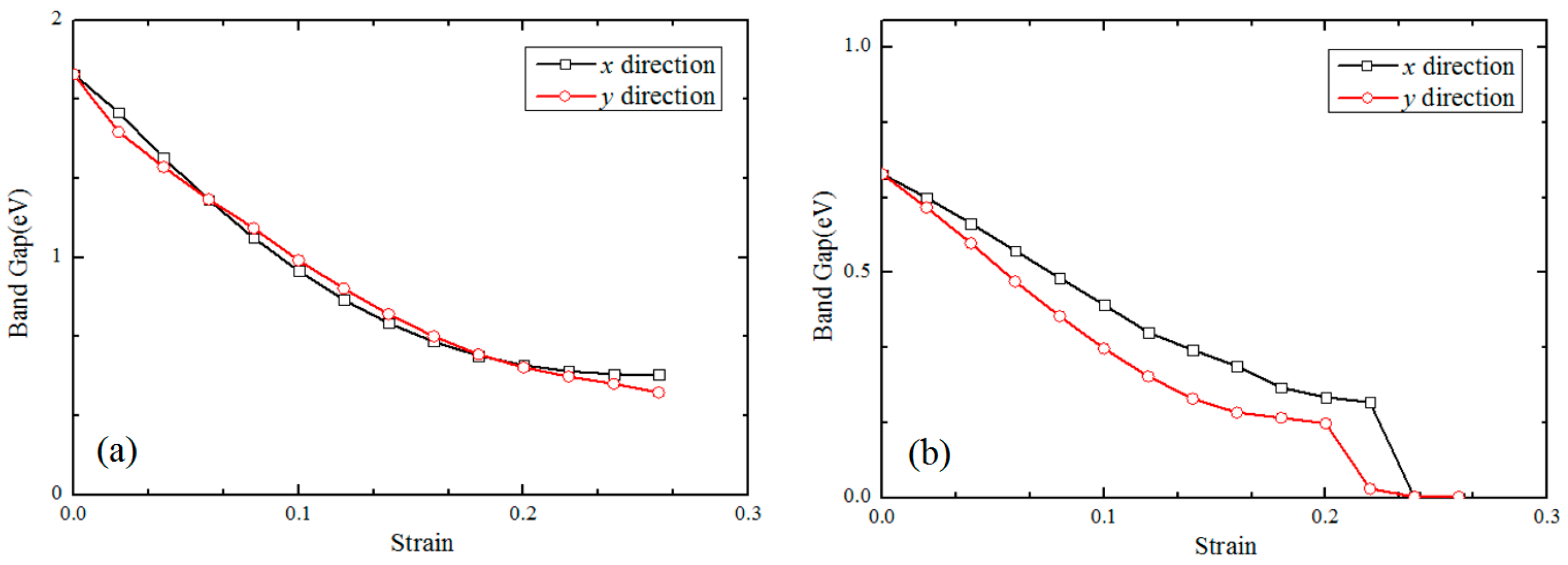
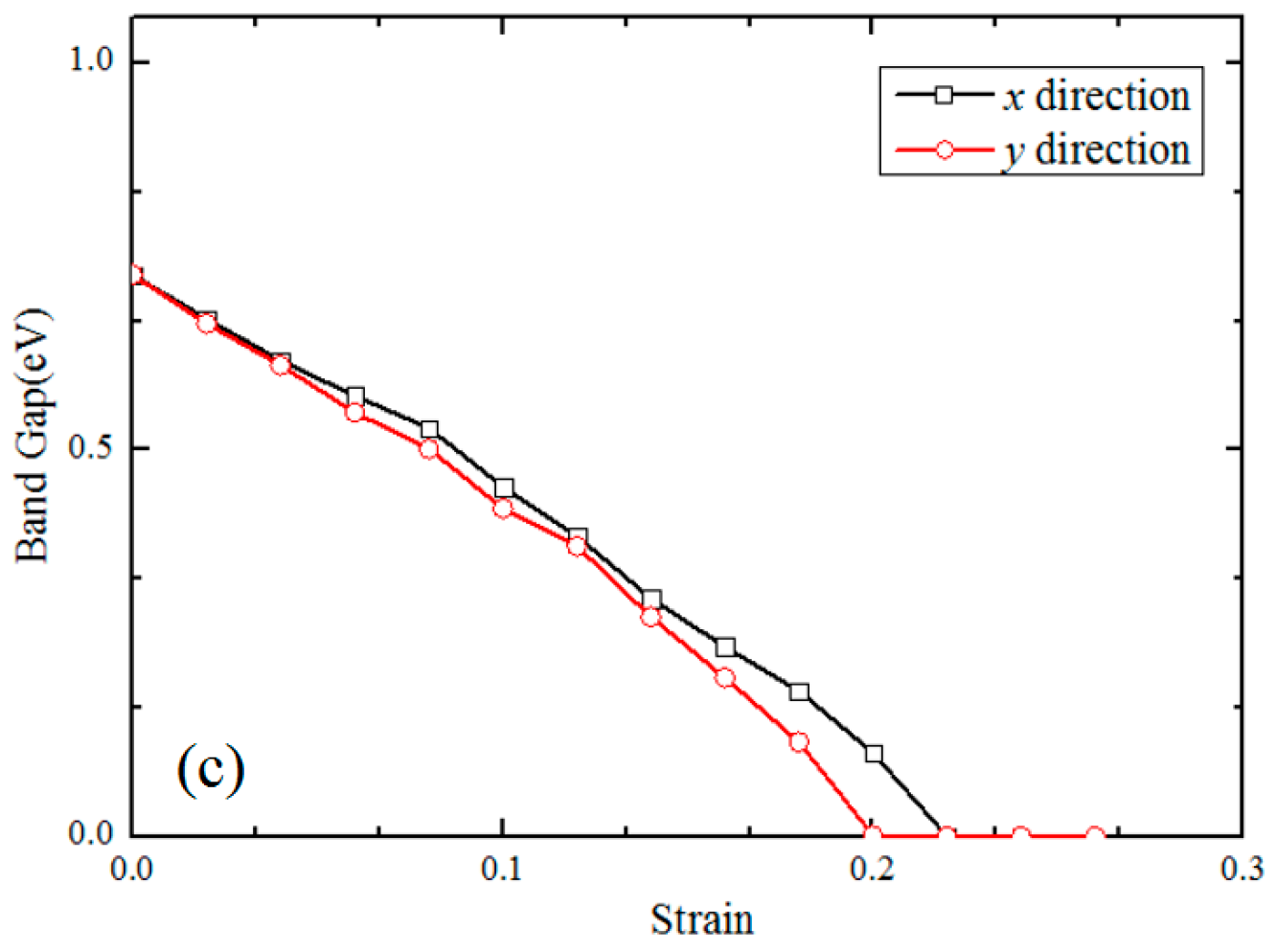
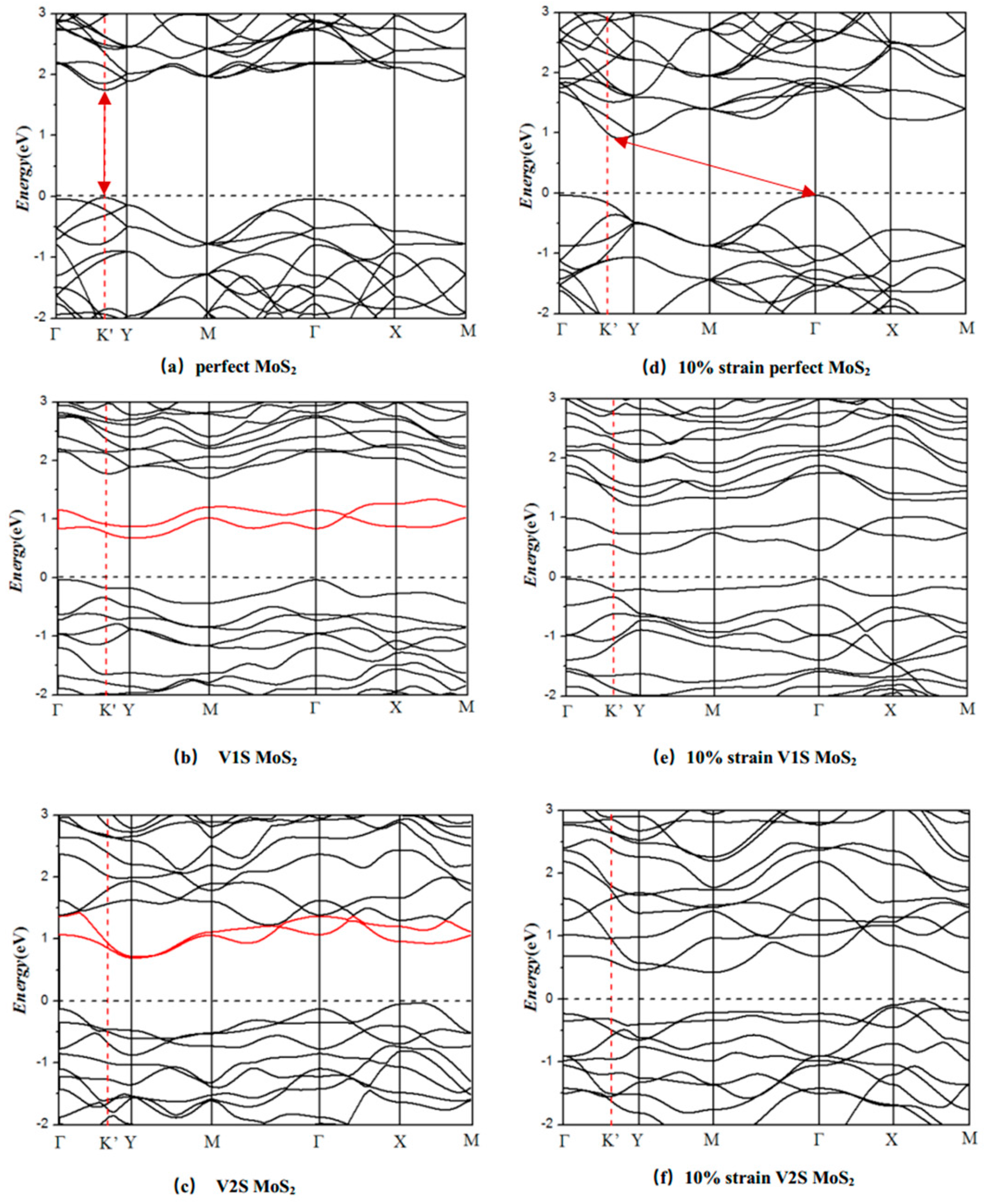
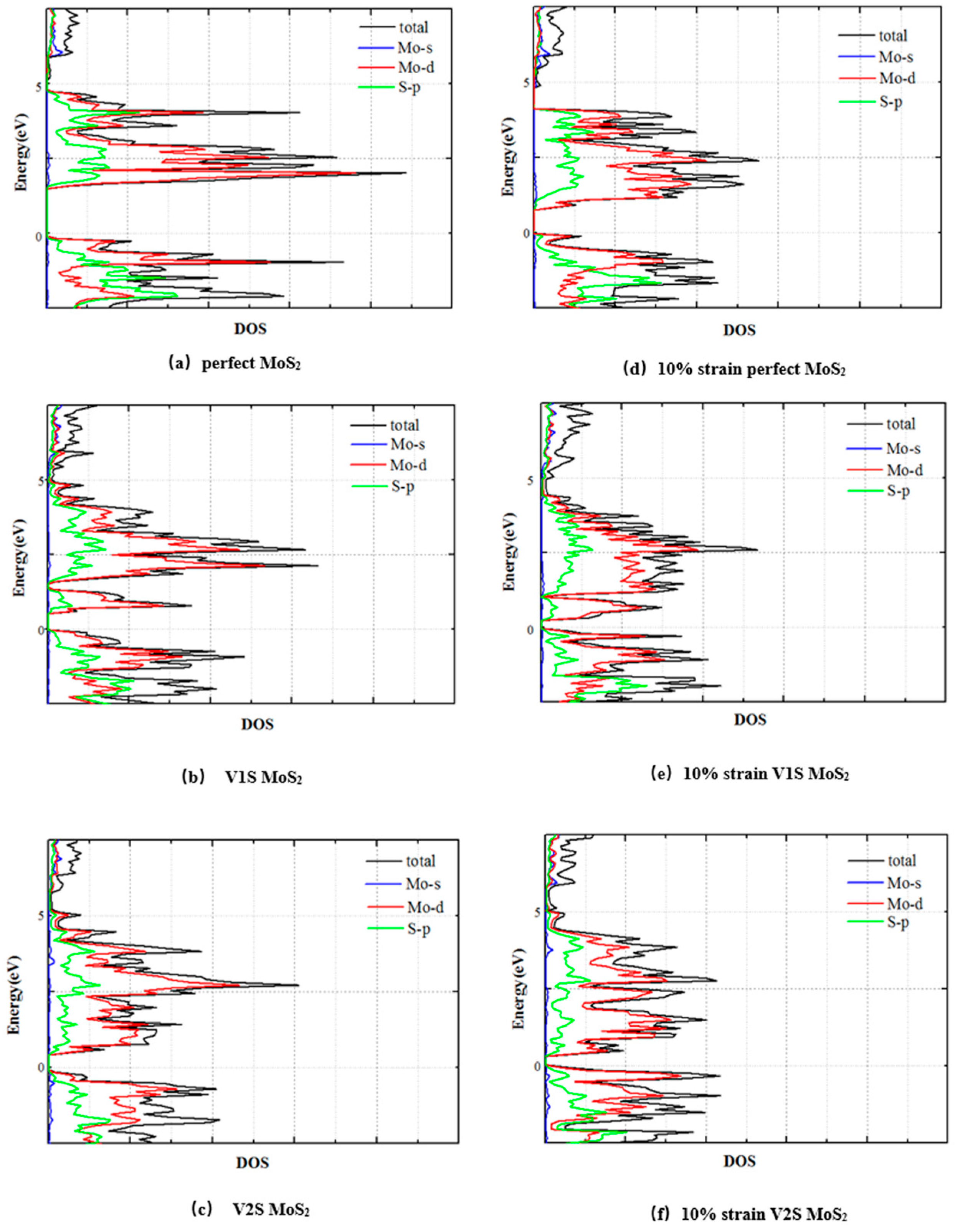
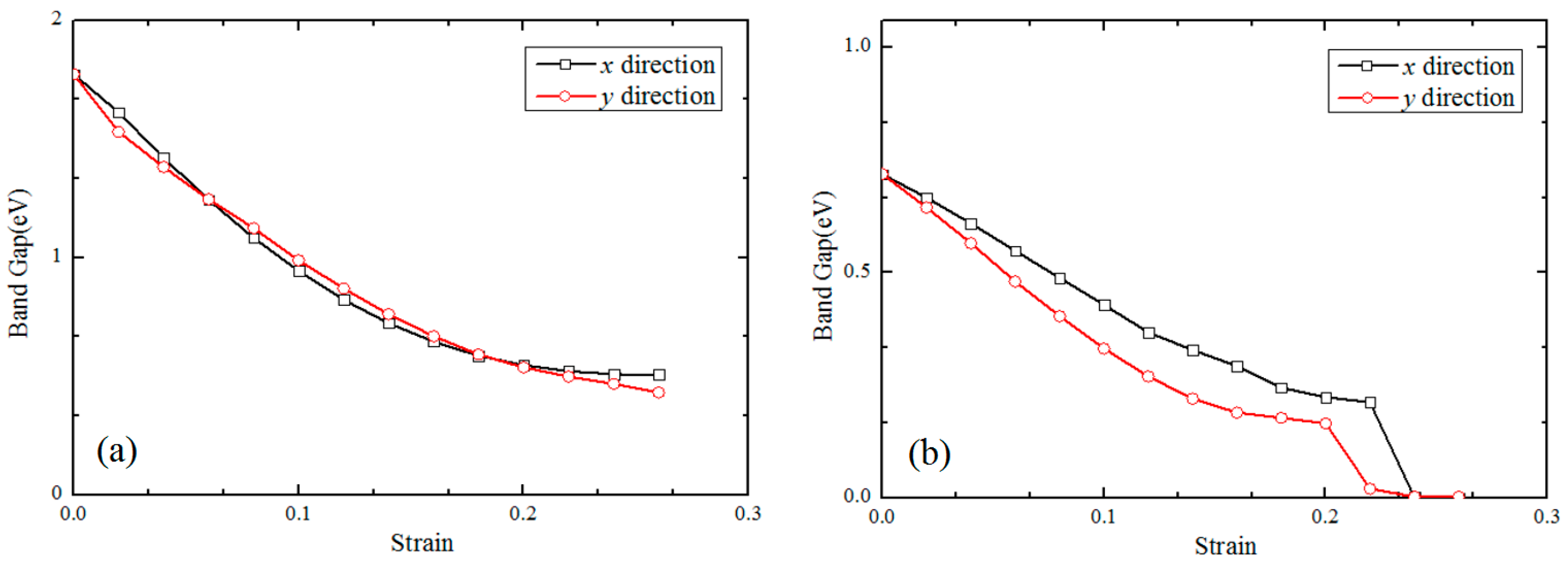
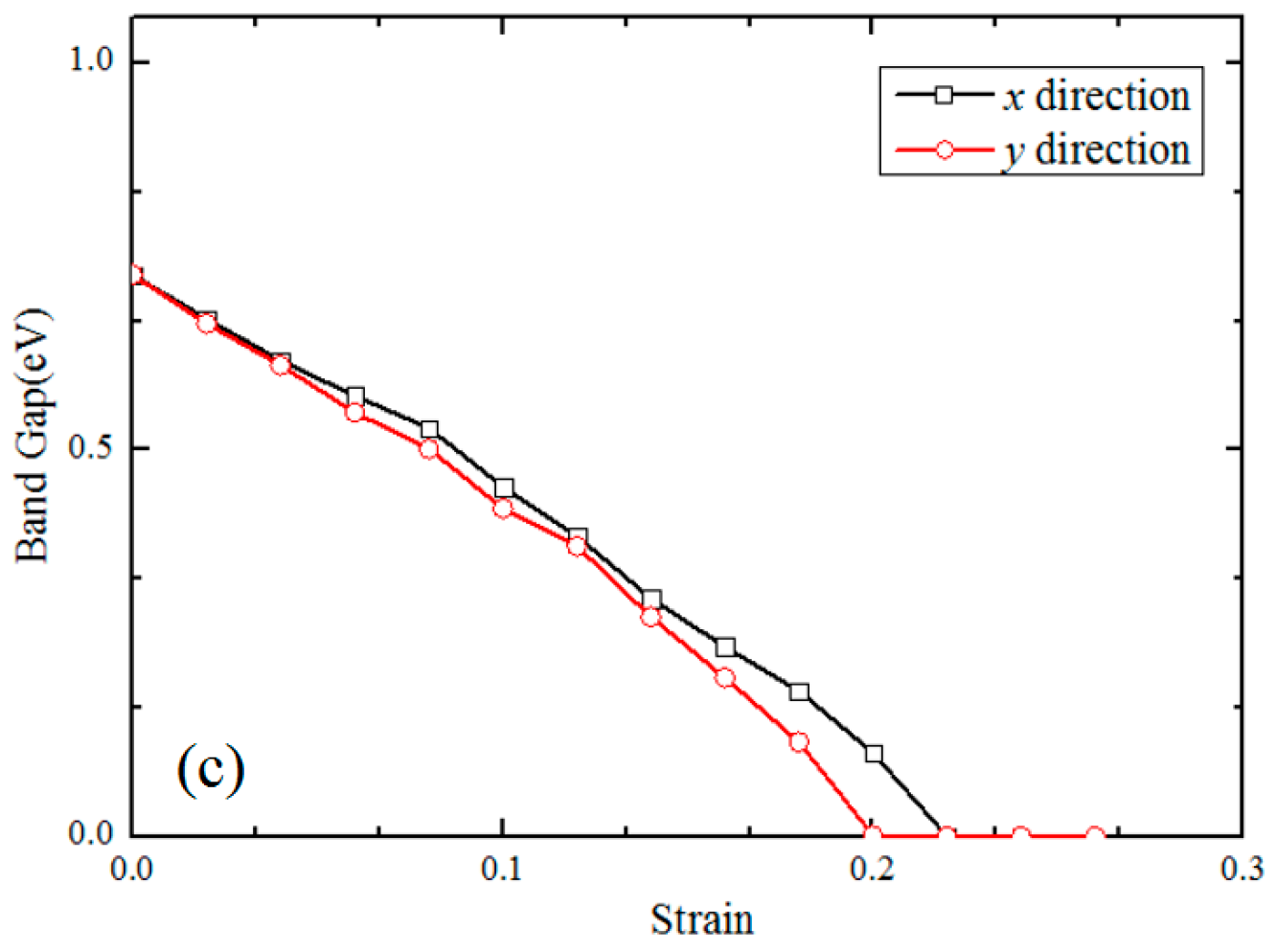
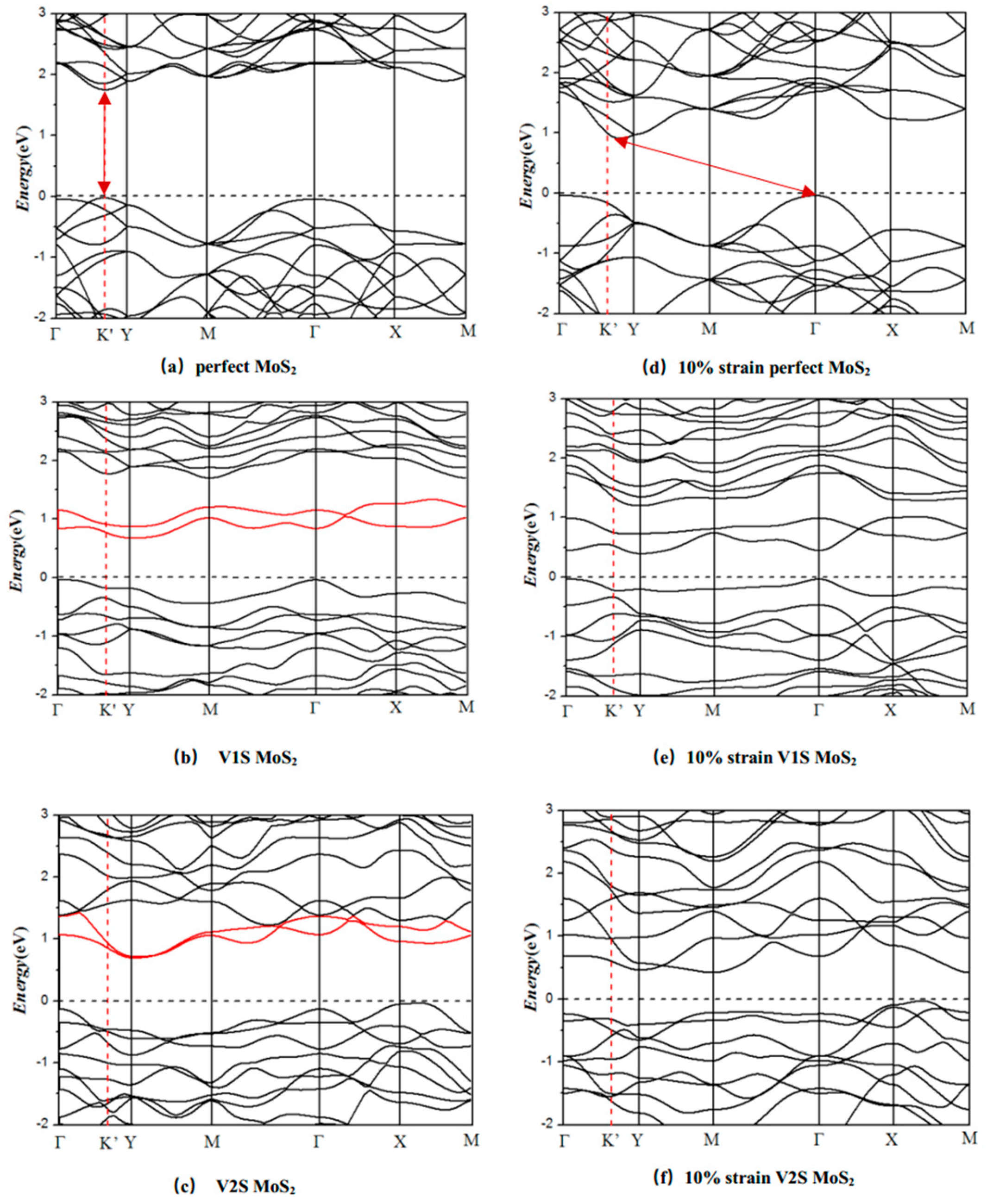
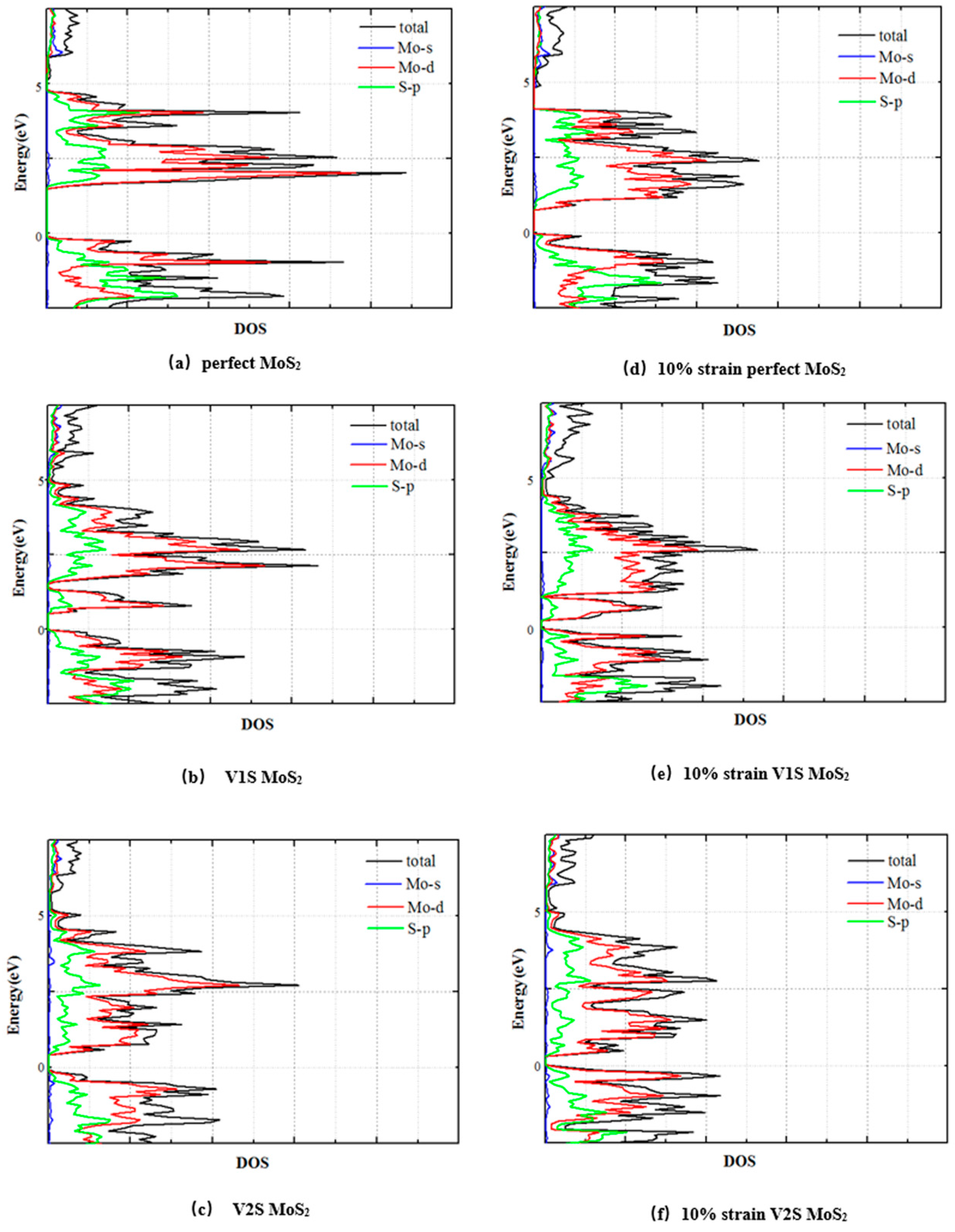
3.2. Electronic Properties
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Topsakal, M.; Cahangirov, S.; Ciraci, S. The response of mechanical and electronic properties of graphane to the elastic strain. Appl. Phys. Lett. 2010, 96, 091912. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.; Wu, X.; Guo, W.; Zeng, X.C. Strain-dependent electronic and magnetic properties of MoS2 monolayer, bilayer, nanoribbons and nanotubes. Phys. Chem. Chem. Phys. 2012, 14, 13035–13040. [Google Scholar] [CrossRef] [PubMed]
