
Clinical Practice Guidelines for the Diagnosis and Management of Hereditary Fructose Intolerance



Abstract
1. Introduction
1.1. Epidemiology
1.2. Pathophysiology
1.3. Clinic
1.4. Diagnosis
1.5. Treatment
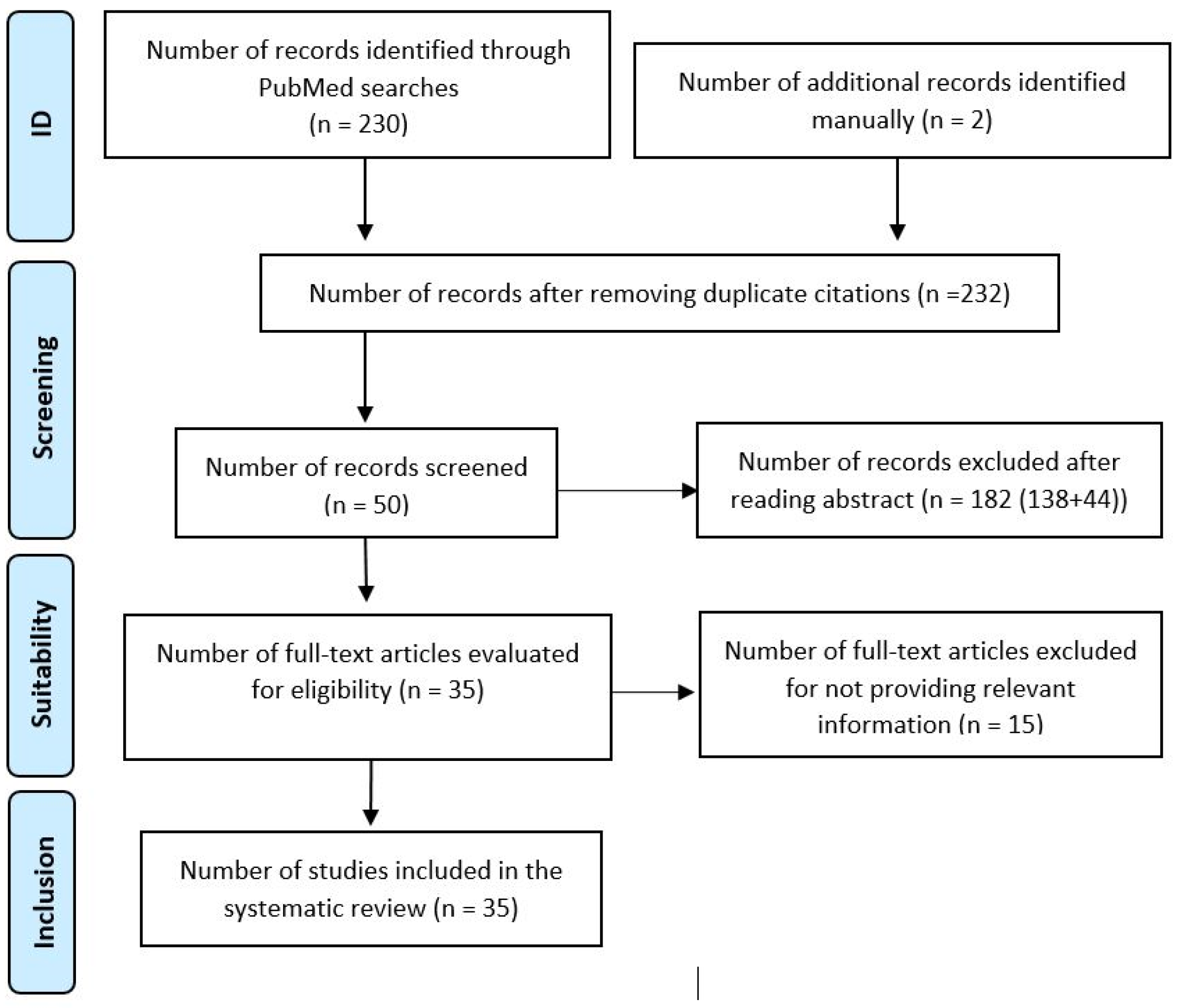
2. Material and Methods
- 1.
- Initial Search: A general search with the term ‘hereditary fructose intolerance’ yielded 1089 publications in PubMed. Fourteen articles were included for a comprehensive understanding, including aspects such as symptomatology, differential diagnosis, and treatment options. This step was carried out between 1 December 2022 and 31 January 2023.
