Whitening Agents from Reseda luteola L. and Their Chemical Characterization Using Combination of CPC, UPLC-HRMS and NMR

 ,
,
Abstract
:1. Introduction
2. Material and Methods
2.1. Plant Material
2.2. Materials
2.3. Plant Extraction
2.3.1. Soxhlet Extraction
2.3.2. Liquid-Liquid Extraction (LLE)
2.4. Biological Activity Assays
2.4.1. Instrumentation
2.4.2. DPPH Radical Scavenging Assay
2.4.3. Tyrosinase Assay
2.4.4. Lipoxygenase Assay
2.5. High-Performance-Liquid-Chromatography
2.6. Centrifugal Partition Chromatography Purification
2.7. UPLC-ESI-HRMS Analyses of Fractions
2.8. 1H NMR Analysis of Isolated Compounds
3. Results and Discussion
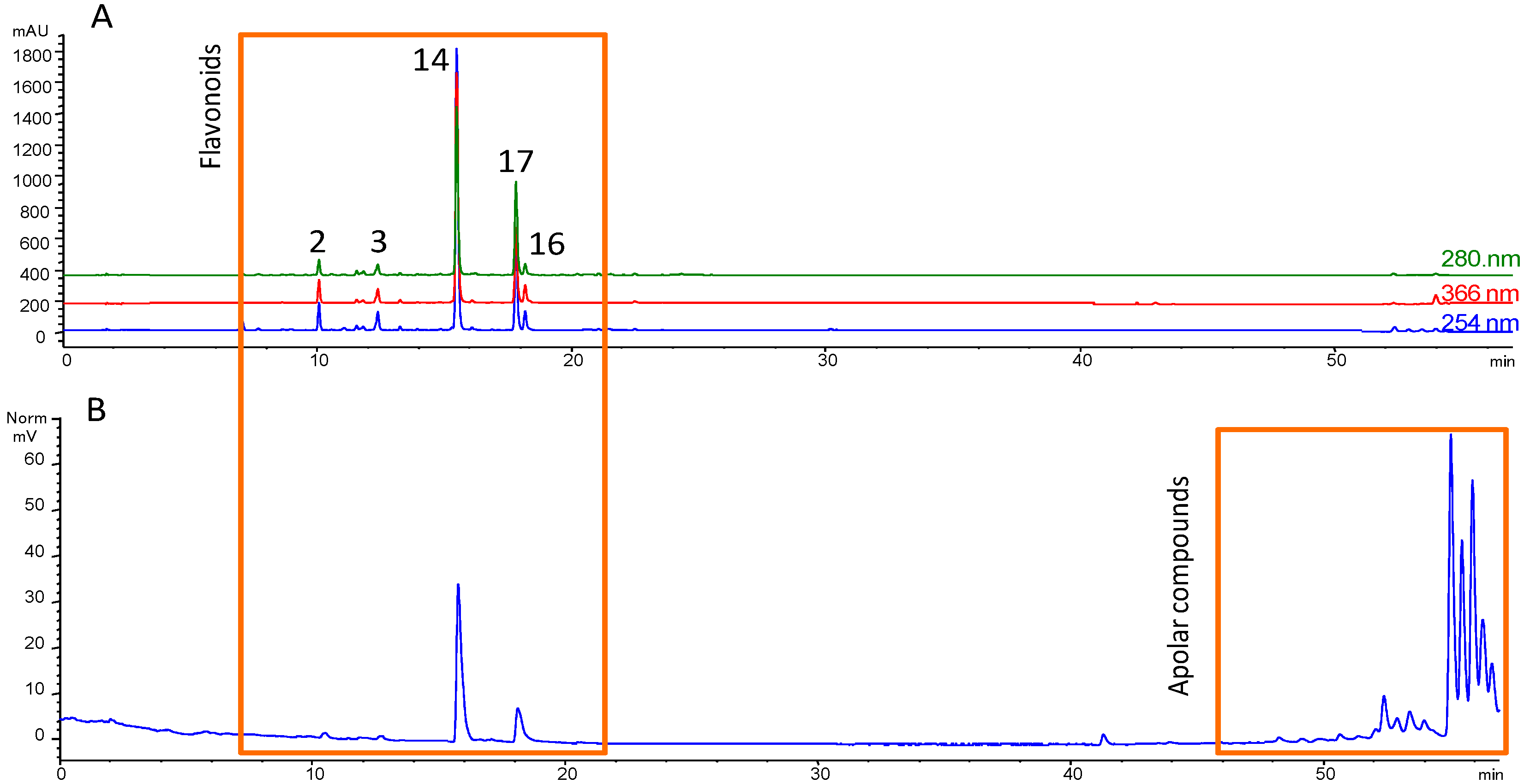
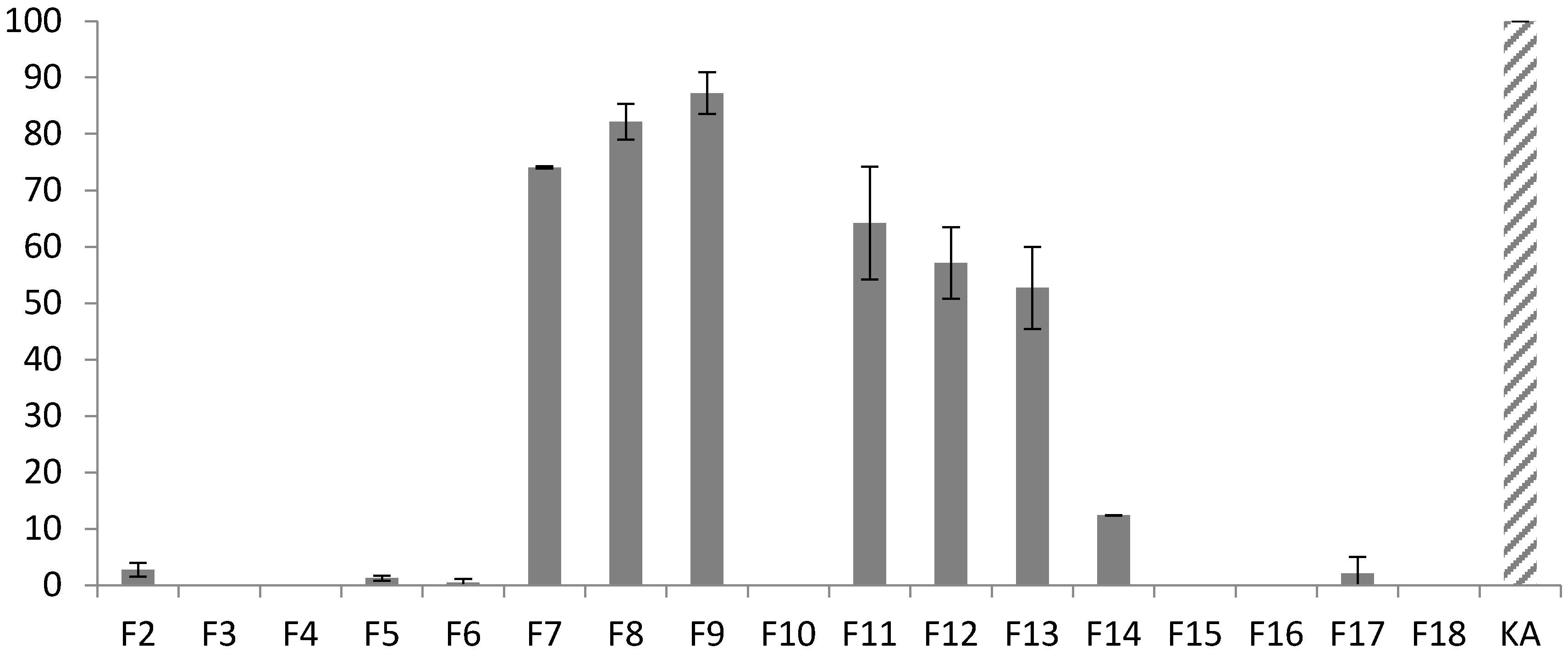
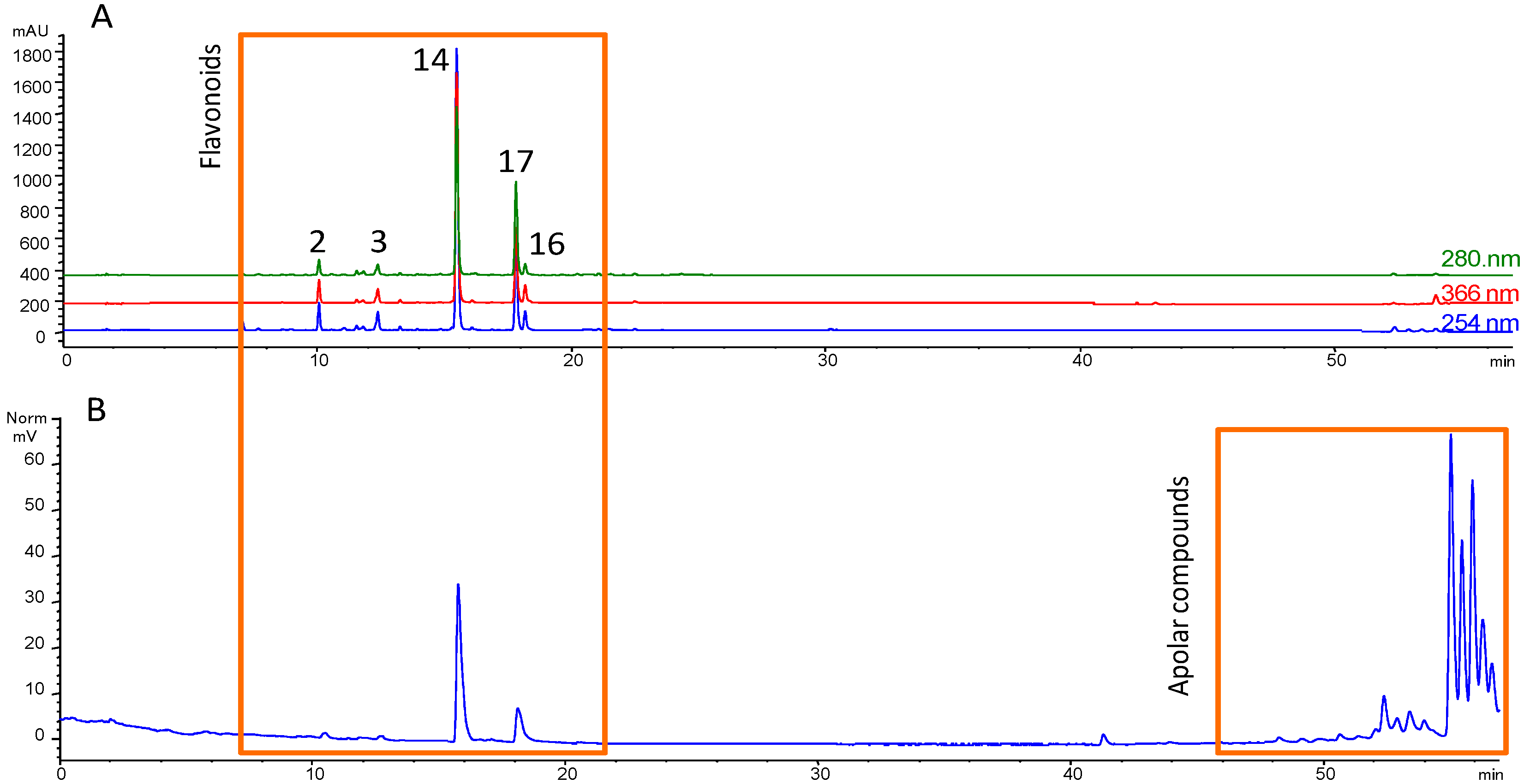
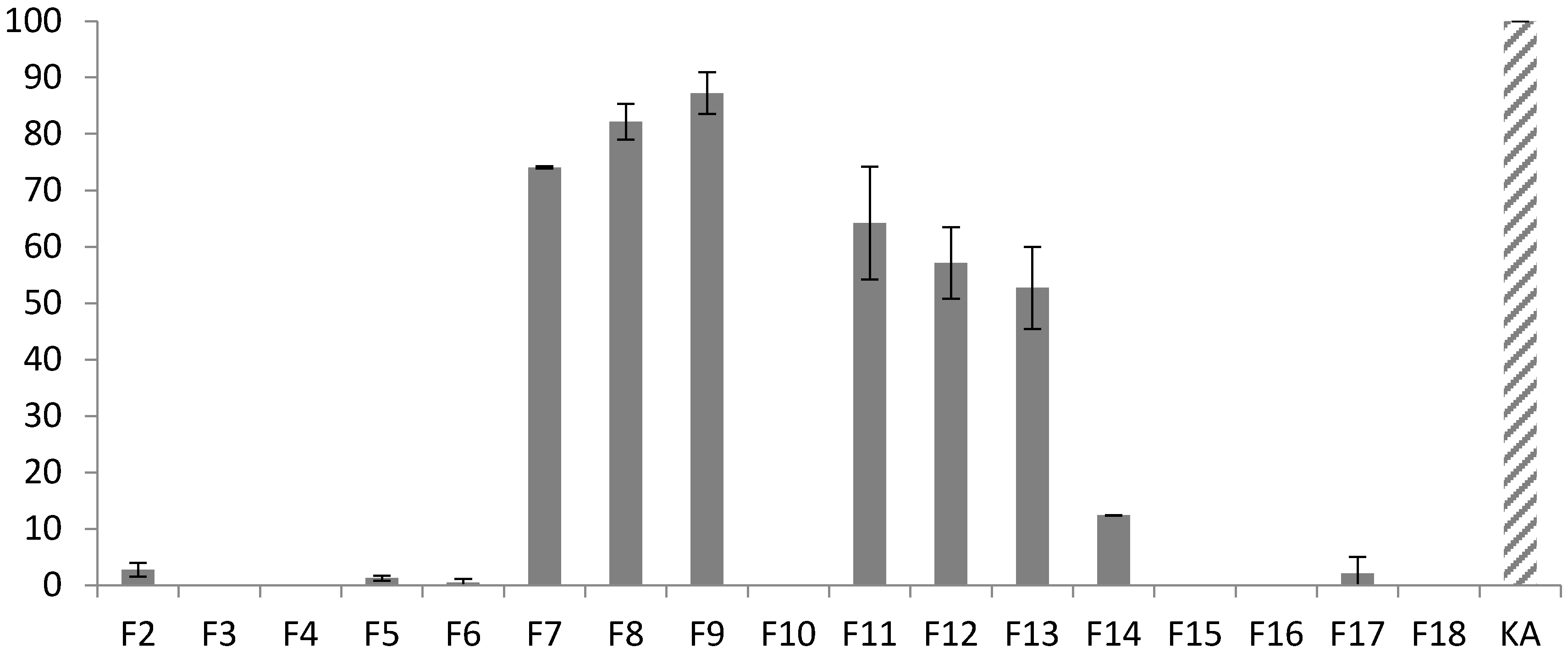
3.1. Fractionation Strategy
3.2. Putative Identification of Compounds