- Scalise, E.; Houssa, M.; Pourtois, G.; Afanas’ev, V.V.; Stesmans, A. First-principles study of strained 2D MoS2. Physica E 2014, 56, 416–421. [Google Scholar] [CrossRef]
- Tao, P.; Guo, H.; Yang, T.; Zhang, Z. Strain-induced magnetism in MoS2 monolayer with defects. J. Appl. Phys. 2014, 115, 054305. [Google Scholar] [CrossRef]
- Cooper, R.C.; Lee, C.; Marianetti, C.A.; Wei, X.; Hone, J.; Kysar, J.W. Nonlinear elastic behavior of two-dimensional molybdenum disulfide. Phys. Rev. B 2013, 87, 035423. [Google Scholar]
- Bertolazzi, S.; Brivio, J.; Kis, A. Stretching and breaking of ultrathin MoS2. ACS Nano 2011, 5, 9703–9709. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Edmonds, K.W.; Gallagher, B.L.; Rushforth, A.W.; Makarovsky, O.; Patanè, A.; Campion, R.P.; Foxon, C.T.; Novak, V.; Jungwirth, T. High Curie temperatures at low compensation in the ferromagnetic semiconductor (Ga,Mn)As. Phys. Rev. B 2013, 87, 121301. [Google Scholar] [CrossRef]