- 2.
- Systematic Search: Terms related to prevalent pathologies (kidney disease, liver disease, hypoglycemia, failure to thrive) were combined with ‘hereditary fructose intolerance’ in a systematic search. Using the Boolean operators AND and OR, the most appropriate combination of terms was created to yield the best results. The combination was as follows: (hereditary fructose intolerance) AND ((kidney disease) OR (liver disease) OR (hypoglycemia) OR (failure to thrive)). A total of 346 results was obtained in PubMed. Before proceeding to the selection of articles, the inclusion and exclusion criteria were defined:
- Inclusion criteria: Any paper related to the pathology associated with hereditary fructose intolerance in humans, including studies, reviews, case series, editorials, guidelines, etc.
- Exclusion criteria: Unusual manifestations, diseases not related to hereditary fructose intolerance, and, finally, pathology in animals.
A total of 230 articles was obtained after applying inclusion and exclusion criteria; 48 articles were selected for further analysis. Fifteen articles were discarded for not adding relevant information. This step was conducted between 1 February 2022 and 31 March 2023. - 3.
- Manual Search: Based on references from the selected studies, 2 additional articles were included, bringing the total to 35 empirical articles published between 1961 and 2023. This step took place during the month of May 2023.
3. Results
- 1.
- Renal pathology:
- Proximal tubular dysfunction.
- Nephrolithiasis/nephrocalcinosis.
- 2.
- Growth retardation:
- Impaired growth in children.
- 3.
- Hepatopathy:
- Acute manifestations.
- Fatty liver.
- 4.
- Irritable bowel syndrome.
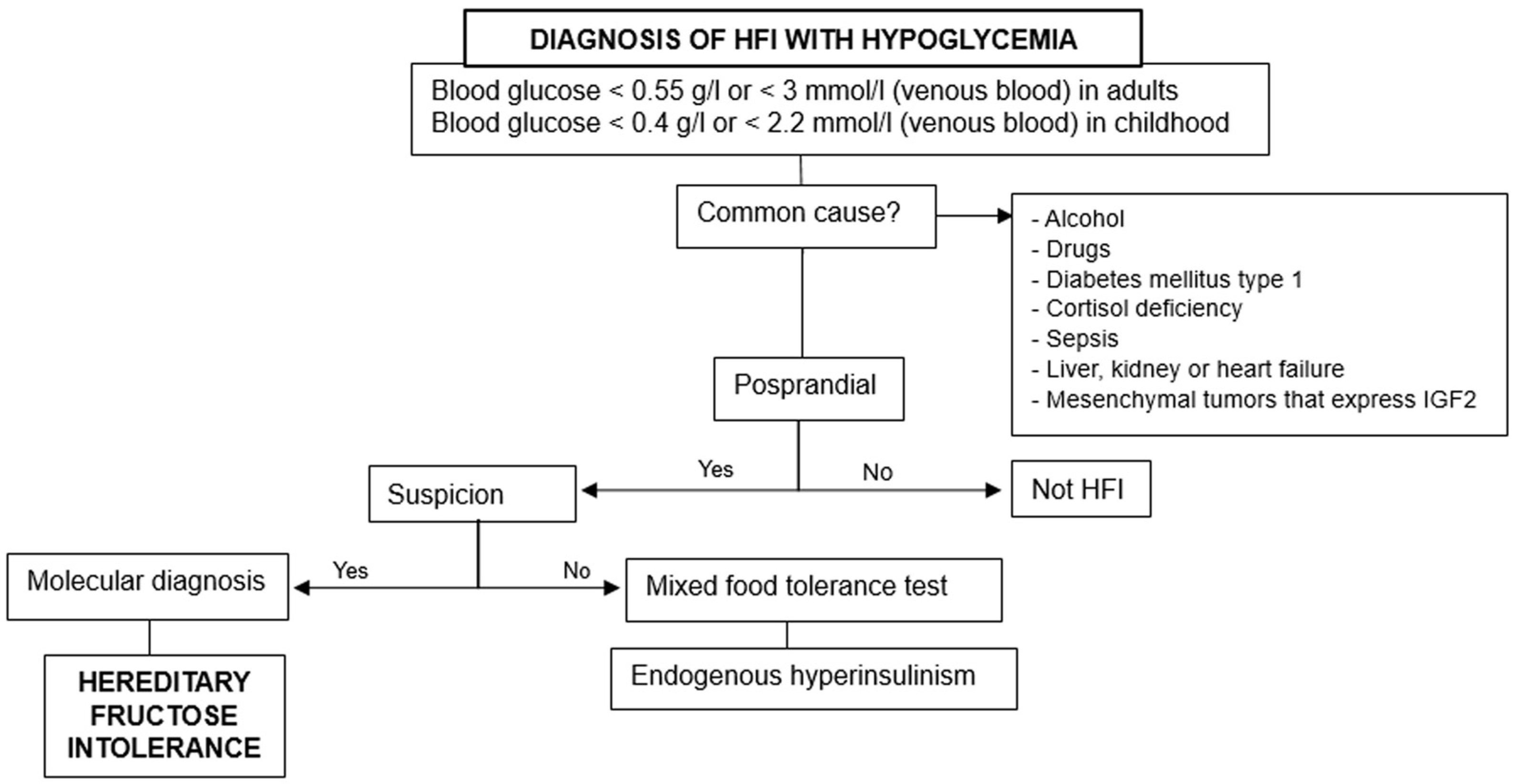
3.1. Hypoglycemia
- 1.
