
Chloramphenicol Derivatization in Its Primary Hydroxyl Group with Basic Amino Acids Leads to New Pharmacophores with High Antimicrobial Activity

 and
and 
Abstract
:1. Introduction
2. Results
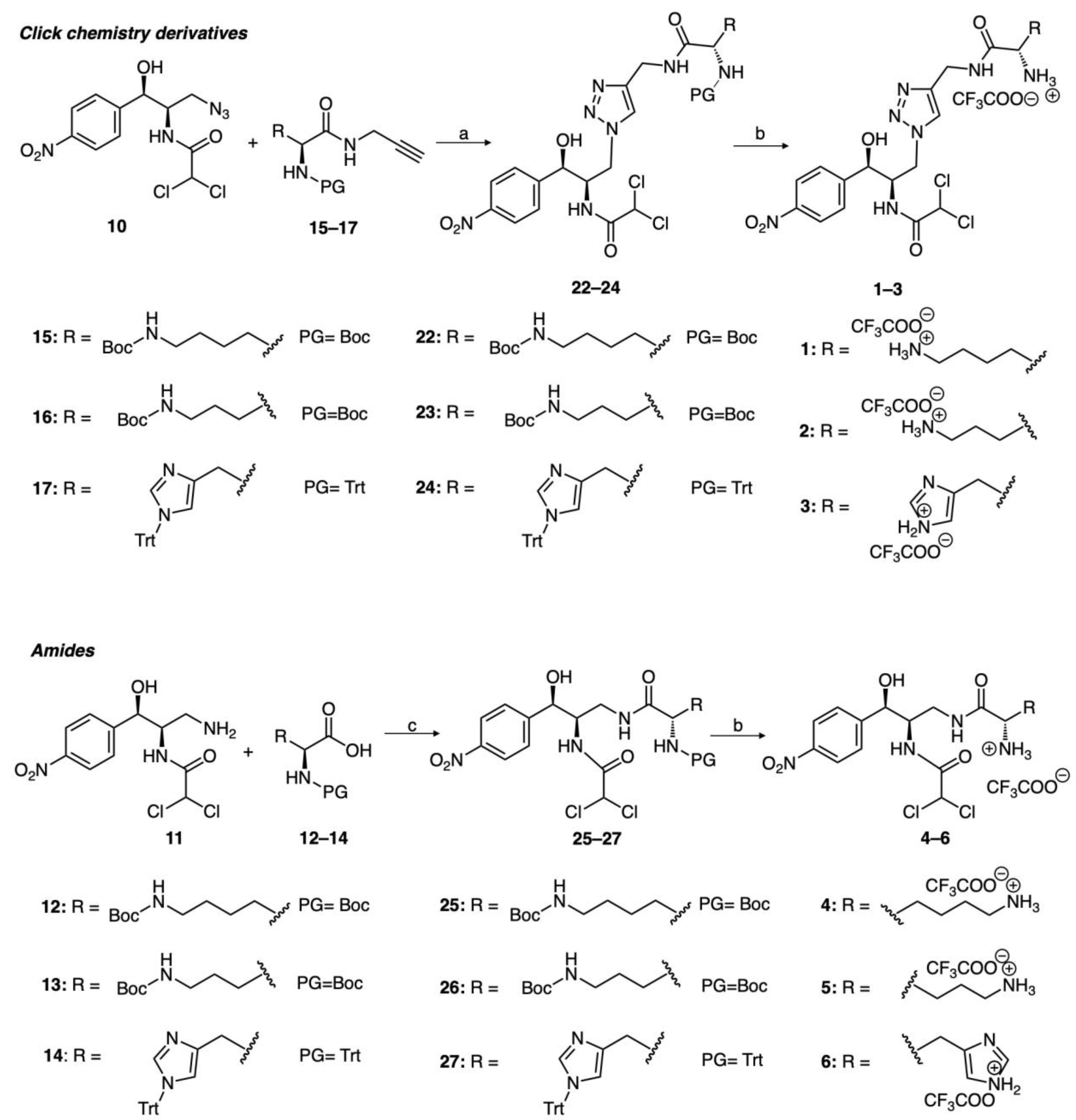
2.1. Chemical Synthesis
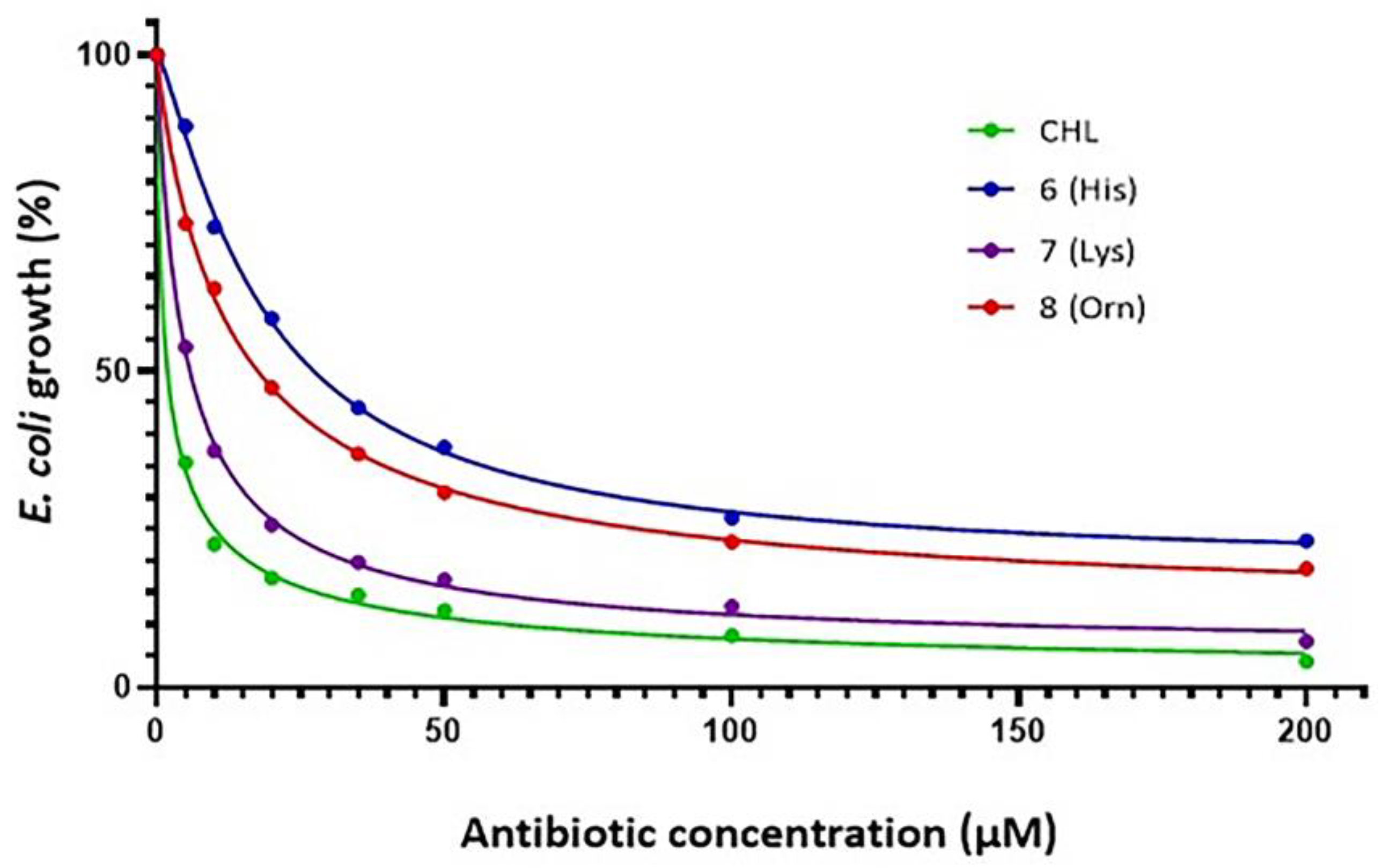
2.2. Antibacterial Activity
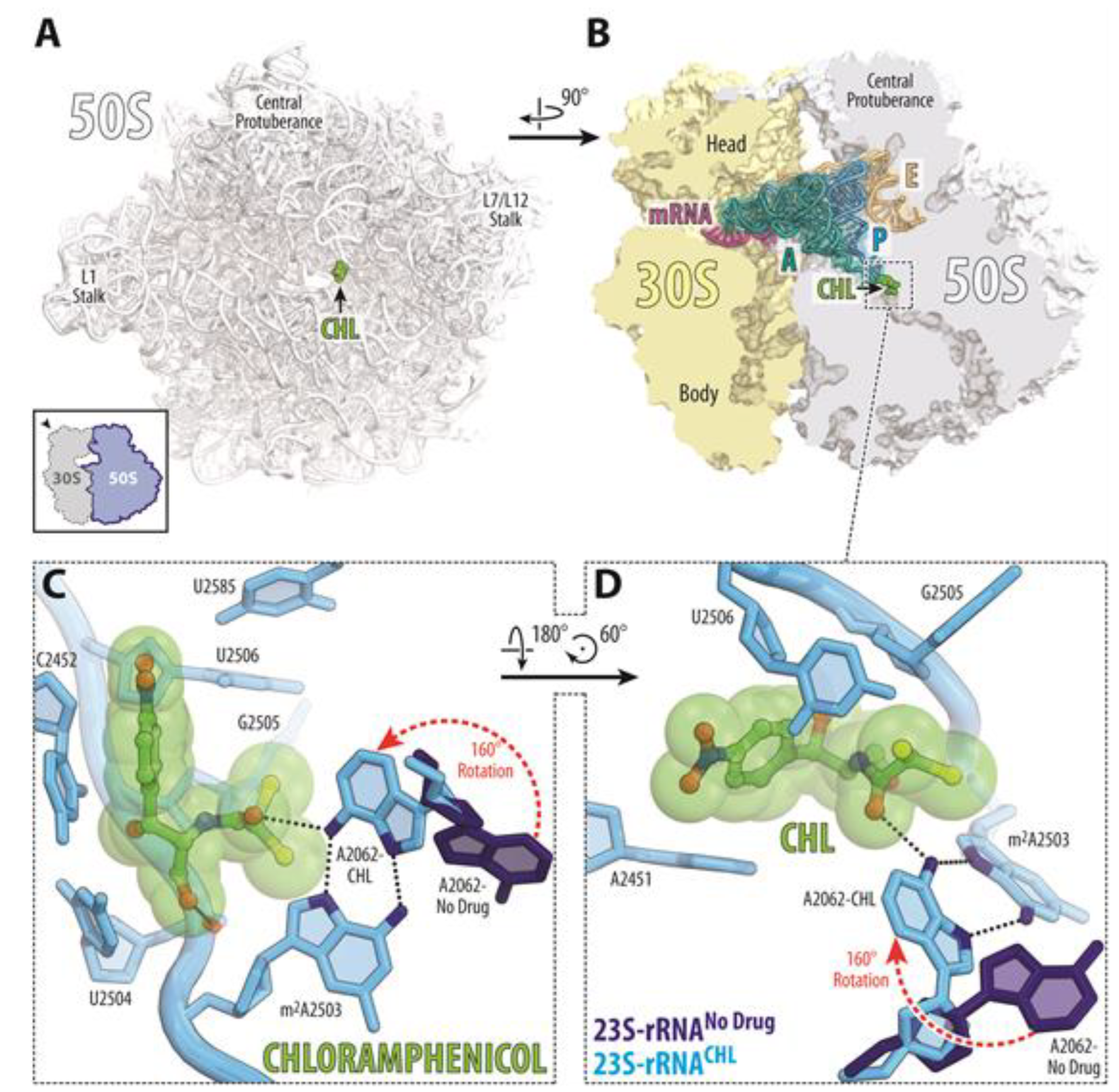
2.3. Protein Synthesis Inhibition
2.3.1. Cell-Free Transcription–Translation Inhibition
2.3.2. Puromycin Reaction
3. Discussion
4. Materials and Methods
4.1. Chemical Synthesis
4.2. Bacterial Strains
4.3. Biochemical Preparations
4.4. Inhibition of Translation Using an E. coli-Based In Vitro Cell-Free Expression System
4.5. EC50 Determination
4.6. Competition in [14C]-Chloramphenicol Binding
4.7. Puromycin Reaction
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Costa, V.M.; King, C.E.; Kalan, L.; Morar, M.; Sung, W.W.L.; Schwarz, C.; Froese, D.; Zazula, G.; Calmels, F.; Debruyne, R.; et al. Antibiotic Resistance Is Ancient. Nature 2011, 477, 457–461. [Google Scholar] [CrossRef]
- Bhullar, K.; Waglechner, N.; Pawlowski, A.; Koteva, K.; Banks, E.D.; Johnston, M.D.; Barton, H.A.; Wright, G.D. Antibiotic Resistance Is Prevalent in an Isolated Cave Microbiome. PLoS ONE 2012, 7, e34953. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. 20-Valent Pneumococcal Conjugate Vaccine: A Review of Its Use in Adults. Drugs 2022, 82, 989–999. [Google Scholar] [CrossRef]
- De Oliveira, D.M.P.; Forde, B.M.; Kidd, T.J.; Harris, P.N.A.; Schembri, M.A.; Beatson, S.A.; Paterson, D.L.; Walker, M.J. Antimicrobial Resistance in ESKAPE Pathogens. Clin. Microbiol. Rev. 2020, 33, e00181-19. [Google Scholar] [CrossRef] [PubMed]