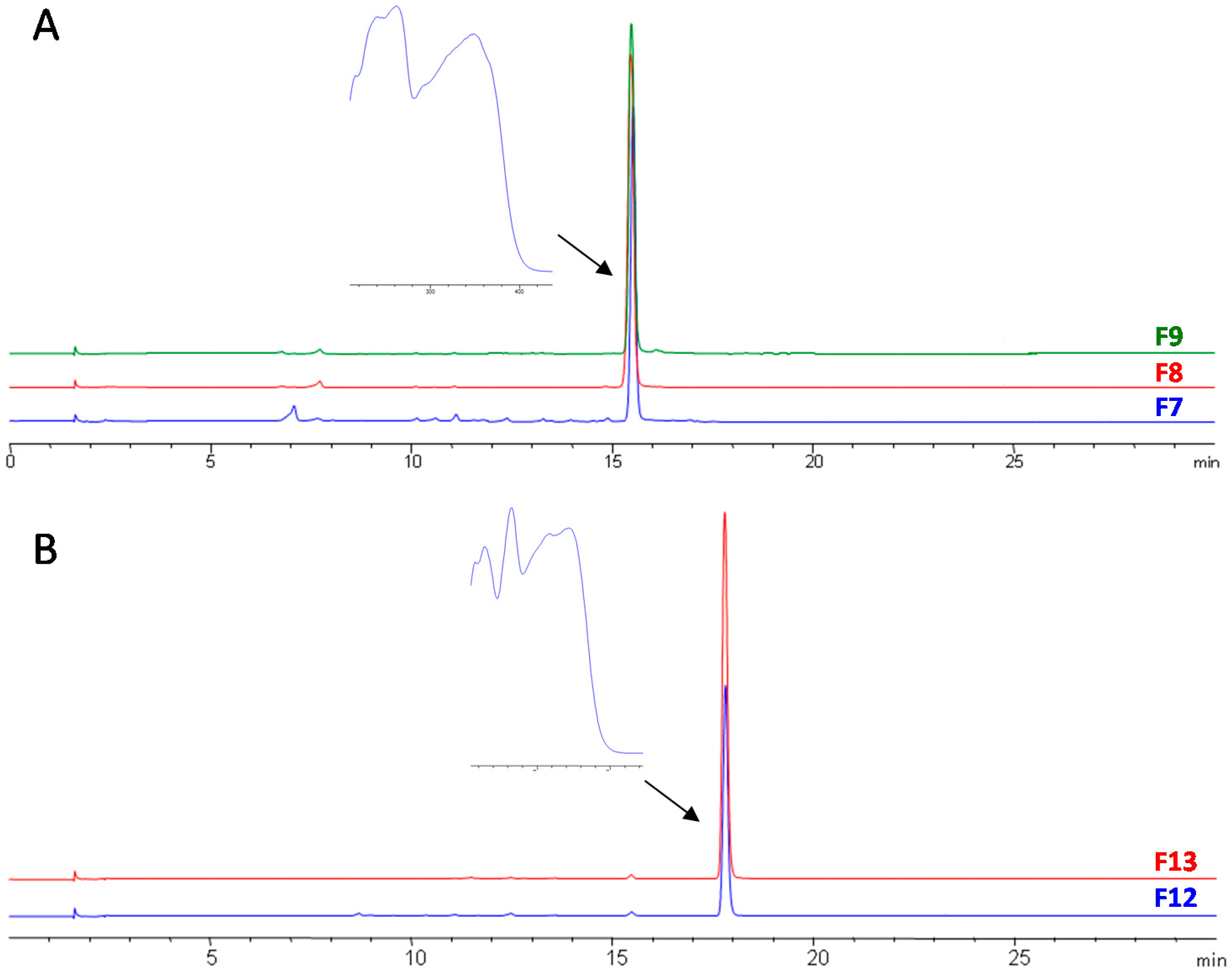
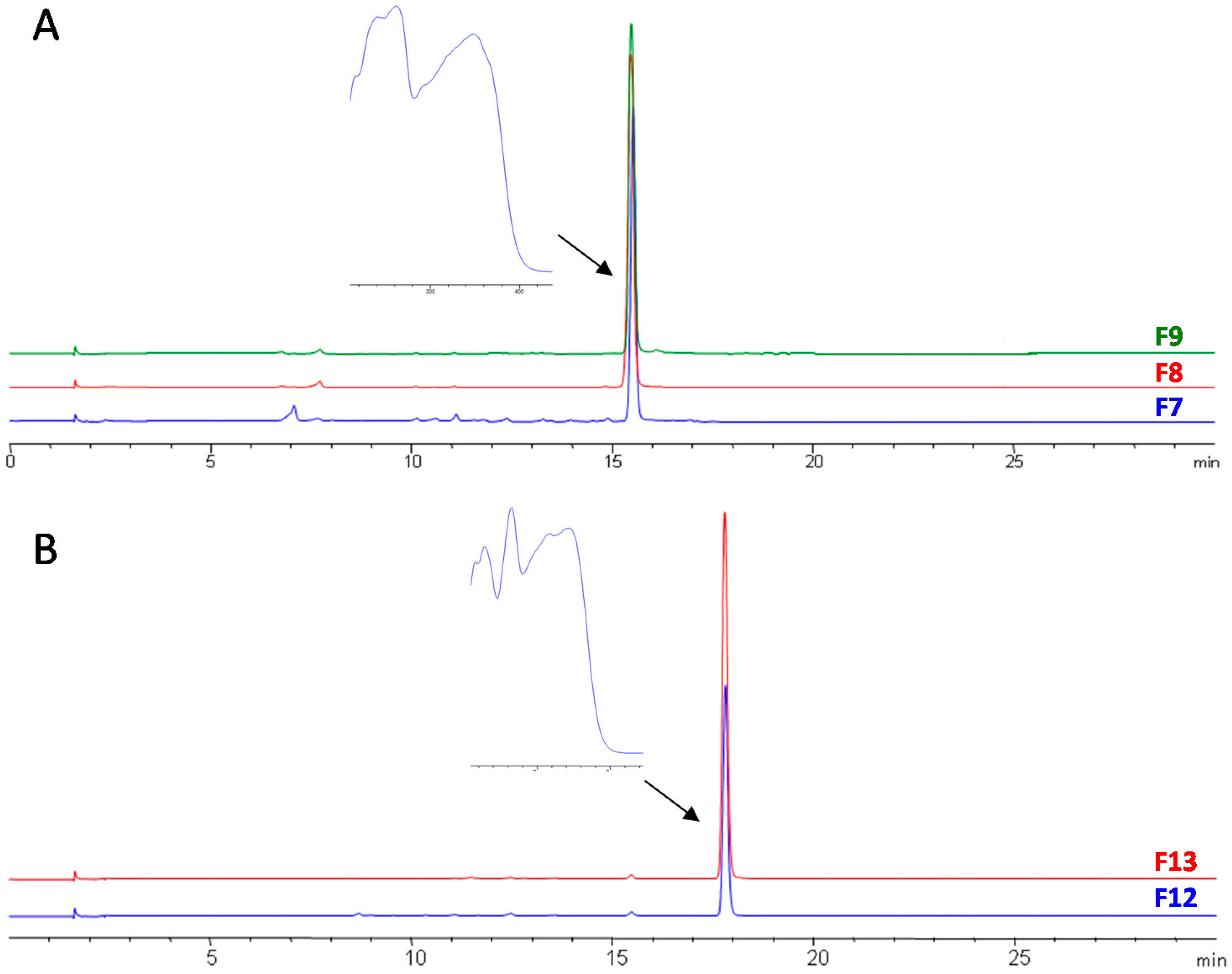
3.3. Final Purification and Identification of Active Compounds
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Burger, P.; Landreau, A.; Azoulay, S.; Michel, T.; Fernandez, X. Skin whitening cosmetics: Feedback and challenges in the development of natural skin lighteners. Cosmetics 2016, 3, 36. [Google Scholar] [CrossRef]
- Chang, T.-S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef] [PubMed]
- Couteau, C.; Coiffard, L. Overview of skin whitening agents: Drugs and cosmetic products. Cosmetics 2016, 3, 27. [Google Scholar] [CrossRef]
- Regulation (EC) No 1223/2009 of the European Parliament and of the Council of 30 November 2009 on Cosmetic Products. Available online: http://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2009:342:0059:0209:en:PDF (accessed on 21 September 2016).
