
The Metabolic and Lipidomic Fingerprint of Torin1 Exposure in Mouse Embryonic Fibroblasts Using Untargeted Metabolomics
 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Chemicals
2.2. Cell Line Generation, Treatment, Viability, and Western Blot Analysis
2.3. Exposure Parameters, Sample Collection, and Sample Preparation
2.4. Instrumental Analysis
2.5. Data Analysis
2.6. Annotation of Metabolites
3. Results and Discussion
3.1. Cell Viability and Western Blot Analysis
3.2. Data Quality Management
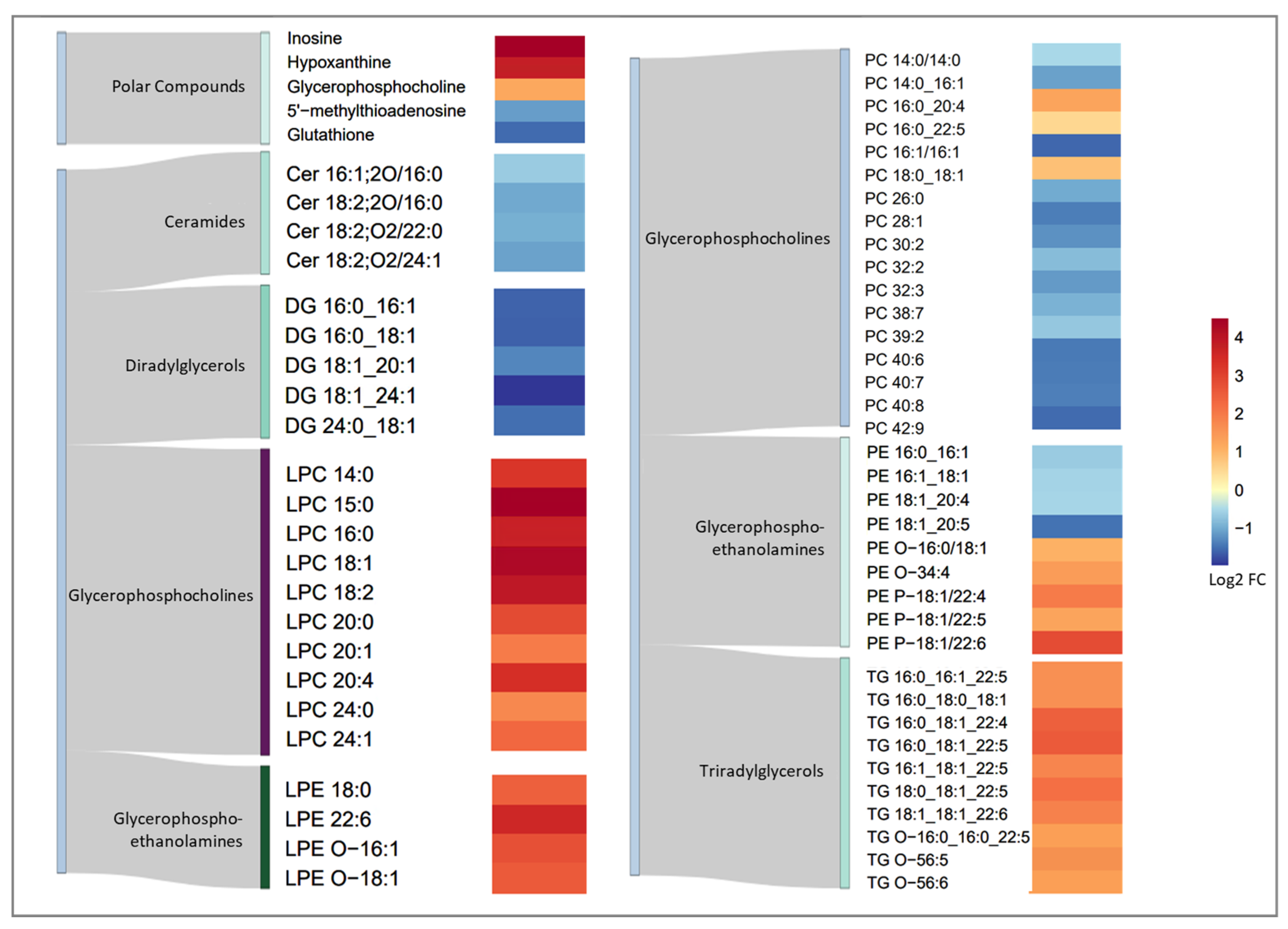
3.3. Metabolic Fingerprint of Torin1 Exposure in MEF Cells
3.4. Metabolic Pathway Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fiehn, O. Metabolomics—The Link between Genotypes and Phenotypes. In Functional Genomics; Town, C., Ed.; Springer: Dordrecht, The Netherlands, 2002; pp. 155–171. ISBN 978-94-010-0448-0. [Google Scholar]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. “Metabonomics”: Understanding the Metabolic Responses of Living Systems to Pathophysiological Stimuli via Multivariate Statistical Analysis of Biological NMR Spectroscopic Data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef]
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic Functional Analysis of the Yeast Genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Atkinson, A.J.; Colburn, W.A.; DeGruttola, V.G.; DeMets, D.L.; Downing, G.J.; Hoth, D.F.; Oates, J.A.; Peck, C.C.; Schooley, R.T.; Spilker, B.A.; et al. Biomarkers and Surrogate Endpoints: Preferred Definitions and Conceptual Framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Kang, S.A.; Chang, J.W.; Liu, Q.; Zhang, J.; Gao, Y.; Reichling, L.J.; Sim, T.; Sabatini, D.M.; Gray, N.S. An ATP-Competitive Mammalian Target of Rapamycin Inhibitor Reveals Rapamycin-Resistant Functions of MTORC1. J. Biol. Chem. 2009, 284, 8023–8032. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Chang, J.W.; Wang, J.; Kang, S.A.; Thoreen, C.C.; Markhard, A.; Hur, W.; Zhang, J.; Sim, T.; Sabatini, D.M.; et al. Discovery of 1-(4-(4-Propionylpiperazin-1-Yl)-3-(Trifluoromethyl)Phenyl)-9-(Quinolin-3-Yl)Benzo[h][1,6]Naphthyridin-2(1H)-One as a Highly Potent, Selective Mammalian Target of Rapamycin (MTOR) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2010, 53, 7146–7155. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. MTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef]
- Wong, P.-M.; Puente, C.; Ganley, I.G.; Jiang, X. The ULK1 Complex: Sensing Nutrient Signals for Autophagy Activation. Autophagy 2013, 9, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for Taxonomy-Based Analysis of Pathways and Genomes. Nucleic Acids Res. 2023, 51, D587–D592. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Torrence, M.E.; MacArthur, M.R.; Hosios, A.M.; Valvezan, A.J.; Asara, J.M.; Mitchell, J.R.; Manning, B.D. The MTORC1-Mediated Activation of ATF4 Promotes Protein and Glutathione Synthesis Downstream of Growth Signals. Elife 2021, 10, e63326. [Google Scholar] [CrossRef]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. MTORC1 Induces Purine Synthesis through Control of the Mitochondrial Tetrahydrofolate Cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Hosios, A.M.; Wilkinson, M.E.; McNamara, M.C.; Kalafut, K.C.; Torrence, M.E.; Asara, J.M.; Manning, B.D. MTORC1 Regulates a Lysosome-Dependent Adaptive Shift in Intracellular Lipid Species. Nat. Metab. 