- Wright, P.M.; Seiple, I.B.; Myers, A.G. The Evolving Role of Chemical Synthesis in Antibacterial Drug Discovery. Angew. Chem. Int. Ed. 2014, 53, 8840–8869. [Google Scholar] [CrossRef]
- Mitcheltree, M.J.; Pisipati, A.; Syroegin, E.A.; Silvestre, K.J.; Klepacki, D.; Mason, J.D.; Terwilliger, D.W.; Testolin, G.; Pote, A.R.; Wu, K.J.Y.; et al. A Synthetic Antibiotic Class Overcoming Bacterial Multidrug Resistance. Nature 2021, 599, 507–512. [Google Scholar] [CrossRef]
- Zhang, J.; Lair, C.; Roubert, C.; Amaning, K.; Barrio, M.B.; Benedetti, Y.; Cui, Z.; Xing, Z.; Li, X.; Franzblau, S.G.; et al. Discovery of Natural-Product-Derived Sequanamycins as Potent Oral Anti-Tuberculosis Agents. Cell 2023, 186, 1013–1025. [Google Scholar] [CrossRef]
- Tereshchenkov, A.G.; Dobosz-Bartoszek, M.; Osterman, I.A.; Marks, J.; Sergeeva, V.A.; Kasatsky, P.; Komarova, E.S.; Stavrianidi, A.N.; Rodin, I.A.; Konevega, A.L.; et al. Binding and Action of Amino Acid Analogs of Chloramphenicol upon the Bacterial Ribosome. J. Mol. Biol. 2018, 430, 842–852. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, J.A.; Tereshchenkov, A.G.; Nazarov, P.A.; Lukianov, D.A.; Skvortsov, D.A.; Polshakov, V.I.; Vasilieva, B.F.; Efremenkova, O.V.; Kaiumov, M.Y.; Paleskava, A.; et al. Conjugates of Chloramphenicol Amine and Berberine as Antimicrobial Agents. Antibiotics 2022, 12, 15. [Google Scholar] [CrossRef]
- Ehrlich, J.; Bartz, Q.R.; Smith, R.M.; Joslyn, D.A.; Burkholder, P.R. Chloromycetin, a New Antibiotic from a Soil Actinomycete. Science 1947, 106, 417. [Google Scholar] [CrossRef]
