
Direct Infusion Mass Spectrometry to Rapidly Map Metabolic Flux of Substrates Labeled with Stable Isotopes

 ,
, _Verhoeven-Duif.png) , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture Conditions
2.2. Isotope Tracing in Organoids
2.3. DI-HRMS-Based Metabolomics
2.3.1. Sample Preparation
2.3.2. Direct Infusion–High-Resolution Mass Spectrometry
2.3.3. Data Processing
2.4. Targeted TCA Analysis
2.4.1. Standard and Sample Preparation
2.4.2. Chromatographic Separation
2.4.3. Metabolite Detection
2.4.4. Software/Statistical Analysis
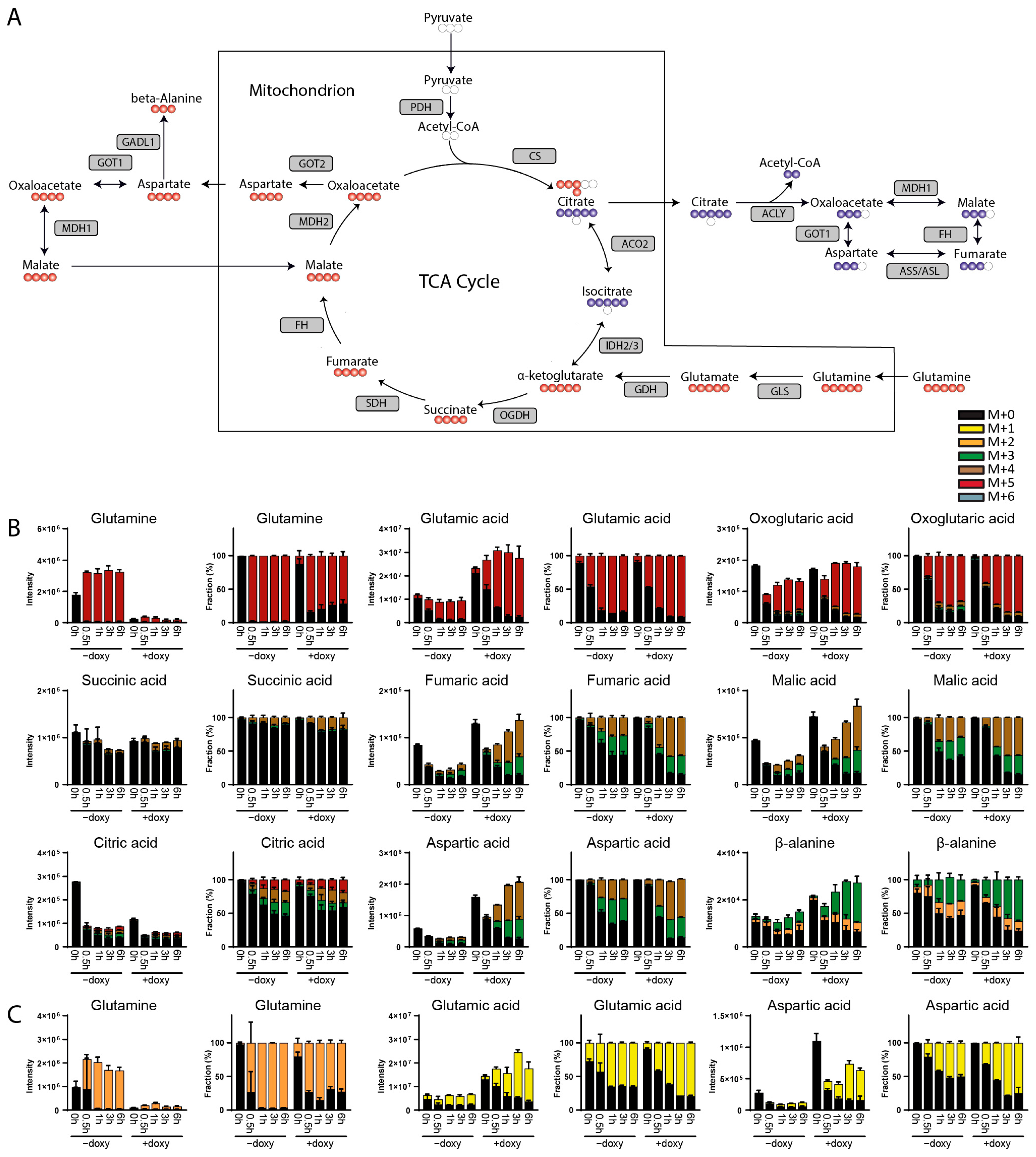
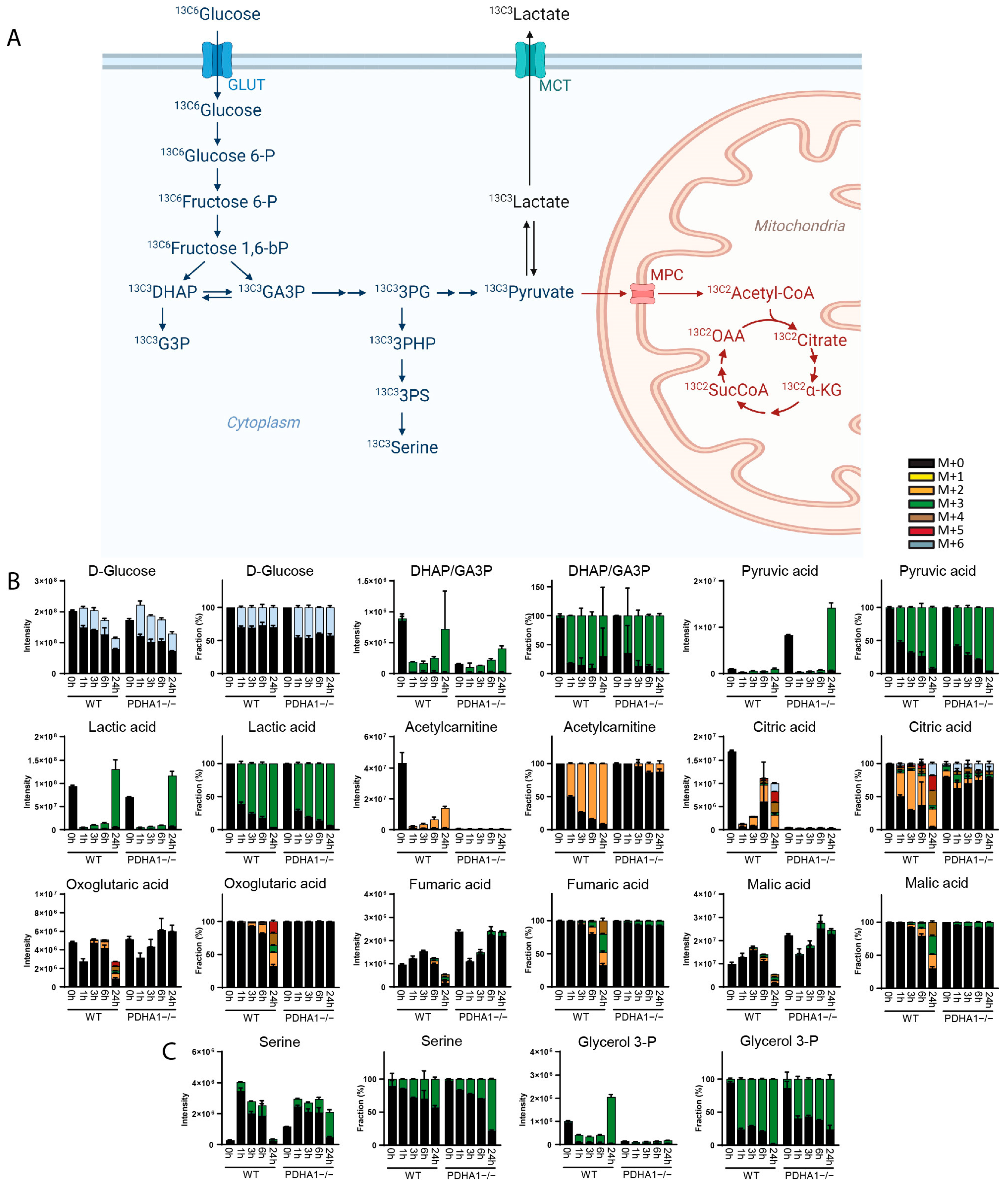
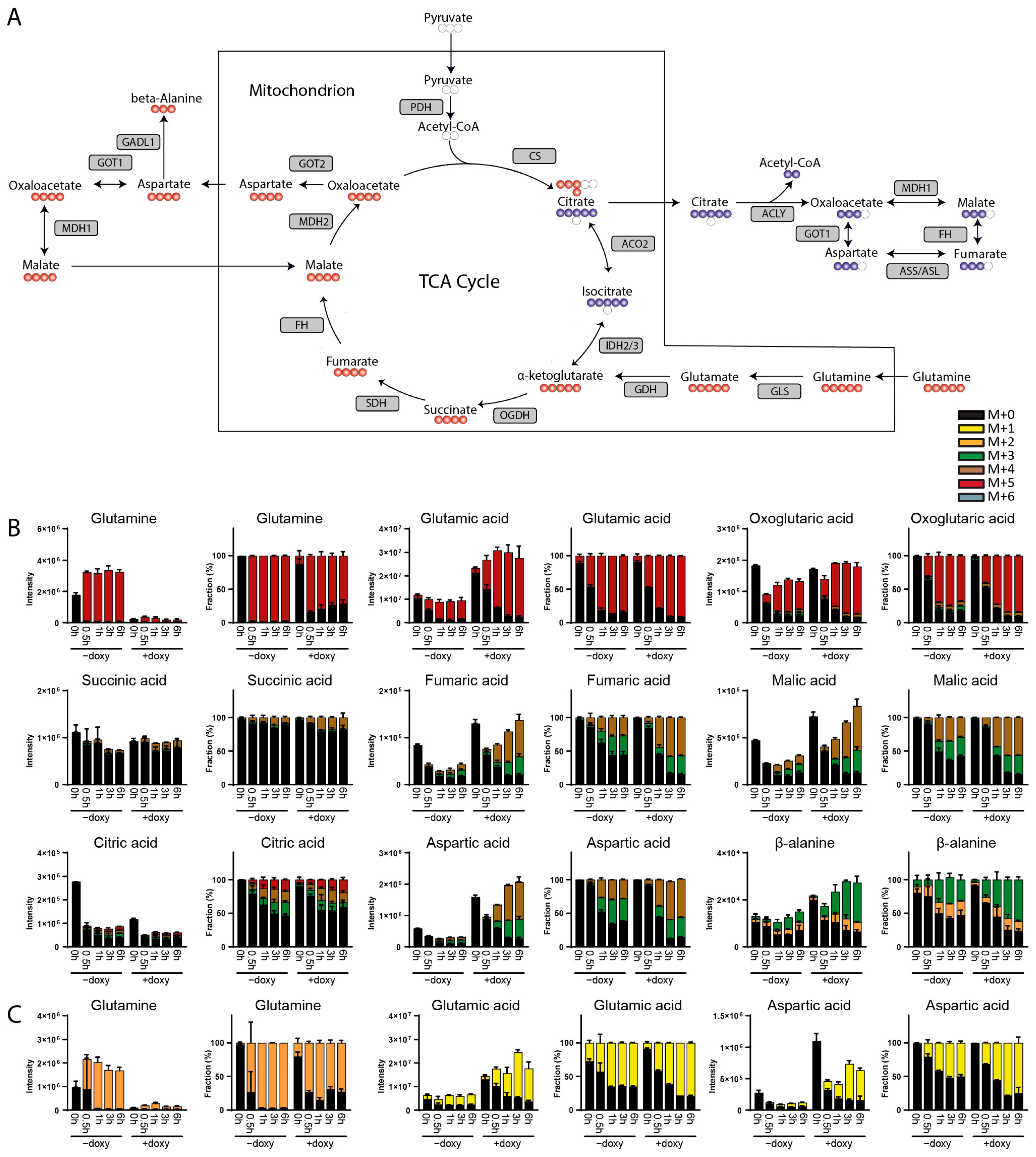
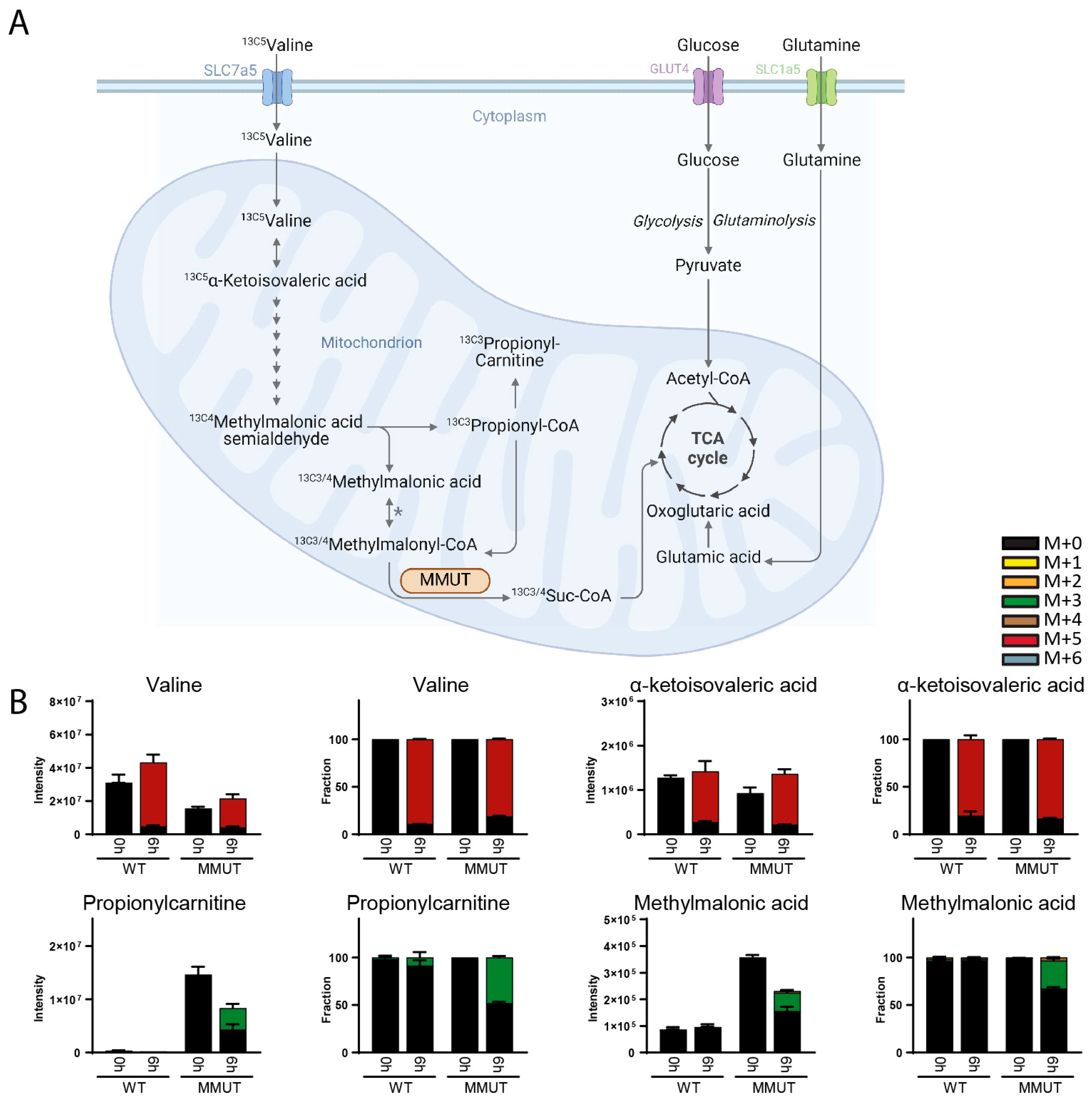
3. Results
3.1. Method Development
3.2. Method Validation
4. Discussion
5. Conclusions
6. Limitations of This Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cossu, M.; Pintus, R.; Zaffanello, M.; Mussap, M.; Serra, F.; Marcialis, M.A.; Fanos, V. Metabolomic Studies in Inborn Errors of Metabolism: Last Years and Future Perspectives. Metabolites 2023, 13, 447. [Google Scholar] [CrossRef] [PubMed]