2022, 4, 1792–1811. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Broadhurst, D.I.; Wilson, M.; Wishart, D.S. Translational Biomarker Discovery in Clinical Metabolomics: An Introductory Tutorial. Metabolomics 2013, 9, 280–299. [Google Scholar] [CrossRef] [PubMed]
- Broadhurst, D.I.; Kell, D.B. Statistical Strategies for Avoiding False Discoveries in Metabolomics and Related Experiments. Metabolomics 2006, 2, 171–196. [Google Scholar] [CrossRef]
- Patti, G.J. Separation Strategies for Untargeted Metabolomics. J. Sep. Sci. 2011, 34, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Iturrospe, E.; da Silva, K.M.; van de Lavoir, M.; Robeyns, R.; Cuykx, M.; Vanhaecke, T.; van Nuijs, A.L.N.; Covaci, A. Mass Spectrometry-Based Untargeted Metabolomics and Lipidomics Platforms to Analyze Cell Culture Extracts. In Mass Spectrometry for Metabolomics; González-Domínguez, R., Ed.; Springer: New York, NY, USA, 2023; pp. 189–206. ISBN 978-1-0716-2699-3. [Google Scholar]
- da Silva, K.M.; Iturrospe, E.; Heyrman, J.; Koelmel, J.P.; Cuykx, M.; Vanhaecke, T.; Covaci, A.; van Nuijs, A.L.N. Optimization of a Liquid Chromatography-Ion Mobility-High Resolution Mass Spectrometry Platform for Untargeted Lipidomic and Application to HepaRG Cell Extracts. Talanta 2021, 235, 122808. [Google Scholar] [CrossRef] [PubMed]
- Iturrospe, E.; Da Silva, K.M.; Talavera Andújar, B.; Cuykx, M.; Boeckmans, J.; Vanhaecke, T.; Covaci, A.; van Nuijs, A.L.N. An Exploratory Approach for an Oriented Development of an Untargeted Hydrophilic Interaction Liquid Chromatography-Mass Spectrometry Platform for Polar Metabolites in Biological Matrices. J. Chromatogr. A 2021, 1637, 461807. [Google Scholar] [CrossRef] [PubMed]
- Xu, J. Preparation, Culture, and Immortalization of Mouse Embryonic Fibroblasts. Curr. Protoc. Mol. Biol. 2005, 70, 28.1.1–28.1.8. [Google Scholar] [CrossRef] [PubMed]
- Essen BioScience. User Guide: Incucyte® Cytotox Dyes—For Detection of Cell Membrane Integrity Disruption; Essen BioScience: Ann Arbor, MI, USA, 2020. [Google Scholar]
- Zhou, J.; Tan, S.-H.; Nicolas, V.; Bauvy, C.; Yang, N.-D.; Zhang, J.; Xue, Y.; Codogno, P.; Shen, H.-M. Activation of Lysosomal Function in the Course of Autophagy via MTORC1 Suppression and Autophagosome-Lysosome Fusion. Cell Res. 2013, 23, 508–523. [Google Scholar] [CrossRef]
- Jiang, P.; Mizushima, N. LC3- and P62-Based Biochemical Methods for the Analysis of Autophagy Progression in Mammalian Cells. Methods 2015, 75, 13–18. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.-A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a Mammalian Homologue of Yeast Apg8p, Is Localized in Autophagosome Membranes after Processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef]