- Santosh, K.C.; Longo, R.C.; Addou, R.; Wallace, R.M.; Cho, K. Impact of intrinsic atomic defects on the electronic structure of MoS2 monolayers. Nanotechnology 2014, 25, 375703. [Google Scholar]
- Wang, W.; Li, L.; Yang, C.; Soler-Crespo, R.A.; Meng, Z.; Li, M.; Zhang, X.; Keten, S.; Espinosa, H.D. Plasticity resulted from phase transformation for monolayer molybdenum disulfide film during nanoindentation simulations. Nanotechnology 2017, 28, 164005. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wan, Y.; Tu, L.; Yang, Y.; Lou, J. The effect of VMoS3 point defect on the elastic properties of monolayer MoS2 with REBO potentials. Nanoscale Res. Lett. 2016, 11, 155. [Google Scholar] [CrossRef] [PubMed]
- Spirko, J.A.; Neiman, M.L.; Oelker, A.M.; Klier, K. Electronic structure and reactivity of defect MoS2 II. Bonding and activation of hydrogen on surface defect sites and clusters. Surf. Sci. 2004, 572, 191–205. [Google Scholar] [CrossRef]
- Defo, R.K.; Fang, S.; Shirodkar, S.N.; Tritsaris, G.A.; Dimoulas, A.; Kaxiras, E. Strain dependence of band gaps and exciton energies in pure and mixed transition-metal dichalcogenides. Phy. Rev. B 2016, 94, 155310. [Google Scholar] [CrossRef]
- Maniadaki, A.E.; Kopidakis, G.; Remediakis, I.N. Strain engineering of electronic properties of transition metal dichalcogenide monolayers. Solid State Commun. 2016, 227, 33–39. [Google Scholar] [CrossRef]
- Coehoorn, R.; Haas, C.; de Groot, R.A. Electronic structure of MoSe2, MoS2, and WSe2. II. The nature of the optical band gaps. Phys. Rev. B. 1987, 35, 6203. [Google Scholar] [CrossRef]
- Lebègue, S.; Eriksson, O. Electronic structure of two-dimensional crystals from ab initio theory. Phy. Rev. B 2009, 79, 115409. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169. [Google Scholar] [CrossRef]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Kang, J.; Shao, Z.; Zhang, X.; Chang, S.; Wang, G.; Qin, S.; Li, J. Mechanical and electronic properties of monolayer MoS2 under elastic strain. Phys. Lett. A 2012, 376, 1166–1170. [Google Scholar] [CrossRef]
- Lu, N.; Guo, H.; Li, L.; Dai, J.; Wang, L.; Mei, W.-N.; Wu, X.; Zeng, X.C. MoS2/MX2 heterobilayers: Bandgap engineering via tensile strain or external electrical field. Nanoscale 2014, 6, 2879–2886. [Google Scholar] [CrossRef] [PubMed]
- Krasheninnikov, A.V. Strain fields and electronic structure of vacancy-type defects in graphene from first-principles simulation. In Nanodevices and Nanomaterials for Ecological Security; Spriger Nature: New York, NY, USA, 2012. [Google Scholar]
- Van der Zande, A.M.; Huang, P.Y.; Chenet, D.A.; Berkelbach, T.C.; You, Y.; Lee, G.-H.; Heinz, T.F.; Reichman, D.R.; Muller, D.A.; Hone, J.C. Grains and grain boundaries in highly crystalline monolayer molybdenum disulphide. Nat. Mater. 2013, 12, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Ansari1, R.; Malakpour, S.; Faghihnasiri, M.; Ajori, S. Characterization of the mechanical properties of monolayer molybdenum disulfide nanosheets using first principles. J. Nanotechnol. Eng. Med. 2014, 4, 034501. [Google Scholar] [CrossRef]
- Gan, Y.; Zhao, H. Chirality effect of mechanical and electronic properties of monolayer MoS2 with vacancies. Phys. Lett. A 2014, 378, 2910–2914. [Google Scholar] [CrossRef]
- Priezjev, N. Interfacial friction between semiflexible polymers and crystalline surfaces. J. Chem. Phys. 2012, 136, 224702. [Google Scholar] [CrossRef] [PubMed]