- Schroeter, W. Hematologic Side Effects of Chloramphenicol. Neuropadiatrie 1974, 5, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Eliakim-Raz, N.; Lador, A.; Leibovici-Weissman, Y.; Elbaz, M.; Paul, M.; Leibovici, L. Efficacy and Safety of Chloramphenicol: Joining the Revival of Old Antibiotics? Systematic Review and Meta-Analysis of Randomized Controlled Trials. J. Antimicrob. Chemother. 2014, 70, 979–996. [Google Scholar] [CrossRef] [PubMed]
- Bulkley, D.; Innis, C.A.; Blaha, G.; Steitz, T.A. Revisiting the Structures of Several Antibiotics Bound to the Bacterial Ribosome. Proc. Natl. Acad. Sci. USA 2010, 107, 17158–17163. [Google Scholar] [CrossRef] [PubMed]
- Dunkle, J.A.; Xiong, L.; Mankin, A.S.; Cate, J.H.D. Structures of the Escherichia Coli Ribosome with Antibiotics Bound near the Peptidyl Transferase Center Explain Spectra of Drug Action. Proc. Natl. Acad. Sci. USA 2010, 107, 17152–17157. [Google Scholar] [CrossRef]
- Dinos, G.P.; Athanassopoulos, C.M.; Missiri, D.A.; Giannopoulou, P.C.; Vlachogiannis, I.A.; Papadopoulos, G.E.; Papaioannou, D.; Kalpaxis, D.L. Chloramphenicol Derivatives as Antibacterial and Anticancer Agents: Historic Problems and Current Solutions. Antibiotics 2016, 5, 20. [Google Scholar] [CrossRef]
- Svetlov, M.S.; Plessa, E.; Chen, C.W.; Bougas, A.; Krokidis, M.G.; Dinos, G.P.; Polikanov, Y.S. High-Resolution Crystal Structures of Ribosome-Bound Chloramphenicol and Erythromycin Provide the Ultimate Basis for Their Competition. RNA 2019, 25, 600–606. [Google Scholar] [CrossRef]