- Iturrospe, E.; da Silva, K.M.; Robeyns, R.; van de Lavoir, M.; Boeckmans, J.; Vanhaecke, T.; van Nuijs, A.L.N.; Covaci, A. Metabolic Signature of Ethanol-Induced Hepatotoxicity in HepaRG Cells by Liquid Chromatography–Mass Spectrometry-Based Untargeted Metabolomics. J. Proteome Res. 2022, 21, 1153–1166. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.E.; Kruger, M.C.; Cooper, G.J.S.; Poppitt, S.D.; Fraser, K. Tissue-Specific Sample Dilution: An Important Parameter to Optimise Prior to Untargeted Lc-Ms Metabolomics. Metabolites 2019, 9, 124. [Google Scholar] [CrossRef]
- Broadhurst, D.; Goodacre, R.; Reinke, S.N.; Kuligowski, J.; Wilson, I.D.; Lewis, M.R.; Dunn, W.B. Guidelines and Considerations for the Use of System Suitability and Quality Control Samples in Mass Spectrometry Assays Applied in Untargeted Clinical Metabolomic Studies. Metabolomics 2018, 14, 72. [Google Scholar] [CrossRef]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. ProteoWizard: Open Source Software for Rapid Proteomics Tools Development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A Lipidome Atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- DeFelice, B.C.; Mehta, S.S.; Samra, S.; Čajka, T.; Wancewicz, B.; Fahrmann, J.F.; Fiehn, O. Mass Spectral Feature List Optimizer (MS-FLO): A Tool To Minimize False Positive Peak Reports in Untargeted Liquid Chromatography–Mass Spectroscopy (LC-MS) Data Processing. Anal. Chem. 2017, 89, 3250–3255. [Google Scholar] [CrossRef]
- Klåvus, A.; Kokla, M.; Noerman, S.; Koistinen, V.M.; Tuomainen, M.; Zarei, I.; Meuronen, T.; Häkkinen, M.R.; Rummukainen, S.; Farizah Babu, A.; et al. “Notame”: Workflow for Non-Targeted LC–MS Metabolic Profiling. Metabolites 2020, 10, 135. [Google Scholar] [CrossRef]
- Di Guida, R.; Engel, J.; Allwood, J.W.; Weber, R.J.M.; Jones, M.R.; Sommer, U.; Viant, M.R.; Dunn, W.B. Non-Targeted UHPLC-MS Metabolomic Data Processing Methods: A Comparative Investigation of Normalisation, Missing Value Imputation, Transformation and Scaling. Metabolomics 2016, 12, 93. [Google Scholar] [CrossRef]
- De Livera, A.M.; Olshansky, M.; Speed, T.P. Statistical Analysis of Metabolomics Data BT—Metabolomics Tools for Natural Product Discovery: Methods and Protocols; Roessner, U., Dias, D.A., Eds.; Humana Press: Totowa, NJ, USA, 2013; pp. 291–307. ISBN 978-1-62703-577-4. [Google Scholar]
- Li, B.; Tang, J.; Yang, Q.; Cui, X.; Li, S.; Chen, S.; Cao, Q.; Xue, W.; Chen, N.; Zhu, F. Performance Evaluation and Online Realization of Data-Driven Normalization Methods Used in LC/MS Based Untargeted Metabolomics Analysis. Sci. Rep. 2016, 6, 38881. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Wang, J.; Su, M.; Jia, E.; Chen, S.; Chen, T.; Ni, Y. Missing Value Imputation Approach for Mass Spectrometry-Based Metabolomics Data. Sci. Rep. 2018, 8, 663. [Google Scholar] [CrossRef] [PubMed]
- Ejigu, B.A.; Valkenborg, D.; Baggerman, G.; Vanaerschot, M.; Witters, E.; Dujardin, J.C.; Burzykowski, T.; Berg, M. Evaluation of Normalization Methods to Pave the Way towards Large-Scale LC-MS-Based Metabolomics Profiling Experiments. Omi. A J. Integr. Biol. 2013, 17, 473–485. [Google Scholar] [CrossRef]