- Rule out the previous common causes and others based on fasting, postprandial, or exercise-induced hypoglycemia.
- 2.
- Consider hereditary fructose intolerance if triggered by food ingestion.
- 3.
- Perform molecular diagnosis for confirmation, especially when there are suggestive data [18].
3.2. Kidney Pathology
3.3. Growth Delay
3.4. Liver Pathology
3.5. Irritable Bowel Syndrome
4. Discussion
5. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Lee, H.J.; Cha, J.Y. Recent insights into the role of ChREBP in intestinal fructose absorption and metabolism. BMB Rep. 2018, 51, 429–436. [Google Scholar] [CrossRef]
- Vantyghem, M.C.; Mention, C.; Dobbelaere, D.; Douillard, C. Hypoglycemia and endocrine effects of adults’ inborn errors of metabolism. Ann. Endocrinol. 2009, 70, 25–42. [Google Scholar] [CrossRef]
- Aldag, E.; Fan, E.M.; Marshall, I.; Christensen, R.D.; Shayota, B.J.; Meznarich, J.A. An Infant with Hereditary Fructose Intolerance and a Novel Presentation of Disseminated Intravascular Coagulopathy Following Pyloromyotomy. J. Pediatr. Hematol. Oncol. 2022, 44, 409–411. [Google Scholar] [CrossRef]
- Buziau, A.M.; Schalkwijk, C.G.; Stehouwer, C.D.A.; Tolan, D.R.; Brouwers, M.C.G.J. Recent advances in the pathogenesis of hereditary fructose intolerance: Implications for its treatment and the understanding of fructose-induced non-alcoholic fatty liver disease. Cell. Mol. Life Sci. 2020, 77, 1709–1719. [Google Scholar] [CrossRef] [PubMed]
- Di Dato, F.; Spadarella, S.; Puoti, M.G.; Caprio, M.G.; Pagliardini, S.; Zuppaldi, C.; Vallone, G.; Fecarotta, S.; Esposito, G.; Iorio, R.; et al. Daily fructose traces intake and liver injury in children with hereditary fructose intolerance. Nutrients 2019, 11, 2397. [Google Scholar] [CrossRef] [PubMed]
- El-Shabrawi, M.H.F.; Kamal, N.M. Medical management of chronic liver diseases in children (Part I): Focus on curable or potentially curable diseases. Pediatr. Drugs 2011, 13, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Guery, M.J.; Douillard, C.; Marcelli-Tourvieille, S.; Dobbelaere, D.; Wemeau, J.L.; Vantyghem, M.C. Doctor, my son is so tired... about a case of hereditary fructose intolerance. Ann. Endocrinol. 2007, 68, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, F.C.; Sperb-Ludwig, F.; Schwartz, I.V.D. Epidemiological aspects of hereditary fructose intolerance: A database study. Hum. Mutat. 2021, 42, 1548–1566. [Google Scholar] [CrossRef]
- Stormon, M.O.; Cutz, E.; Furuya, K.; Bedford, M.; Yerkes, L.; Tolan, D.R.; Feigenbaum, A. A six-month-old infant with liver steatosis. J. Pediatr. 2004, 144, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Parliament. Decision No 1295/1999/EC of the European Parliament and of the Council. D of the Eur Communities 2003, 8, 1–6. Available online: https://eur-lex.europa.eu/legal-content/en/TXT/?uri=CELEX:31999D1295 (accessed on 5 January 2024).