- Kamakshi, R. Fairness via formulations: A review of cosmetic skin-lightening ingredients. J. Cosmet. Sci. 2012, 63, 43–54. [Google Scholar] [PubMed]
- Webb, D.A. Flora Europaea; Tutin, T.G., Heywood, V.H., Burges, N.A., Moore, D.M., Valentine, D.H., Walters, S.M., Eds.; Cambridge University Press: Cambridge, UK, 2010; Volume 5. [Google Scholar]
- Angelini, L.G.; Bertoli, A.; Rolandelli, S.; Pistelli, L. Agronomic potential of Reseda luteola L. as new crop for natural dyes in textiles production. Ind. Crops Prod. 2003, 17, 199–207. [Google Scholar] [CrossRef]
- Ferreira, E.S.; Hulme, A.N.; McNab, H.; Quye, A. The natural constituents of historical textile dyes. Chem. Soc. Rev. 2004, 33, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Guinot, P.; Rogé, A.; Gargadennec, A.; Garcia, M.; Dupont, D.; Lecoeur, E.; Candelier, L.; Andary, C. Dyeing plants screening: An approach to combine past heritage and present development. Colo. Technol. 2006, 122, 93–101. [Google Scholar] [CrossRef]
- Cerrato, A.; De Santis, D.; Moresi, M. Production of luteolin extracts from Reseda luteola and assessment of their dyeing properties. J. Sci. Food Agric. 2002, 82, 1189–1199. [Google Scholar] [CrossRef]
- Colorants Végétaux. Available online: http://www.critt-horticole.com/activite/colorants-vegetaux/references/ (accessed on 7 February 2017).
- Cristea, D.; Bareau, I.; Vilarem, G. Identification and quantitative HPLC analysis of the main flavonoids present in weld (Reseda luteola L.). Dyes Pigments 2003, 57, 267–272. [Google Scholar] [CrossRef]
- Moiteiro, C.; Gaspar, H.; Rodrigues, A.I.; Lopes, J.F.; Carnide, V. HPLC quantification of dye flavonoids in Reseda luteola L. from Portugal. J. Sep. Sci. 2008, 31, 3683–3687. [Google Scholar] [CrossRef] [PubMed]
- Villela, A.; van der Klift, E.J.C.; Mattheussens, E.S.G.M.; Derksen, G.C.H.; Zuilhof, H.; van Beek, T.A. Fast chromatographic separation for the quantitation of the main flavone dyes in Reseda luteola (weld). J. Chromatogr. A 2011, 1218, 8544–8550. [Google Scholar] [CrossRef] [PubMed]
- Friesen, J.B.; McAlpine, J.B.; Chen, S.-N.; Pauli, G.F. Countercurrent separation of natural products: An update. J. Nat. Prod. 2015, 78, 1765–1796. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Destandau, E.; Elfakir, C. New advances in countercurrent chromatography and centrifugal partition chromatography: Focus on coupling strategy. Anal. Bioanal. Chem. 2014, 406, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Koleva, I.I.; van Beek, T.A.; Linssen, J.P.H.; de Groot, A.; Evstatieva, L.N. Screening of plant extracts for antioxidant activity: A comparative study on three testing methods. Phytochem. Anal. PCA 2002, 13, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Ferrer, A.; Rodríguez-López, J.N.; García-Cánovas, F.; García-Carmona, F. Tyrosinase: A comprehensive review of its mechanism. Biochim. Biophys. Acta 1995, 1247, 1–11. [Google Scholar] [CrossRef]
- Rackova, L.; Oblozinsky, M.; Kostalova, D.; Kettmann, V.; Bezakova, L. Free radical scavenging activity and lipoxygenase inhibition of Mahonia aquifolium extract and isoquinoline alkaloids. J. Inflamm. Lond. Engl. 2007, 4, 15. [Google Scholar] [CrossRef] [PubMed]
- Seelinger, G.; Merfort, I.; Schempp, C.M. Anti-oxidant, anti-inflammatory and anti-allergic activities of luteolin. Planta Med. 2008, 74, 1667–1677. [Google Scholar] [CrossRef] [PubMed]
- Seo, B.; Yun, J.; Lee, S.; Kim, M.; Hwang, K.; Kim, J.; Min, K.R.; Kim, Y.; Moon, D. Barbarin as a new tyrosinase inhibitor from Barbarea orthocerus. Planta Med. 1999, 65, 683–686. [Google Scholar] [CrossRef] [PubMed]