- van den Berg, R.A.; Hoefsloot, H.C.J.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, Scaling, and Transformations: Improving the Biological Information Content of Metabolomics Data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.; Viroli, C.; Gormley, I.C. Infinite Mixtures of Infinite Factor Analysers. Bayesian Anal. 2020, 15, 937–963. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.A. MixOmics: An R Package for ‘omics Feature Selection and Multiple Data Integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]
- Gaude, E.; Chignola, F.; Spiliotopoulos, D.; Spitaleri, A.; Ghitti, M.; M Garcia-Manteiga, J.; Mari, S.; Musco, G. muma, An R Package for Metabolomics Univariate and Multivariate Statistical Analysis. Curr. Metabolomics 2013, 1, 180–189. [Google Scholar] [CrossRef]
- Vinaixa, M.; Samino, S.; Saez, I.; Duran, J.; Guinovart, J.J.; Yanes, O. A Guideline to Univariate Statistical Analysis for LC/MS-Based Untargeted Metabolomics-Derived Data. Metabolites 2012, 2, 775–795. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- Kind, T.; Tsugawa, H.; Cajka, T.; Ma, Y.; Lai, Z.; Mehta, S.S.; Wohlgemuth, G.; Barupal, D.K.; Showalter, M.R.; Arita, M.; et al. Identification of Small Molecules Using Accurate Mass MS/MS Search. Mass Spectrom. Rev. 2018, 37, 513–532. [Google Scholar] [CrossRef]
- MoNA—MassBank of North America. Available online: https://mona.fiehnlab.ucdavis.edu/ (accessed on 1 April 2023).
- Phinney, K.W.; Ballihaut, G.; Bedner, M.; Benford, B.S.; Camara, J.E.; Christopher, S.J.; Davis, W.C.; Dodder, N.G.; Eppe, G.; Lang, B.E.; et al. Development of a Standard Reference Material for Metabolomics Research. Anal. Chem. 2013, 85, 11732–11738. [Google Scholar] [CrossRef] [PubMed]
- Koelmel, J.P.; Kroeger, N.M.; Ulmer, C.Z.; Bowden, J.A.; Patterson, R.E.; Cochran, J.A.; Beecher, C.W.W.; Garrett, T.J.; Yost, R.A. LipidMatch: An Automated Workflow for Rule-Based Lipid Identification Using Untargeted High-Resolution Tandem Mass Spectrometry Data. BMC Bioinform. 2017, 18, 331. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Angelidou, G.; Lange, M.; Hoffmann, R.; Fedorova, M. LipidHunter Identifies Phospholipids by High-Throughput Processing of LC-MS and Shotgun Lipidomics Datasets. Anal. Chem. 2017, 89, 8800–8807. [Google Scholar] [CrossRef] [PubMed]
- da Silva, K.M.; van de Lavoir, M.; Robeyns, R.; Iturrospe, E.; Verheggen, L.; Covaci, A.; van Nuijs, A.L.N. Guidelines and Considerations for Building Multidimensional Libraries for Untargeted MS-Based Metabolomics. Metabolomics 2022, 19, 4. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Wu, X.; Feng, Y. Fragmentation Patterns of Five Types of Phospholipids by Ultra-High-Performance Liquid Chromatography Electrospray Ionization Quadrupole Time-of-Flight Tandem Mass Spectrometry. Anal. Methods 2016, 8, 1319–1332. [Google Scholar] [CrossRef]