- Marks, J.; Kannan, K.; Roncase, E.J.; Klepacki, D.; Kefi, A.; Orelle, C.; Vázquez-Laslop, N.; Mankin, A.S. ContextSpecific Inhibition of Translation by Ribosomal Antibiotics Targeting the Peptidyl Transferase Center. Proc. Natl. Acad. Sci. USA 2016, 113, 12150–12155. [Google Scholar] [CrossRef]
- Choi, J.; Marks, J.; Zhang, J.; Chen, D.H.; Wang, J.; Vázquez-Laslop, N.; Mankin, A.S.; Puglisi, J.D. Dynamics of the Context-Specific Translation Arrest by Chloramphenicol and Linezolid. Nat. Chem. Biol. 2020, 16, 310–317. [Google Scholar] [CrossRef]
- Chen, H.; Liu, C.; Chen, D.; Madrid, K.; Peng, S.; Dong, X.; Zhang, M.; Gu, Y. Bacteria-Targeting Conjugates Based on Antimicrobial Peptide for Bacteria Diagnosis and Therapy. Mol. Pharm. 2015, 12, 2505–2516. [Google Scholar] [CrossRef]
- Dong, F.; Li, L.; Lin, L.; He, D.; Chen, J.; Wei, W.; Wei, D. Transesterification Synthesis of Chloramphenicol Esters with the Lipase from Bacillus Amyloliquefaciens. Molecules 2017, 22, 1523. [Google Scholar] [CrossRef]
- Louzoun Zada, S.; Green, K.D.; Shrestha, S.K.; Herzog, I.M.; Garneau-Tsodikova, S.; Fridman, M. Derivatives of Ribosome-Inhibiting Antibiotic Chloramphenicol Inhibit the Biosynthesis of Bacterial Cell Wall. ACS Infect. Dis. 2018, 4, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Khairullina, Z.Z.; Tereshchenkov, A.G.; Zavyalova, S.A.; Komarova, E.S.; Lukianov, D.A.; Tashlitsky, V.N.; Osterman, I.A.; Sumbatyan, N.V. Interaction of Chloramphenicol Cationic Peptide Analogues with the Ribosome. Biochemistry 2020, 85, 1443–1457. [Google Scholar] [CrossRef] [PubMed]