- Alfadhel, M.; Babiker, A. Inborn errors of metabolism associated with hyperglycaemic ketoacidosis and diabetes mellitus: Narrative review. Sudan. J. Paediatr. 2018, 18, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Erez, A.; Shchelochkov, O.A.; Plon, S.E.; Scaglia, F.; Lee, B. Insights into the pathogenesis and treatment of cancer from inborn errors of metabolism. Am. J. Hum. Genet. 2011, 88, 402–421. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Fan, T.W.; Lane, A.N.; Higashi, R.M. Quantification of Isotopologues of Amino Acids by Multiplexed Stable Isotope-Resolved Metabolomics Using Ultrahigh-Resolution Mass Spectrometry Coupled with Direct Infusion. Methods Mol. Biol. 2019, 2030, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Hackman, G.L.; Saha, A.; Rathore, A.S.; Collins, M.; Friedman, C.; Yi, S.S.; Matsuda, F.; DiGiovanni, J.; Lodi, A.; et al. Metabolomics-based phenotypic screens for evaluation of drug synergy via direct-infusion mass spectrometry. iScience 2022, 25, 104221. [Google Scholar] [CrossRef]
- Giavalisco, P.; Hummel, J.; Lisec, J.; Inostroza, A.C.; Catchpole, G.; Willmitzer, L. High-resolution direct infusion-based mass spectrometry in combination with whole 13C metabolome isotope labeling allows unambiguous assignment of chemical sum formulas. Anal. Chem. 2008, 80, 9417–9425. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.J.; Martin, H.G.; Myers, S.; Rodriguez, S.; Baidoo, E.E.; Keasling, J.D. Advances in analysis of microbial metabolic fluxes via (13)C isotopic labeling. Mass Spectrom. Rev. 2009, 28, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Haijes, H.A.; Willemsen, M.; Van der Ham, M.; Gerrits, J.; Pras-Raves, M.L.; Prinsen, H.; Van Hasselt, P.M.; De Sain-van der Velden, M.G.M.; Verhoeven-Duif, N.M.; Jans, J.J.M. Direct Infusion Based Metabolomics Identifies Metabolic Disease in Patients’ Dried Blood Spots and Plasma. Metabolites 2019, 9, 12. [Google Scholar] [CrossRef]
- Noronha, A.; Modamio, J.; Jarosz, Y.; Guerard, E.; Sompairac, N.; Preciat, G.; Danielsdottir, A.D.; Krecke, M.; Merten, D.; Haraldsdottir, H.S.; et al. The Virtual Metabolic Human database: Integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 2019, 47, D614–D624. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 2019, 28, 1947–1951. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef] [PubMed]
- Dexter, J.P.; Ward, P.S.; Dasgupta, T.; Hosios, A.M.; Gunawardena, J.; Vander Heiden, M.G. Lack of evidence for substrate channeling or flux between wildtype and mutant isocitrate dehydrogenase to produce the oncometabolite 2-hydroxyglutarate. J. Biol. Chem. 2018, 293, 20051–20061. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lu, J.; Kulkarni, S.; Zhang, W.; Gorka, J.E.; Mandel, J.A.; Goetzman, E.S.; Prochownik, E.V. Metabolic and oncogenic adaptations to pyruvate dehydrogenase inactivation in fibroblasts. J. Biol. Chem. 2019, 294, 5466–5486. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, K.N.; Egnatchik, R.A.; Calvaruso, M.A.; Wasti, A.T.; Padanad, M.S.; Boroughs, L.K.; Ko, B.; Hensley, C.T.; Acar, M.; Hu, Z.; et al. Metabolic plasticity maintains proliferation in pyruvate dehydrogenase deficient cells. Cancer Metab. 2015, 3, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Ko, B.; Hensley, C.T.; Jiang, L.; Wasti, A.T.; Kim, J.; Sudderth, J.; Calvaruso, M.A.; Lumata, L.; Mitsche, M.; et al. Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 2014, 56, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Zielinski, D.C.; Jamshidi, N.; Corbett, A.J.; Bordbar, A.; Thomas, A.; Palsson, B.O. Systems biology analysis of drivers underlying hallmarks of cancer cell metabolism. Sci. Rep. 2017, 7, 41241. [Google Scholar] [CrossRef]