- Lange, M.; Angelidou, G.; Ni, Z.; Criscuolo, A.; Schiller, J.; Blüher, M.; Fedorova, M. AdipoAtlas: A Reference Lipidome for Human White Adipose Tissue. Cell Rep. Med. 2021, 2, 100407. [Google Scholar] [CrossRef] [PubMed]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, C.T.; Bansal, S.; Booth, B.; DeStefano, A.J.; Rose, M.J.; Sailstad, J.; Shah, V.P.; Skelly, J.P.; Swann, P.G.; Weiner, R. Quantitative Bioanalytical Methods Validation and Implementation: Best Practices for Chromatographic and Ligand Binding Assays. Pharm. Res. 2007, 24, 1962–1973. [Google Scholar] [CrossRef] [PubMed]
- Naz, S.; Vallejo, M.; García, A.; Barbas, C. Method Validation Strategies Involved in Non-Targeted Metabolomics. J. Chromatogr. A 2014, 1353, 99–105. [Google Scholar] [CrossRef]
- Trygg, J.; Holmes, E.; Lundstedt, T. Chemometrics in Metabonomics. J. Proteome Res. 2007, 6, 469–479. [Google Scholar] [CrossRef]
- Gika, H.G.; Theodoridis, G.A.; Wingate, J.E.; Wilson, I.D. Within-Day Reproducibility of an HPLC−MS-Based Method for Metabonomic Analysis: Application to Human Urine. J. Proteome Res. 2007, 6, 3291–3303. [Google Scholar] [CrossRef] [PubMed]
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Köfeler, H.; et al. Update on LIPID MAPS Classification, Nomenclature, and Shorthand Notation for MS-Derived Lipid Structures. J. Lipid Res. 2020, 61, 1539–1555. [Google Scholar] [CrossRef] [PubMed]
- Rose, T.D.; Köhler, N.; Falk, L.; Klischat, L.; Lazareva, O.E.; Pauling, J.K. Lipid Network and Moiety Analysis for Revealing Enzymatic Dysregulation and Mechanistic Alterations from Lipidomics Data. Brief. Bioinform. 2023, 24, bbac572. [Google Scholar] [CrossRef] [PubMed]
- Nagan, N.; Zoeller, R.A. Plasmalogens: Biosynthesis and Functions. Prog. Lipid Res. 2001, 40, 199–229. [Google Scholar] [CrossRef] [PubMed]
- Schooneveldt, Y.L.; Paul, S.; Calkin, A.C.; Meikle, P.J. Ether Lipids in Obesity: From Cells to Population Studies. Front. Physiol. 2022, 13, 841278. [Google Scholar] [CrossRef] [PubMed]
- Dean, J.M.; Lodhi, I.J. Structural and Functional Roles of Ether Lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Harvald, E.B.; Olsen, A.S.B.; Færgeman, N.J. Autophagy in the Light of Sphingolipid Metabolism. Apoptosis 2015, 20, 658–670. [Google Scholar] [CrossRef]
- de la Monte, S.M. Triangulated Mal-Signaling in Alzheimer’s Disease: Roles of Neurotoxic Ceramides, ER Stress, and Insulin Resistance Reviewed. J. Alzheimers. Dis. 2012, 30 (Suppl. S2), S231–S249. [Google Scholar] [CrossRef]
- Arboleda, G.; Morales, L.C.; Benítez, B.; Arboleda, H. Regulation of Ceramide-Induced Neuronal Death: Cell Metabolism Meets Neurodegeneration. Brain Res. Rev. 2009, 59, 333–346. [Google Scholar] [CrossRef]
- Samad, F.; Badeanlou, L.; Shah, C.; Yang, G. Adipose Tissue and Ceramide Biosynthesis in the Pathogenesis of Obesity. Adv. Exp. Med. Biol. 2011, 721, 67–86. [Google Scholar] [CrossRef]
- Chavez, J.A.; Summers, S.A. A Ceramide-Centric View of Insulin Resistance. Cell Metab. 2012, 15, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. An Emerging Role of MTOR in Lipid Biosynthesis. Curr. Biol. 2009, 19, R1046–R1052. [Google Scholar] [CrossRef]
- Guri, Y.; Colombi, M.; Dazert, E.; Hindupur, S.K.; Roszik, J.; Moes, S.; Jenoe, P.; Heim, M.H.; Riezman, I.; Riezman, H.; et al. MTORC2 Promotes Tumorigenesis via Lipid Synthesis. Cancer Cell 2017, 32, 807–823.e12. [Google Scholar] [CrossRef] [PubMed]