- Orelle, C.; Carlson, S.; Kaushal, B.; Almutairi, M.M.; Liu, H.; Ochabowicz, A.; Quan, S.; Pham, V.C.; Squires, C.L.; Murphy, B.T.; et al. Tools for Characterizing Bacterial Protein Synthesis Inhibitors. Antimicrob. Agents Chemother. 2013, 57, 5994–6004. [Google Scholar] [CrossRef]
- Tsirogianni, A.; Kournoutou, G.G.; Bougas, A.; Poulou-Sidiropoulou, E.; Dinos, G.; Athanassopoulos, C.M. New Chloramphenicol Derivatives with a Modified Dichloroacetyl Tail as Potential Antimicrobial Agents. Antibiotics 2021, 10, 394. [Google Scholar] [CrossRef]
- Dinos, G.; Wilson, D.N.; Teraoka, Y.; Szaflarski, W.; Fucini, P.; Kalpaxis, D.; Nierhaus, K.H. Dissecting the Ribosomal Inhibition Mechanisms of Edeine and Pactamycin: The Universally Conserved Residues G693 and C795 Regulate P-Site RNA Binding. Mol. Cell 2004, 13, 113–124. [Google Scholar] [CrossRef]
- Mamos, P.; Krokidis, M.G.; Papadas, A.; Karahalios, P.; Starosta, A.L.; Wilson, D.N.; Kalpaxis, D.L.; Dinos, G.P. On the Use of the Antibiotic Chloramphenicol to Target Polypeptide Chain Mimics to the Ribosomal Exit Tunnel. Biochimie 2013, 95, 1765–1772. [Google Scholar] [CrossRef]
- Kostopoulou, O.N.; Kouvela, E.C.; Magoulas, G.E.; Garnelis, T.; Panagoulias, I.; Rodi, M.; Papadopoulos, G.; Mouzaki, A.; Dinos, G.P.; Papaioannou, D.; et al. Conjugation with Polyamines Enhances the Antibacterial and Anticancer Activity of Chloramphenicol. Nucleic Acids Res. 2014, 42, 8621–8634. [Google Scholar] [CrossRef] [PubMed]
- Syroegin, E.A.; Aleksandrova, E.V.; Polikanov, Y.S. Insights into the Ribosome Function from the Structures of Non-Arrested Ribosome-Nascent Chain Complexes. Nat. Chem. 2023, 15, 143–153. [Google Scholar] [CrossRef]