- Böker, T.; Severin, R.; Müller, A.; Janowitz, C.; Manzke, R.; Voß, D.; Krüger, P.; Mazur, A.; Pollmann, J. Band structure of MoS2, MoSe2, and α-MoTe2: Angle-resolved photoelectron spectroscopy and ab initio calculations. Phys. Rev. B 2001, 64, 235305. [Google Scholar] [CrossRef]
- Remskar, M.; Mrzel, A.; Virsek, M.; Godec, M.; Krause, M.; Kolitsch, A.; Singh, A.; Seabaugh, A. The MoS2 nanotubes with defect-controlled electric properties. Nanoscale Res. Lett. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.-P.; Ma, F.; Huang, Y.-H.; Xu, K.-W. Strain effects on electronic states and lattice vibration of monolayer MoS2. Physica E 2015, 71, 1–6. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model Type | a | b | c | d | e |
|---|---|---|---|---|---|
| PL | 2.415 | 2.415 | 2.415 | 2.415 | 2.415 |
| V1S | 2.430 | 2.450 | 2.398 | 2.402 | 2.371 |
| V2S | 2.428 | 2.453 | 2.400 | 2.405 | / |
| E (GPa) | (GPa) | |||
|---|---|---|---|---|
| x | y | x | y | |
| Perfect | 315 | 335 | 33.0 | 17.7 |
| V1S | 272 | 290 | 22.1 | 17.1 |
| V2S | 227 | 241 | 16.1 | 15.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, W.; Yang, C.; Bai, L.; Li, M.; Li, W. First-Principles Study on the Structural and Electronic Properties of Monolayer MoS2 with S-Vacancy under Uniaxial Tensile Strain. Nanomaterials 2018, 8, 74. https://doi.org/10.3390/nano8020074
Wang W, Yang C, Bai L, Li M, Li W. First-Principles Study on the Structural and Electronic Properties of Monolayer MoS2 with S-Vacancy under Uniaxial Tensile Strain. Nanomaterials. 2018; 8(2):74. https://doi.org/10.3390/nano8020074
Chicago/Turabian StyleWang, Weidong, Chenguang Yang, Liwen Bai, Minglin Li, and Weibing Li. 2018. "First-Principles Study on the Structural and Electronic Properties of Monolayer MoS2 with S-Vacancy under Uniaxial Tensile Strain" Nanomaterials 8, no. 2: 74. https://doi.org/10.3390/nano8020074