- Albers, E. Metabolic Characteristics and Importance of the Universal Methionine Salvage Pathway Recycling Methionine from 5′-Methylthioadenosine. IUBMB Life 2009, 61, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Villa, E.; Sahu, U.; O’Hara, B.P.; Ali, E.S.; Helmin, K.A.; Asara, J.M.; Gao, P.; Singer, B.D.; Ben-Sahra, I. MTORC1 Stimulates Cell Growth through SAM Synthesis and M6A MRNA-Dependent Control of Protein Synthesis. Mol. Cell 2021, 81, 2076–2093.e9. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Zhang, H.; Rinna, A. Glutathione: Overview of Its Protective Roles, Measurement, and Biosynthesis. Mol. Aspects Med. 2009, 30, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Chen, Y.; Gibson, S.B. Regulation of Autophagy by Reactive Oxygen Species (ROS): Implications for Cancer Progression and Treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative Stress and Autophagy: The Clash between Damage and Metabolic Needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Desideri, E.; Filomeni, G.; Ciriolo, M.R. Glutathione Participates in the Modulation of Starvation-Induced Autophagy in Carcinoma Cells. Autophagy 2012, 8, 1769–1781. [Google Scholar] [CrossRef]
- Ryu, A.H.; Eckalbar, W.L.; Kreimer, A.; Yosef, N.; Ahituv, N. Use Antibiotics in Cell Culture with Caution: Genome-Wide Identification of Antibiotic-Induced Changes in Gene Expression and Regulation. Sci. Rep. 2017, 7, 7533. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polar ESI (+) | Polar ESI (−) | Apolar ESI (+) | Apolar ESI (−) | ||
|---|---|---|---|---|---|
| Batch 1 | |||||
| PLS-DA | R2 | 0.999 | 0.998 | 0.998 | 0.989 |
| Q2 | 0.991 | 0.933 | 0.977 | 0.979 | |
| R2PERM | 0.002 | 0.001 | 0.001 | 0.002 | |
| Q2PERM | 0.002 | 0.001 | 0.001 | 0.002 | |
| RF | AUC | 1 | 1 | 1 | 1 |
| Batch 2 | |||||
| PLS-DA | R2 | 0.995 | 0.992 | 0.984 | 0.977 |
| Q2 | 0.978 | 0.923 | 0.976 | 0.962 | |
| R2PERM | 0.002 | 0.001 | 0.001 | 0.001 | |
| Q2PERM | 0.002 | 0.001 | 0.001 | 0.001 | |
| RF | AUC | 1 | 1 | 1 | 1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robeyns, R.; Sisto, A.; Iturrospe, E.; da Silva, K.M.; van de Lavoir, M.; Timmerman, V.; Covaci, A.; Stroobants, S.; van Nuijs, A.L.N. The Metabolic and Lipidomic Fingerprint of Torin1 Exposure in Mouse Embryonic Fibroblasts Using Untargeted Metabolomics. Metabolites 2024, 14, 248. https://doi.org/10.3390/metabo14050248
Robeyns R, Sisto A, Iturrospe E, da Silva KM, van de Lavoir M, Timmerman V, Covaci A, Stroobants S, van Nuijs ALN. The Metabolic and Lipidomic Fingerprint of Torin1 Exposure in Mouse Embryonic Fibroblasts Using Untargeted Metabolomics. Metabolites. 2024; 14(5):248. https://doi.org/10.3390/metabo14050248
Chicago/Turabian StyleRobeyns, Rani, Angela Sisto, Elias Iturrospe, Katyeny Manuela da Silva, Maria van de Lavoir, Vincent Timmerman, Adrian Covaci, Sigrid Stroobants, and Alexander L. N. van Nuijs. 2024. "The Metabolic and Lipidomic Fingerprint of Torin1 Exposure in Mouse Embryonic Fibroblasts Using Untargeted Metabolomics" Metabolites 14, no. 5: 248. https://doi.org/10.3390/metabo14050248