- Polikanov, Y.S.; Melnikov, S.V.; Söll, D.; Steitz, T.A. Structural Insights into the Role of RRNA Modifications in Protein Synthesis and Ribosome Assembly. Nat. Struct. Mol. Biol. 2015, 22, 342–344. [Google Scholar] [CrossRef]
- Blaha, G.; Stelzl, U.; Spahn, C.M.T.; Agrawal, R.K.; Frank, J.; Nierhaus, K.H. Preparation of Functional Ribosomal Complexes and Effect of Buffer Conditions on TRNA Positions Observed by Cryoelectron Microscopy. Methods Enzymol. 2000, 317, 292–309. [Google Scholar] [CrossRef]
- Triana-Alonso, F.J.; Dabrowski, M.; Wadzack, J.; Nierhaus, K.H. Self-Coded 3′-Extension of Run-off Transcripts Produces Aberrant Products during In Vitro Transcription with T7 RNA Polymerase. J. Biol. Chem. 1995, 270, 6298–6307. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, M.A.; Tastan, A.O.; Patzke, S.; Blaha, G.; Spahn, C.M.T.; Wilson, D.N.; Nierhaus, K.H. Codon-Anticodon Interaction at the P Site Is a Prerequisite for TRNA Interaction with the Small Ribosomal Subunit. J. Biol. Chem. 2002, 277, 19095–19105. [Google Scholar] [CrossRef] [PubMed]
- Rheinberger, H.J.; Geigenmüller, U.; Wedde, M.; Nierhaus, K.H. Parameters for the Preparation of Escherichia Coli Ribosomes and Ribosomal Subunits Active in TRNA Binding. Methods Enzymol. 1988, 164, 658–670. [Google Scholar] [CrossRef]
- Dinos, G.; Kalpaxis, D.L.; Wilson, D.N.; Nierhaus, K.H. Deacylated TRNA Is Released from the E Site upon A Site Occupation but before GTP Is Hydrolyzed by EF-Tu. Nucleic Acids Res. 2005, 33, 5291–5296. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibiotic | EC50 (μΜ) | EC50 (μgr/mL) |