- Kjaer, A.; Gmelin, R. Isothiocyanates XXVIII. A new isothiocyanate glucoside (glucobarbarin) furnishing (−)-5-phenyl-2-oxazolidinethione upon enzymic hydrolysis. Acta Chem. Scand. 1957, 11, 906–907. [Google Scholar] [CrossRef]
- Marques, R.; Sousa, M.M.; Oliveira, M.C.; Melo, M.J. Characterization of weld (Reseda luteola L.) and spurge flax (Daphne gnidium L.) by high-performance liquid chromatography-diode array detection-mass spectrometry in Arraiolos historical textiles. J. Chromatogr. A 2009, 1216, 1395–1402. [Google Scholar] [CrossRef] [PubMed]
- Peggie, D.A.; Hulme, A.N.; McNab, H.; Quye, A. Towards the identification of characteristic minor components from textiles dyed with weld (Reseda luteola L.) and those dyed with Mexican cochineal (Dactylopius coccus Costa). Microchim. Acta 2008, 162, 371–380. [Google Scholar] [CrossRef]
- Schmidt, J. Negative ion electrospray high-resolution tandem mass spectrometry of polyphenols. J. Mass Spectrom. 2016, 51, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Batirov, E.K.; Tadzhibaev, M.M.; Malikov, V.M. Flavonoids of Reseda luteola. Chem. Nat. Compd. 1979, 15, 643–644. [Google Scholar] [CrossRef]
- Geiger, H.; Krumbein, B. Uber zwei weitere Luteolinglykoside aus Reseda luteola L. Z. Naturforschung C J. Biosci. 1973, 28, 773. [Google Scholar]
- Benmerache, A.; Berrehal, D.; Khalfallah, A.; Kabouche, A.; Semra, Z.; Kabouche, Z. Antioxidant, antibacterial activities and flavonoids of Reseda phyteuma L. Pharm. Lett. 2012, 4, 1878–1882. [Google Scholar]
- Woelfle, U.; Simon-Haarhaus, B.; Merfort, I.; Schempp, C.M. Reseda luteola L. extract displays antiproliferative and pro-apoptotic activities that are related to its major flavonoids. Phytother. Res. 2010, 24, 1033–1036. [Google Scholar] [PubMed]
- Lee, J.Y.; Chang, E.J.; Kim, H.J.; Park, J.H.; Choi, S.W. Antioxidative flavonoids from leaves of Carthamus tinctorius. Arch. Pharm. Res. 2002, 25, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Svehliková, V.; Bennett, R.N.; Mellon, F.A.; Needs, P.W.; Piacente, S.; Kroon, P.A.; Bao, Y. Isolation, identification and stability of acylated derivatives of apigenin 7-O-glucoside from chamomile (Chamomilla recutita [L.] Rauschert). Phytochemistry 2004, 65, 2323–2332. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.-B.; Yu, Z.-Y.; Wang, X.; Zhao, J.; Geng, Y.-L.; Liu, J.-H.; Duan, L.I.U. Preparative isolation and purification of flavonoids from the flower of Chrysanthemum morifolium Ramat. by High-speed Counter-current Chromatography. Chem. Ind. For. Prod. 2014, 34, 17–22. [Google Scholar] [CrossRef]
- Wang, X.; Cheng, C.; Sun, Q.; Li, F.; Liu, J.; Zheng, C. Isolation and purification of four flavonoid constituents from the flowers of Paeonia suffruticosa by high-speed counter-current chromatography. J. Chromatogr. A 2005, 1075, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, J.; Sun, Y.; Li, S.; Wang, H. Purification of caffeic acid, chlorogenic acid and luteolin from Caulis Lonicerae by high-speed counter-current chromatography. Sep. Purif. Technol. 2008, 63, 721–724. [Google Scholar] [CrossRef]
- Zhao, X.-H.; Han, F.; Li, Y.-L.; Zhou, G.-Y.; Yue, H.-L. Semi-preparative separation and purification of three flavonoids from Pedicularis longiflora var. tubiformis (Klotzsch) PC Tsoong by HSCCC. J. Liq. Chromatogr. Relat. Technol. 2013, 36, 1751–1761. [Google Scholar]
- Yang, F.; Quan, J.; Zhang, T.Y.; Ito, Y. Multidimensional counter-current chromatographic system and its application. J. Chromatogr. A 1998, 803, 298–301. [Google Scholar] [CrossRef]