- Corless, J.A.; Allsup, D.J.; Deeble, T.J.; Delaney, J.C. Self Assessment Questions: A pulmonary mass and hyperviscosity Abdominal pain in a 22 year old woman. Postgrad. Med. J. 2000, 76, 582–586. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, A.Y.; Hughes, J.J.; Dempsey, A.P.; Schatz, K.S.; Wang, T.; Gunay-Aygun, M. Pitfalls in the diagnosis of hereditary fructose intolerance. Pediatrics 2020, 146, e20193324. [Google Scholar] [CrossRef]
- Riveros, M.J.; Parada, A.; Pettinelli, P. Fructose consumption and its implications for health; Fructose malabsorption and nonalcoholic fatty liver. Nutr. Hosp. 2014, 29, 491–499. [Google Scholar] [PubMed]
- Demirbas, D.; Brucker, W.J.; Berry, G.T. Inborn Errors of Metabolism with Hepatopathy: Metabolism Defects of Galactose, Fructose, and Tyrosine. Pediatr. Clin. N. Am. 2018, 65, 337–352. [Google Scholar] [CrossRef]
- Kim, M.S.; Moon, J.S.; Kim, M.J.; Seong, M.W.; Park, S.S.; Ko, J.S. Hereditary Fructose Intolerance Diagnosed in Adulthood. Gut Liver 2021, 15, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Byers, H.M.; Diaz-Kuan, A.; Vos, M.B.; Hall, P.L.; Tortorelli, S.; Singh, R.; Wallenstein, M.B.; Allain, M.; Dimmock, D.P.; et al. Acute liver failure in neonates with undiagnosed hereditary fructose intolerance due to exposure from widely available infant formulas. Mol. Genet. Metab. 2018, 123, 428–432. [Google Scholar] [CrossRef]
- Donner, M.G.; Erhardt, A.; Häussinger, D. Stoffwechselerkrankungen der Leber—Teil 2: Glykogenosen, hereditäre Fruktoseintoleranz, Galaktosadie; mie und hepatische Porphyrien. Dtsch. Med. Wochenschr. 2010, 135, 2540–2547. [Google Scholar] [CrossRef]
- Douillard, C.; Mention, K.; Dobbelaere, D.; Wemeau, J.L.; Saudubray, J.M.; Vantyghem, M.C. Hypoglycaemia related to inherited metabolic diseases in adults. Orphanet J. Rare Dis. 2012, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Aldámiz-Echevarría, L.; Heras, J.d.L.; Couce, M.L.; Alcalde, C.; Vitoria, I.; Bueno, M.; Blasco-Alonso, J.; García, M.C.; Ruiz, M.; Suárez, R.; et al. Non-alcoholic fatty liver in hereditary fructose intolerance. Clin. Nutr. 2020, 39, 455–459. [Google Scholar] [CrossRef]
- Ruiz-Pons, M.; de Las Heras, J.; Aldámiz-Echevarría, L. Errores congénitos del metabolismo de la fructosa. In Diagnóstico y Tratamiento de las Enfermedades Metabólicas Hereditarias, 5th ed; Couce, M.L., Aldámiz-Echevarría, L., García-Jiménez, M.C., González Lamuño, D., Eds.; Ergon: Madrid, Spain, 2022; pp. 729–739. [Google Scholar]
- Odièvre, M.; Gentil, C.; Gautier, M.; Alagille, D. Hereditary Fructose Intolerance in Childhood: Diagnosis, Management, and Course in 55 Patients. Am. J. Dis. Child. 1978, 132, 605–608. [Google Scholar] [CrossRef]
- Fiocchi, A.; Dionisi-Vici, C.; Cotugno, G.; Koch, P.; Dahdah, L. Fruit-induced FPIES masquerading as hereditary fructose intolerance. Pediatrics 2014, 134, e602-5. [Google Scholar] [CrossRef]
- Cano, A.; Alcalde, C.; Belanger-Quintana, A.; Cañedo-Villarroya, E.; Ceberio, L.; Chumillas-Calzada, S.; Correcher, P.; Couce, M.L.; García-Arenas, D.; Gómez, I.; et al. Vitamin C and folate status in hereditary fructose intolerance. Eur. J. Clin. Nutr. 2022, 76, 1733–1739. [Google Scholar] [CrossRef]
- Shrestha, D.B.; Budhathoki, P.; Sedhai, Y.R.; Mandal, S.K.; Shikhrakar, S.; Karki, S.; Baniya, R.K.; Kashiouris, M.G.; Qiao, X.; Fowler, A.A. Vitamin C in Critically Ill Patients: An Updated Systematic Review and Meta-Analysis. Nutrients 2021, 13, 3564. [Google Scholar] [CrossRef]
- Odievre, M. Clinical presentation of metabolic liver disease. J. Inherit. Metab. Dis. 1991, 14, 526–530. [Google Scholar] [CrossRef]
- Izquierdo García, E. Role of the Pharmacist in Response to the Social and Health Needs of Patients with Hereditary Fructose Intolerance; Thesis, Universidad Complutense de Madrid, Madrid, Spain, 2019. Available online: https://dialnet.unirioja.es/servlet/tesis?codigo=248344 (accessed on 26 January 2022).
