Targeted Chiral Analysis of Bioactive Arachidonic Acid Metabolites Using Liquid-Chromatography-Mass Spectrometry

Abstract
:1. Introduction

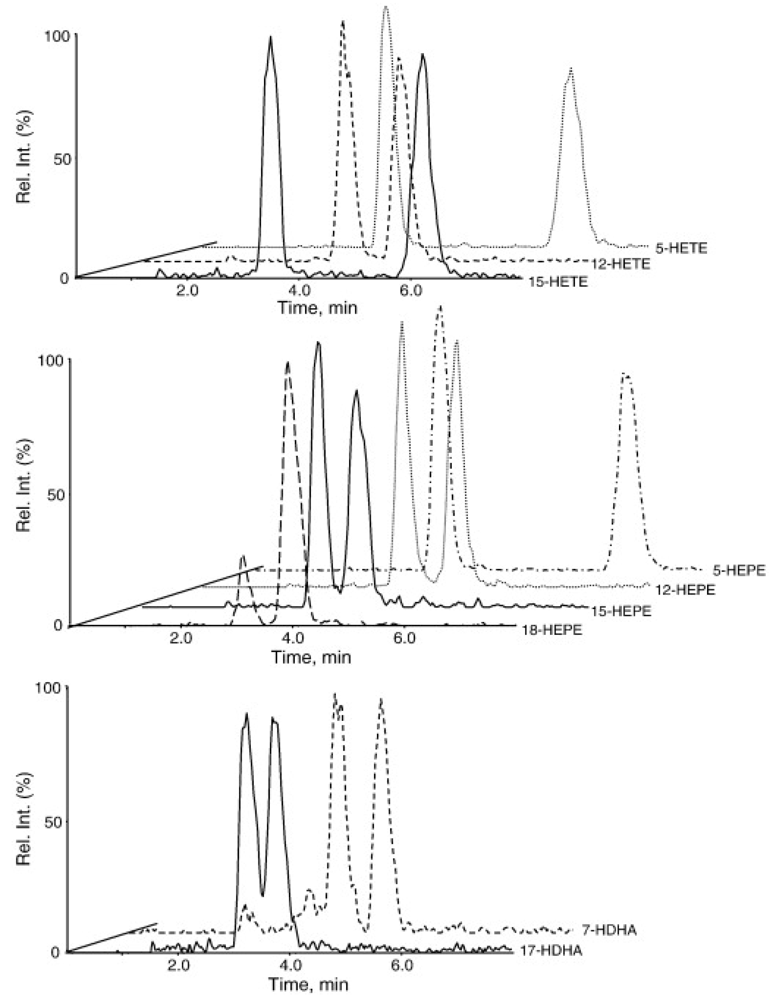
2. Analysis of Arachidonic Acid-Derived Eicosanoids by Targeted LC-MS.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MS Method | Analytes | HPLCcolumn | Derivatization reagent | Starting mobile phase | References |
|---|---|---|---|---|---|
| ECAPCI | HETEs | Chiralpack AD-H | PFB-Br | Hexanes/isopropanol/Methanol (98:1:1) | [103,104,108,109] |
| ECAPCI | EETs | Chiralpack AD-H | PFB-Br | Hexanes/isopropanol(99.6:0.4) | [90] |
| ESI | 5, 12 and 15-HETE | Chiralpack AD-RH | none | Methanol/water/acetic acid (95:5:0.1) | [93] |
3. COX Mediated Metabolism
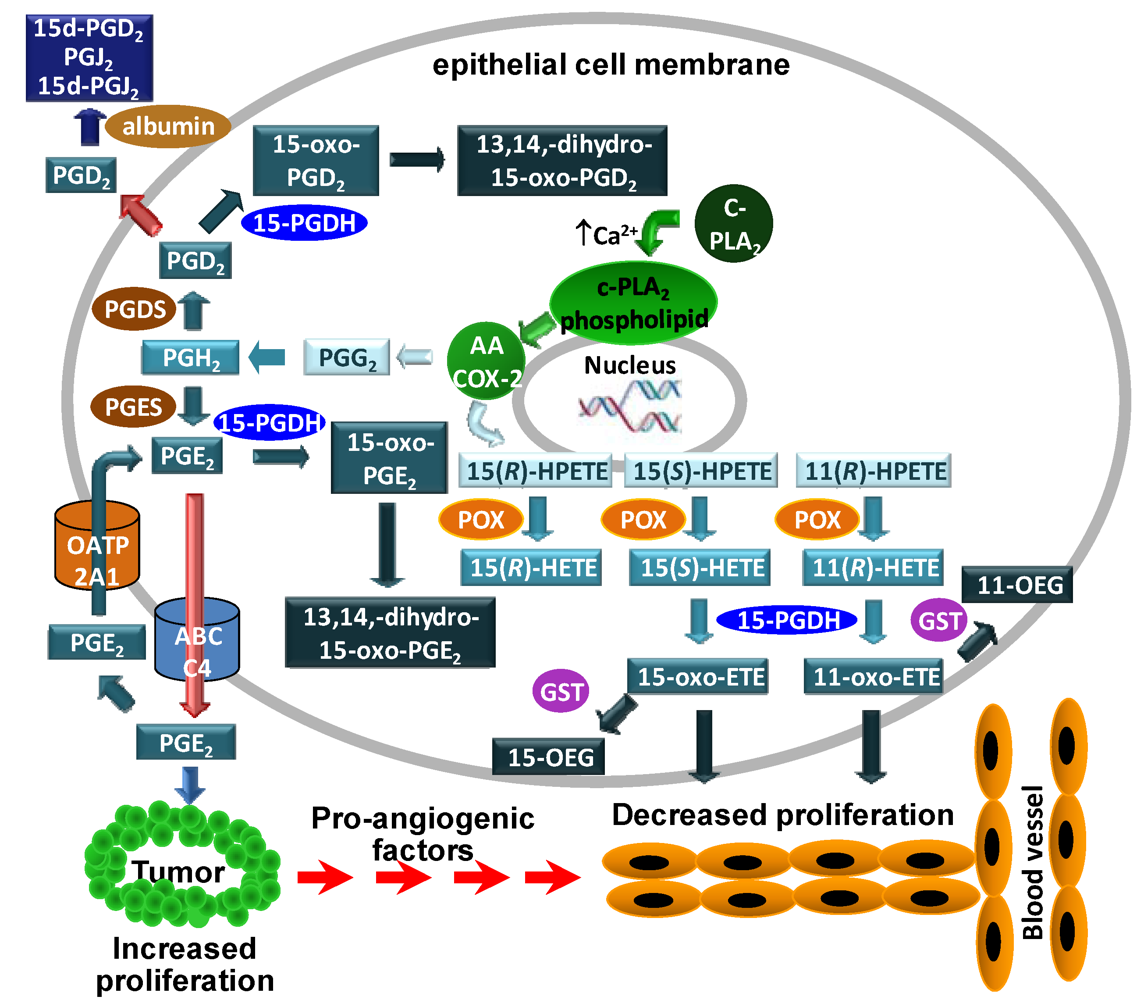
3.1. COX-2 Mediated Metabolism of Arachidonic Acid in Colorectal Adenocarcinoma Cells