|---|---|---|
| Chloramphenicol (CHL) | 1.76 | 0.57 |
| 1 (Lys) | 25.06 | 8.10 |
| 2 (Orn) | 20.36 | 6.58 |
| 3 (His) | 49.21 | 15.90 |
| 4 (Lys) | 29.62 | 9.57 |
| 5 (Orn) | 18.58 | 6.03 |
| 6 (His) | 39.90 | 12.89 |
| 7 (Lys) | 5.01 | 1.62 |
| 8 (Orn) | 13.21 | 4.27 |
| Antibiotic | Ki (μΜ) |
|---|---|
| Chloramphenicol | 1.7 |
| 1 (Lys) | 3.8 |
| 2 (Orn) | 16.8 |
| 3 (His) | 12.3 |
| 4 (Lys) | 22.5 |
| 5 (Orn) | 8.3 |
| 6 (His) | 8.5 |
| 7 (Lys) | 4.7 |
| 8 (Orn) | 2.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsirogianni, A.; Kournoutou, G.G.; Mpogiatzoglou, M.; Dinos, G.; Athanassopoulos, C.M. Chloramphenicol Derivatization in Its Primary Hydroxyl Group with Basic Amino Acids Leads to New Pharmacophores with High Antimicrobial Activity. Antibiotics 2023, 12, 832. https://doi.org/10.3390/antibiotics12050832
Tsirogianni A, Kournoutou GG, Mpogiatzoglou M, Dinos G, Athanassopoulos CM. Chloramphenicol Derivatization in Its Primary Hydroxyl Group with Basic Amino Acids Leads to New Pharmacophores with High Antimicrobial Activity. Antibiotics. 2023; 12(5):832. https://doi.org/10.3390/antibiotics12050832
Chicago/Turabian StyleTsirogianni, Artemis, Georgia G. Kournoutou, Maria Mpogiatzoglou, George Dinos, and Constantinos M. Athanassopoulos. 2023. "Chloramphenicol Derivatization in Its Primary Hydroxyl Group with Basic Amino Acids Leads to New Pharmacophores with High Antimicrobial Activity" Antibiotics 12, no. 5: 832. https://doi.org/10.3390/antibiotics12050832