- An, S.M.; Kim, H.J.; Kim, J.-E.; Boo, Y.C. Flavonoids, taxifolin and luteolin attenuate cellular melanogenesis despite increasing tyrosinase protein levels. Phytother. Res. 2008, 22, 1200–1207. [Google Scholar] [CrossRef] [PubMed]
- Casetti, F.; Jung, W.; Wölfle, U.; Reuter, J.; Neumann, K.; Gilb, B.; Wähling, A.; Wagner, S.; Merfort, I.; Schempp, C.M. Topical application of solubilized Reseda luteola extract reduces ultraviolet B-induced inflammation in vivo. J. Photochem. Photobiol. B 2009, 96, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Funakoshi-Tago, M.; Nakamura, K.; Tago, K.; Mashino, T.; Kasahara, T. Anti-inflammatory activity of structurally related flavonoids, apigenin, luteolin and fisetin. Int. Immunopharmacol. 2011, 11, 1150–1159. [Google Scholar] [CrossRef] [PubMed]
- Seelinger, G.; Merfort, I.; Wölfle, U.; Schempp, C.M. Anti-carcinogenic effects of the flavonoid luteolin. Molecules 2008, 13, 2628–2651. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Gupta, S. Apigenin: A promising molecule for cancer prevention. Pharm. Res. 2010, 27, 962–978. [Google Scholar] [CrossRef] [PubMed]
- Romanová, D.; Vachálková, A.; Cipák, L.; Ovesná, Z.; Rauko, P. Study of antioxidant effect of apigenin, luteolin and quercetin by DNA protective method. Neoplasma 2001, 48, 104–107. [Google Scholar] [PubMed]
- Kubo, I.; Kinst-Hori, I.; Chaudhuri, S.K.; Kubo, Y.; Sánchez, Y.; Ogura, T. Flavonols from Heterotheca inuloides: Tyrosinase inhibitory activity and structural criteria. Bioorg. Med. Chem. 2000, 8, 1749–1755. [Google Scholar] [CrossRef]
- Xie, L.-P.; Chen, Q.-X.; Huang, H.; Wang, H.-Z.; Zhang, R.-Q. Inhibitory effects of some flavonoids on the activity of mushroom tyrosinase. Biochemistry (Mosc.) 2003, 68, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Kukula-Koch, W.; Koch, W.; Stasiak, N.; Głowniak, K.; Asakawa, Y. Quantitative standarization and CPC-based recovery of pharmacologically active components from Polygonum tinctorium Ait. leaf extracts. Ind. Crops Prod. 2015, 69, 324–328. [Google Scholar] [CrossRef]
- Desmedt, B.; Courselle, P.; De Beer, J.O.; Rogiers, V.; Grosber, M.; Deconinck, E.; De Paepe, K. Overview of skin whitening agents with an insight into the illegal cosmetic market in Europe. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 943–950. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Extract | Inhibition (%) | ||
|---|---|---|---|
| DPPH | Tyrosinase | Lipoxygenase | |
| RL1 | 7.00 ± 0.05 | 12.00 ± 0.03 | / |
| RL2 | 7.00 ± 0.05 | 38.00 ± 0.03 | / |
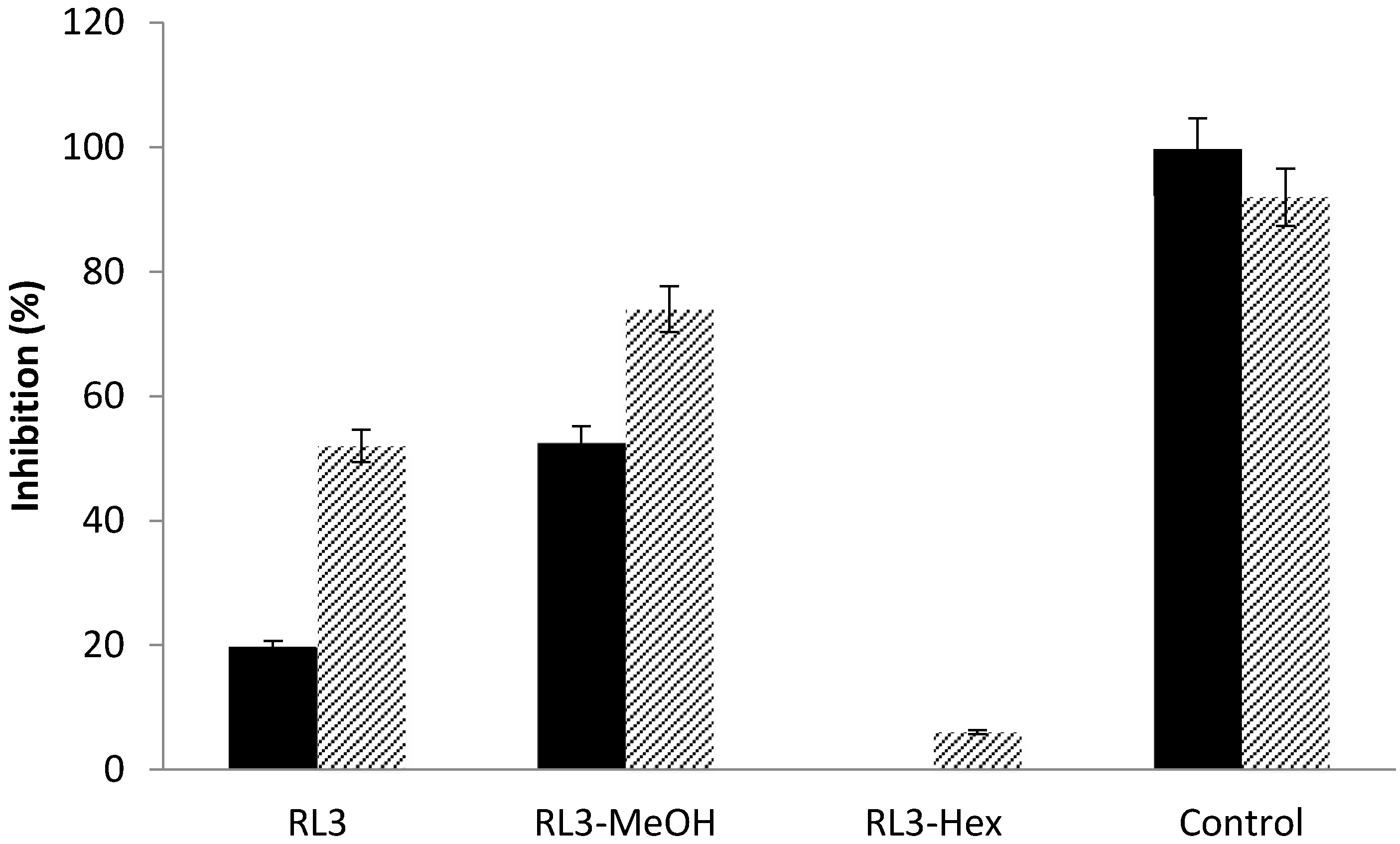
| RL3 | 25.00 ± 0.05 | 45.00 ± 0.01 | 52.00 ± 0.05 |
| RL4 | 53.00 ± 0.05 | 18.00 ± 0.01 | 63.00 ± 0.05 |
| RL5 | 16.00 ± 0.05 | / | / |
| Vitamin C | 99.00 ± 0.05 | - | - |
| Kojic acid | - | 96.00 ± 0.00 | - |
| Quercetin hydrate | - | - | 92.00 ± 0.05 |
| Compound | rt (min) | Positive Mode | Negative Mode | RDBeq | Putative Identification | ||||
|---|---|---|---|---|---|---|---|---|---|
| [M + H]+ m/z | EC | Fragment ions (m/z) | [M − H]− m/z | EC | Fragment Ions (m/z) | ||||
| 1 | 6.53 | 527.0884 | C18H23O18 | 365.0335 | 525.0695 | C18H21O18 | 363.0218 | 8 | Unknown |
| 153.0188 | 299.0564 | ||||||||
| 150.9901 | |||||||||
| 91.0100 | |||||||||
| 2 | 6.62 | 611.1619 | C27H31O16 | 449.1084 | 609.1461 | C27H29O16 | 447.0848 | 14 | Luteolin-3',7-diglucoside |
| 287.0568 | 285.0420 | ||||||||
| 365.0343 | |||||||||
| 3 | 7.67 | 449.1079 | C21H21O11 | 287.0555 | 447.0927 | C21H19O11 | 285.0382 | 12 | Luteolin glycoside |
| 4 | 8.67 | 433.1132 | C21H21O10 | 301.0723 | 431.0975 | C21H19O10 | 268.0375 | 12 | Apigenin-7-O-glucoside |
| 271.0615 | |||||||||