4. LOX Mediated Metabolism
4.1. 5-Lipoxygenases-Mediated Metabolism of Arachidonic Acid in Human Lymphoblastic Cell Line


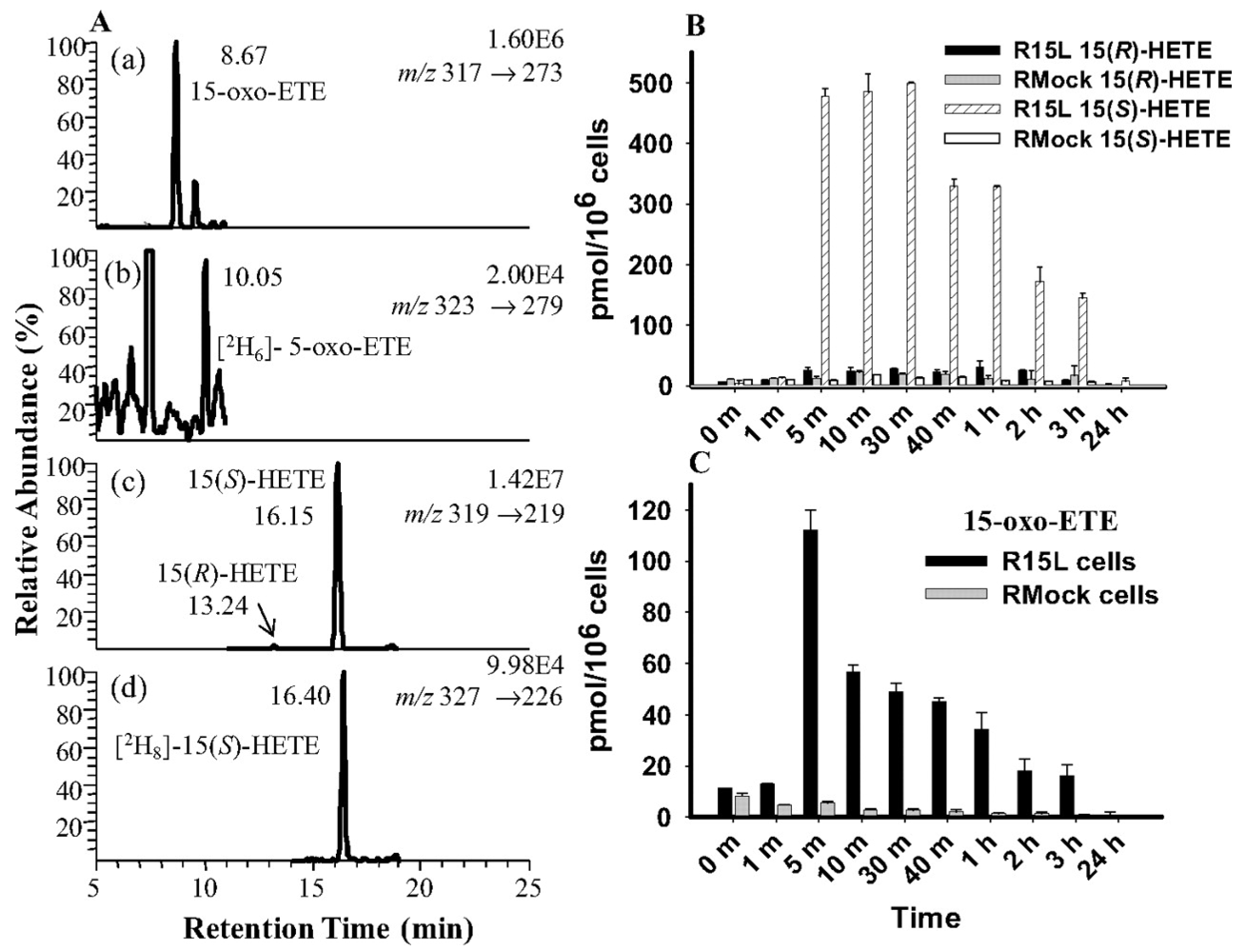
4.2. 15-LOX Mediated Metabolism of Arachidonic Acid in Macrophage Cells


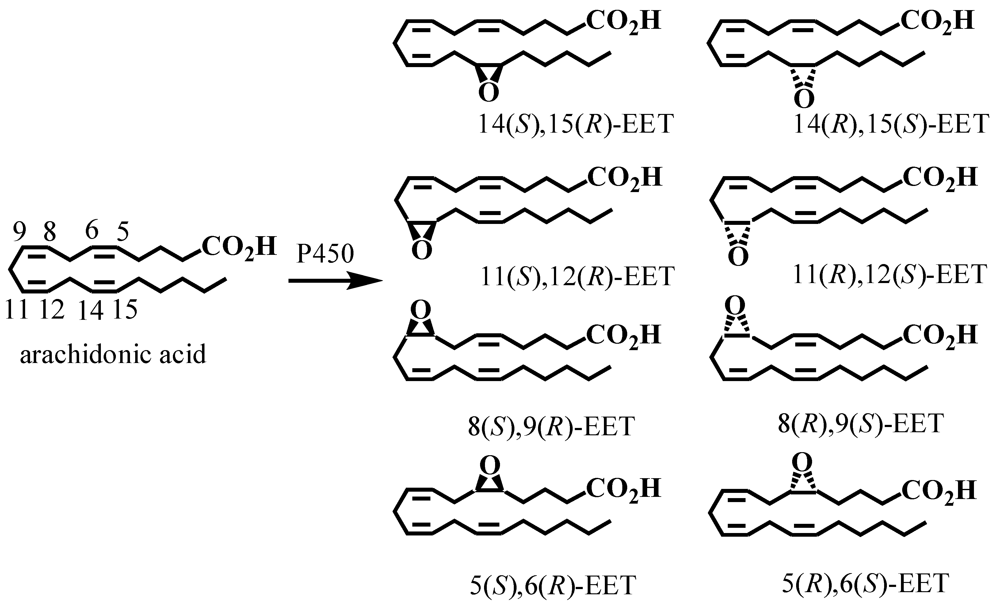
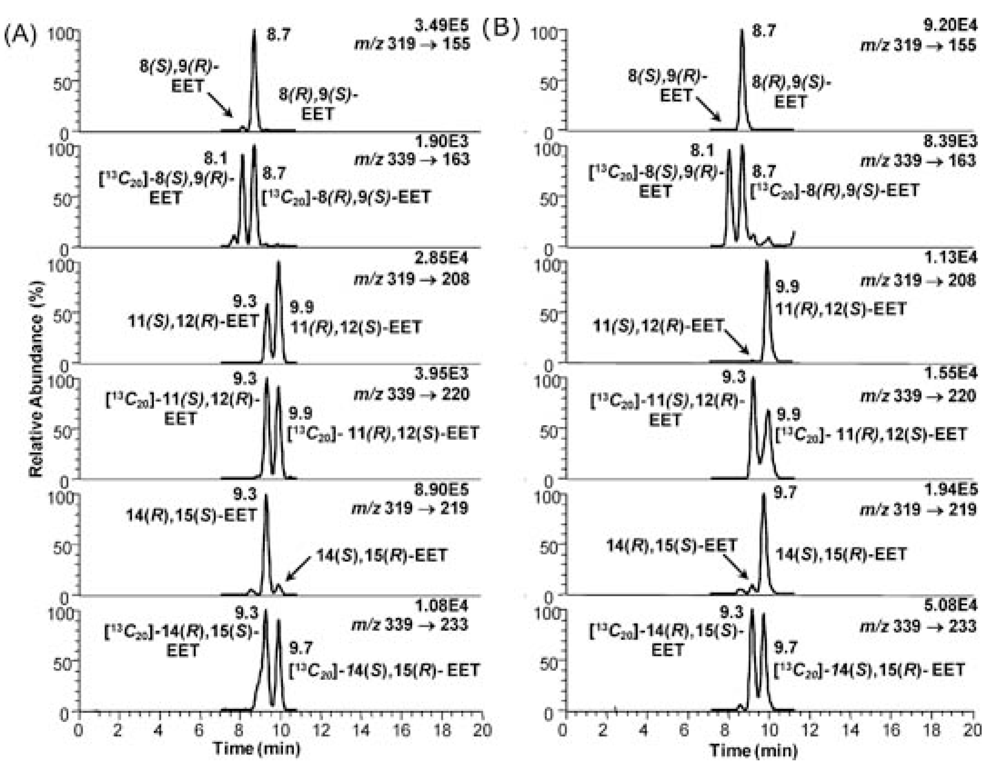
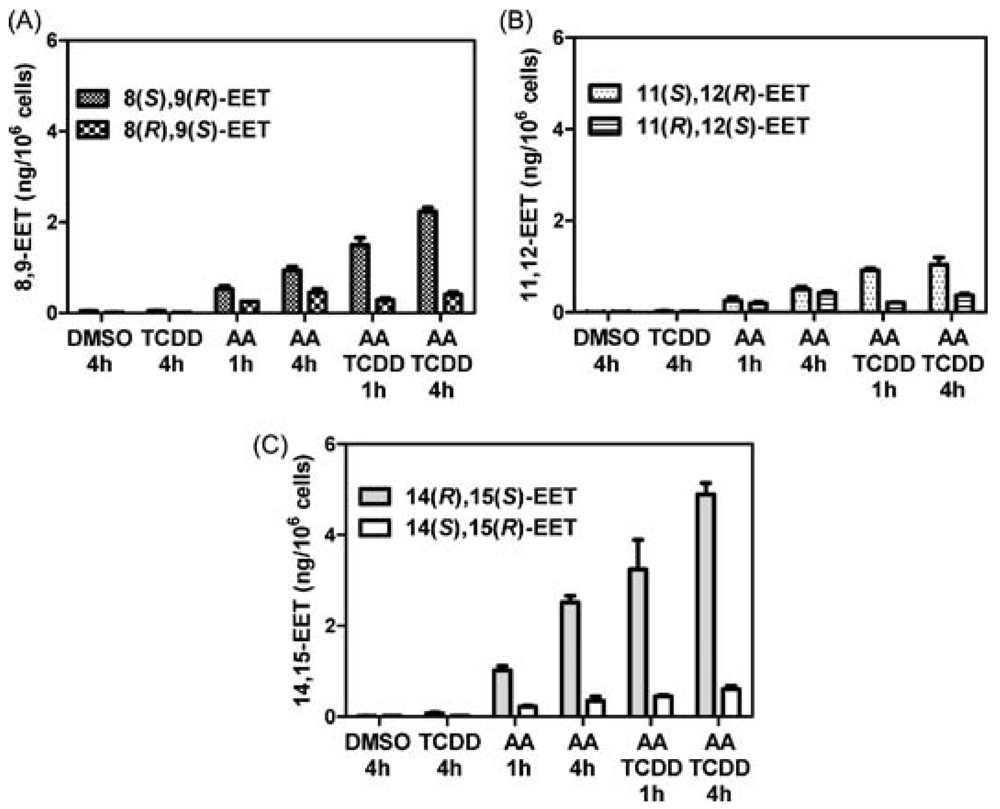
5. CYP-Mediated Metabolism of Arachidonic Acid


6. Summary and Future Directions
Acknowledgments
Conflict of Interest
References and Notes
- Bergstrom, S.; Samuelsson, B. Isolation of prostaglandin E1 from human seminal plasma. Prostaglandins and related factors. 11. J. Biol. Chem. 1962, 237, 3005–3006. [Google Scholar]
- Hamberg, M.; Samuelsson, B. Oxygenation of unsaturated fatty acids by the vesicular gland of sheep. J. Biol. Chem. 1967, 242, 5344–5354. [Google Scholar]