- Maiorana, A.; Sabia, A.; Corsetti, T.; Dionisi-Vici, C. Safety of vaccines administration in hereditary fructose intolerance. Orphanet J. Rare Dis. 2020, 15, 274. [Google Scholar] [CrossRef]
- Huard, K.; Ahn, K.; Amor, P.; Beebe, D.A.; Borzilleri, K.A.; Chrunyk, B.A.; Coffey, S.B.; Cong, Y.; Conn, E.L.; Culp, J.S.; et al. Discovery ofFragment-Derived Small Molecules for in Vivo Inhibition of Ketohexokinase (KHK). J. Med. Chem. 2017, 60, 7835–7849. [Google Scholar] [CrossRef]
- Futatsugi, K.; Smith, A.C.; Tu, M.; Raymer, B.; Ahn, K.; Coffey, S.B.; Dowling, M.S.; Fernando, D.P.; Gutierrez, J.A.; Huard, K.; et al. Discovery of PF-06835919: A Potent Inhibitor of Ketohexokinase (KHK) for the Treatment of Metabolic Disorders Driven by the Overconsumption of Fructose. J. Med. Chem. 2020, 63, 13546–13560. [Google Scholar] [CrossRef]
- Gutierrez, J.A.; Liu, W.; Perez, S.; Xing, G.; Sonnenberg, G.; Kou, K.; Blatnik, M.; Allen, R.; Weng, Y.; Vera, N.B.; et al. Pharmacologic inhibition of ketohexokinase prevents fructose-induced metabolic dysfunction. Mol. Metab. 2021, 48, 101196. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. Int. J. Surg. 2010, 8, 336–341. [Google Scholar] [CrossRef]
- Urrútia, G.; Bonfill, X. La declaración PRISMA: Un paso adelante en la mejora de las publicaciones de la Revista Española de Salud Pública. Rev. Española Salud Pública 2013, 87, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Fructose, A.U. Chemical clinic minutes. Nature 1956, 178, 1325. [Google Scholar]
- Leiva Gea, I.; Ramos, J.M.; Borrás Pérez, V.; López Siguero, J.P. Hipoglucemia. Protoc. Diagnósticos Ter. Pediatría 2019, 1, 171–182. [Google Scholar]
- Morris, R.C. An experimental renal acidification defect in patients with hereditary fructose intolerance. I. Its resemblance to renal tubular acidosis. J. Clin. Investig. 1968, 47, 1389–1398. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.C. An experimental renal acidification defect in patients with hereditary fructose intolerance. II. Its distinction from classic renal tubular acidosis; its resemblance to the renal acidification defect associated with the Fanconi syndrome of children with c. J. Clin. Investig. 1968, 47, 1648–1663. [Google Scholar] [CrossRef] [PubMed]
- Mcinnes, R.R.; Dupont, C.H.; Drummond, K.N. Idiopathic Fanconi Syndrome. Pediatr. Res. 1980, 215, 209–215. [Google Scholar]
- Cochat, P.; Pichault, V.; Bacchetta, J.; Dubourg, L.; Sabot, J.-F.; Saban, C.; Daudon, M.; Liutkus, A. Nephrolithiasis related to inborn metabolic diseases. Pediatr. Nephrol. 2010, 25, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.; Sethi, S.K. Novel techniques and newer markers for the evaluation of proximal tubular dysfunction. Int. Urol. Nephrol. 2011, 43, 1107–1115. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.C.; McSherry, E.; Sebastian, A. Modulation of experimental renal dysfunction of hereditary fructose intolerance by circulating parathyroid hormone. Proc. Natl. Acad. Sci. USA 1971, 68, 132–135. [Google Scholar] [CrossRef]
- Richardson, R.M.A.; Little, J.A.; Patten, R.L.; Goldstein, M.B.; Halperin, M.L. Pathogenesis of acidosis in hereditary fructose intolerance. Metabolism 1979, 28, 1133–1138. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular mechanisms of lipotoxicity and glucotoxicity in nonalcoholic fatty liver disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef]