- Rumping, L.; Tessadori, F.; Pouwels, P.J.W.; Vringer, E.; Wijnen, J.P.; Bhogal, A.A.; Savelberg, S.M.C.; Duran, K.J.; Bakkers, M.J.G.; Ramos, R.J.J.; et al. GLS hyperactivity causes glutamate excess, infantile cataract and profound developmental delay. Hum. Mol. Genet. 2019, 28, 96–104. [Google Scholar] [CrossRef]
- Metallo, C.M.; Walther, J.L.; Stephanopoulos, G. Evaluation of 13C isotopic tracers for metabolic flux analysis in mammalian cells. J. Biotechnol. 2009, 144, 167–174. [Google Scholar] [CrossRef]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef]
- Yoo, H.C.; Park, S.J.; Nam, M.; Kang, J.; Kim, K.; Yeo, J.H.; Kim, J.K.; Heo, Y.; Lee, H.S.; Lee, M.Y.; et al. A Variant of SLC1A5 Is a Mitochondrial Glutamine Transporter for Metabolic Reprogramming in Cancer Cells. Cell Metab. 2020, 31, 267–283.e212. [Google Scholar] [CrossRef]
- Manoli, I.; Sloan, J.L.; Venditti, C.P. Isolated Methylmalonic Acidemia. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: https://www.ncbi.nlm.nih.gov/pubmed/20301409 (accessed on 10 March 2024).