| 153.0190 | |||||||||
| 119.0504 | |||||||||
| 5 | 9.12 | 491.119 | C23H23O12 | 287.0566 | 489.1016 | C23H21O12 | 285.0397 | 13 | Acetylated luteolin-O-glycoside |
| 153.0566 | 429.0814 | ||||||||
| 6 | 9.30 | 449.1084 | C21H21O11 | 287.0558 | 447.0927 | C21H19O11 | 285.0390 | 12 | Luteolin glycoside |
| 153.0184 | |||||||||
| 7 | 9.57 | 365.0328 | C12H13O13 | 286.0471 | 363.0173 | C12H11O13 | 299.0561 | 7 | Unknown |
| 258.0539 | 211.005 | ||||||||
| 153.0193 | 173.0255 | ||||||||
| 151.0023 | |||||||||
| 125.0230 | |||||||||
| 109.0312 | |||||||||
| 8 | 9.91 | 491.119 | C23H23O12 | 287.0558 | 489.1033 | C23H21O12 | 285.0413 | 13 | Acetylated luteolin-O-glycoside |
| 153.0184 | |||||||||
| 9 | 10.04 | 491.119 | C23H23O12 | 287.0558 | 489.1033 | C23H21O12 | 285.0468 | 13 | Acetylated luteolin-O-glycoside |
| 153.0184 | |||||||||
| 10 | 10.21 | 475.124 | C23H23O11 | 271.0617 | 473.1084 | C23H21O11 | 268.0369 | 13 | Acetylated apigenin-O-glycoside |
| 153.0184 | 269.0456 | ||||||||
| 199.1416 | |||||||||
| 11 | 11.00 | 475.1247 | C23H23O11 | 271.0605 | 473.107 | C23H21O11 | 285.0338 | 13 | Acetylated apigenin-O-glycoside |
| 153.0184 | 268.0369 | ||||||||
| 269.0456 | |||||||||
| 12 | 11.40 | 533.1298 | C25H25O13 | 287.0558 | 531.1096 | C25H23O13 | 327.2147 | 14 | Unknown |
| 153.0184 | 285.0399 | ||||||||
| 13 | 11.60 | 287.0561 | C15H11O6 | 269.0455 | 285.0401 | C15H9O6 | 241.0473 | 11 | Luteolin |
| 241.0492 | 217.0513 | ||||||||
| 213.0549 | 199.0341 | ||||||||
| 179.0345 | 175.0378 | ||||||||
| 153.0190 | 151.0019 | ||||||||
| 135.0447 | 133.0285 | ||||||||
| 117.0338 | 107.0126 | ||||||||
| 14 | 12.17 | 555.0931 | C30H19O11 | 403.0839 | 553.0783 | C30H17O11 | 459.0345 | 22 | Unknown |
| 287.0559 | 433.0530 | ||||||||
| 153.0180 | 391.0460 | ||||||||
| 285.0389 | |||||||||
| 15 | 12.43 | 301.0718 | C16H13O6 | 286.0484 | 299.0553 | C16H11O6 | 284.0312 | 11 | O-Methylluteolin |
| 258.0535 | 256.0360 | ||||||||
| 229.0504 | 227.0360 | ||||||||
| 153.0199 | 199.0379 | ||||||||
| 105.0707 | 151.0028 | ||||||||
| 16 | 12.56 | 271.0609 | C15H11O5 | 229.0520 | 269.0451 | C15H9O5 | 225.0554 | 11 | Apigenin |
| 187.0404 | 201.0554 | ||||||||
| 153.0187 | 183.0455 | ||||||||
| 119.0499 | 151.0032 | ||||||||
| 149.0236 | |||||||||
| 117.0343 | |||||||||
| 107.0128 | |||||||||
| 83.0142 | |||||||||
| 17 | 13.02 | 495.1291 | C26H23O10 | 409.0918 | 493.1097 | C26H21O10 | 403.3051 | 16 | Unknown |
| 287.0552 | 285.0408 | ||||||||
| 257.0459 | |||||||||
| 18 | 13.18 | 465.1179 | C25H21O9 | 447.1079 | 463.102 | C25H19O9 | 283.0240 | 16 | Unknown |
| 287.0549 | 255.0293 | ||||||||
| 286.0464 | |||||||||
| 257.0454 | |||||||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burger, P.; Monchot, A.; Bagarri, O.; Chiffolleau, P.; Azoulay, S.; Fernandez, X.; Michel, T. Whitening Agents from Reseda luteola L. and Their Chemical Characterization Using Combination of CPC, UPLC-HRMS and NMR. Cosmetics 2017, 4, 51. https://doi.org/10.3390/cosmetics4040051
Burger P, Monchot A, Bagarri O, Chiffolleau P, Azoulay S, Fernandez X, Michel T. Whitening Agents from Reseda luteola L. and Their Chemical Characterization Using Combination of CPC, UPLC-HRMS and NMR. Cosmetics. 2017; 4(4):51. https://doi.org/10.3390/cosmetics4040051
Chicago/Turabian StyleBurger, Pauline, André Monchot, Olivier Bagarri, Philippe Chiffolleau, Stéphane Azoulay, Xavier Fernandez, and Thomas Michel. 2017. "Whitening Agents from Reseda luteola L. and Their Chemical Characterization Using Combination of CPC, UPLC-HRMS and NMR" Cosmetics 4, no. 4: 51. https://doi.org/10.3390/cosmetics4040051