- DiStefano, J.K. Fructose-mediated effects on gene expression and epigenetic mechanisms associated with NAFLD pathogenesis. Cell. Mol. Life Sci. 2020, 77, 2079–2090. [Google Scholar] [CrossRef]
- Pérez-Carreras, M.; Ibarrola-de-Andrés, C.; Muñoz-Codoceo, C.; López-Martínez, C.; Martín-Algíbez, A. Nonalcoholic steatohepatitis and hepatic adenomatosis: Casual or causal relationship? Rev. Esp. Dis. Dig. 2018, 110, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Paolella, G.; Pisano, P.; Albano, R.; Cannaviello, L.; Mauro, C.; Esposito, G.; Vajro, P. Fatty liver disease and hypertransaminasemia hiding the association of clinically silent Duchenne muscular dystrophy and hereditary fructose intolerance. Ital. J. Pediatr. 2012, 38, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Simons, N.; Debray, F.-G.; Schaper, N.C.; Kooi, M.E.; Feskens, E.J.M.; Hollak, C.E.M.; Lindeboom, L.; Koek, G.H.; Bons, J.A.P.; Lefeber, D.J.; et al. Patients with Aldolase B Deficiency Are Characterized by Increased Intrahepatic Triglyceride Content. J. Clin. Endocrinol. Metab. 2019, 104, 5056–5064. [Google Scholar] [CrossRef] [PubMed]
- Ghannem, L.; Beaufrère, A.; Zucman-Rossi, J.; Paradis, V. Liver adenomatosis and NAFLD developed in the context of hereditary fructose intolerance. Liver Int. 2020, 40, 3125–3126. [Google Scholar] [CrossRef] [PubMed]
- Maggiore, G.; Nastasio, S.; Sciveres, M. Is liver steatosis diagnosis of non-alcoholic fatty liver disease in patients with hereditary fructose intolerance? Clin. Nutr. 2019, 38, 1960–1961. [Google Scholar] [CrossRef]
- Rudolph, B.; Ewart, M.; Levin, T.L.; Douglas, L.C.; Borenstein, S.H.; Thompson, J.F. Mucosal hyperplasia in infant with jejunal web. J. Pediatr. Gastroenterol. Nutr. 2013, 57, e2–e3. [Google Scholar] [CrossRef]
- Lieu, E.L.; Kelekar, N.; Bhalla, P.; Kim, J. Fructose and mannose in inborn errors of metabolism and cancer. Metabolites 2021, 11, 479. [Google Scholar] [CrossRef]
- Izquierdo-García, E.; Escobar-Rodríguez, I.; Moreno-Villares, J.M.; Iglesias-Peinado, I. Social and health care needs in patients with hereditary fructose intolerance in Spain. Endocrinol. Diabetes Nutr. 2020, 67, 253–262. [Google Scholar] [CrossRef]
- Asociación de Afectados por Intolerancia Hereditaria a la Fructosa (deficiencia Aldolasa B). 30 December 2023. Madrid: España. [Guidelines for Hereditary Fructose Intolerance (HFI)]. Available online: https://www.aaihf.com/index.php (accessed on 26 January 2024).


{kind=link}
{kind=link}
{kind=link}
{kind=link}


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Úbeda, F.; Santander, S.; Luesma, M.J. Clinical Practice Guidelines for the Diagnosis and Management of Hereditary Fructose Intolerance. Diseases 2024, 12, 44. https://doi.org/10.3390/diseases12030044
Úbeda F, Santander S, Luesma MJ. Clinical Practice Guidelines for the Diagnosis and Management of Hereditary Fructose Intolerance. Diseases. 2024; 12(3):44. https://doi.org/10.3390/diseases12030044
Chicago/Turabian StyleÚbeda, Félix, Sonia Santander, and María José Luesma. 2024. "Clinical Practice Guidelines for the Diagnosis and Management of Hereditary Fructose Intolerance" Diseases 12, no. 3: 44. https://doi.org/10.3390/diseases12030044
APA StyleÚbeda, F., Santander, S., & Luesma, M. J. (2024). Clinical Practice Guidelines for the Diagnosis and Management of Hereditary Fructose Intolerance. Diseases, 12(3), 44. https://doi.org/10.3390/diseases12030044