- Weindl, D.; Cordes, T.; Battello, N.; Sapcariu, S.C.; Dong, X.; Wegner, A.; Hiller, K. Bridging the gap between non-targeted stable isotope labeling and metabolic flux analysis. Cancer Metab. 2016, 4, 10. [Google Scholar] [CrossRef] [PubMed]
- Lackner, M.; Neef, S.K.; Winter, S.; Beer-Hammer, S.; Nurnberg, B.; Schwab, M.; Hofmann, U.; Haag, M. Untargeted stable isotope-resolved metabolomics to assess the effect of PI3Kbeta inhibition on metabolic pathway activities in a PTEN null breast cancer cell line. Front. Mol. Biosci. 2022, 9, 1004602. [Google Scholar] [CrossRef] [PubMed]
- Bruurs, L.J.; Donker, L.; Zwakenberg, S.; Zwartkruis, F.J.; Begthel, H.; Knisely, A.S.; Posthuma, G.; van de Graaf, S.F.; Paulusma, C.C.; Bos, J.L. ATP8B1-mediated spatial organization of Cdc42 signaling maintains singularity during enterocyte polarization. J. Cell Biol. 2015, 210, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Meerbrey, K.L.; Hu, G.; Kessler, J.D.; Roarty, K.; Li, M.Z.; Fang, J.E.; Herschkowitz, J.I.; Burrows, A.E.; Ciccia, A.; Sun, T.; et al. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 3665–3670. [Google Scholar] [CrossRef]
- Huch, M.; Gehart, H.; van Boxtel, R.; Hamer, K.; Blokzijl, F.; Verstegen, M.M.; Ellis, E.; van Wenum, M.; Fuchs, S.A.; de Ligt, J.; et al. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 2015, 160, 299–312. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meijer, N.W.F.; Zwakenberg, S.; Gerrits, J.; Westland, D.; Ardisasmita, A.I.; Fuchs, S.A.; Verhoeven-Duif, N.M.; Jans, J.J.M.; Zwartkruis, F.J.T. Direct Infusion Mass Spectrometry to Rapidly Map Metabolic Flux of Substrates Labeled with Stable Isotopes. Metabolites 2024, 14, 246. https://doi.org/10.3390/metabo14050246
Meijer NWF, Zwakenberg S, Gerrits J, Westland D, Ardisasmita AI, Fuchs SA, Verhoeven-Duif NM, Jans JJM, Zwartkruis FJT. Direct Infusion Mass Spectrometry to Rapidly Map Metabolic Flux of Substrates Labeled with Stable Isotopes. Metabolites. 2024; 14(5):246. https://doi.org/10.3390/metabo14050246
Chicago/Turabian StyleMeijer, Nils W. F., Susan Zwakenberg, Johan Gerrits, Denise Westland, Arif I. Ardisasmita, Sabine A. Fuchs, Nanda M. Verhoeven-Duif, Judith J. M. Jans, and Fried J. T. Zwartkruis. 2024. "Direct Infusion Mass Spectrometry to Rapidly Map Metabolic Flux of Substrates Labeled with Stable Isotopes" Metabolites 14, no. 5: 246. https://doi.org/10.3390/metabo14050246