- DuBois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; van de Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar]
- Crofford, L.J. COX-1 and COX-2 tissue expression: implications and predictions. J. Rheumatol. Suppl 1997, 49, 15–19. [Google Scholar]
- Pong, S.S.; Hong, S.L.; Levine, L. Prostaglandin production by methylcholanthrene-transformed mouse BALB/3T3. Requirement for protein synthesis. J. Biol. Chem. 1977, 252, 1408–1413. [Google Scholar]
- Hassid, A.; Levine, L. Induction of fatty acid cyclooxygenase activity in canine kidney cells (MDCK) by benzo(a)pyrene. J. Biol. Chem. 1977, 252, 6591–6593. [Google Scholar]
- Hla, T.; Neilson, K. Human cyclooxygenase-2 cDNA. Proc. Natl. Acad. Sci. USA 1992, 89, 7384–7388. [Google Scholar]
- Tazawa, R.; Xu, X.M.; Wu, K.K.; Wang, L.H. Characterization of the genomic structure, chromosomal location and promoter of human prostaglandin H synthase-2 gene. Biochem. Biophys. Res. Commun. 1994, 203, 190–199. [Google Scholar] [CrossRef]
- Rizzo, M.T. Cyclooxygenase-2 in oncogenesis. Clinica Chimica Acta 2011, 412, 671–687. [Google Scholar] [CrossRef]
- Eberhart, C.E.; Coffey, R.J.; Radhika, A.; Giardiello, F.M.; Ferrenbach, S.; DuBois, R.N. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994, 107, 1183–1188. [Google Scholar]
- Zimmermann, K.C.; Sarbia, M.; Weber, A.A.; Borchard, F.; Gabbert, H.E.; Schror, K. Cyclooxygenase-2 expression in human esophageal carcinoma. Cancer Res. 1999, 59, 198–204. [Google Scholar]
- Tucker, O.N.; Dannenberg, A.J.; Yang, E.K.; Zhang, F.; Teng, L.; Daly, J.M.; Soslow, R.A.; Masferrer, J.L.; Woerner, B.M.; Koki, A.T.; Fahey, T.J., III. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999, 59, 987–990. [Google Scholar]
- Goulet, A.C.; Einsphar, J.G.; Alberts, D.S.; Beas, A.; Burk, C.; Bhattacharyya, A.; Bangert, J.; Harmon, J.M.; Fujiwara, H.; Koki, A.; Nelson, M.A. Analysis of cyclooxygenase 2 (COX-2) expression during malignant melanoma progression. Cancer Biol. Ther. 2003, 2, 713–718. [Google Scholar]
- Richardsen, E.; Uglehus, R.D.; Due, J.; Busch, C.; Busund, L.T. COX-2 is overexpressed in primary prostate cancer with metastatic potential and may predict survival. A comparison study between COX-2, TGF-beta, IL-10 and Ki67. Cancer Epidemiol. 2010, 34, 316–322. [Google Scholar] [CrossRef]
- Mrena, J.; Wiksten, J.P.; Kokkola, A.; Nordling, S.; Ristimaki, A.; Haglund, C. COX-2 is associated with proliferation and apoptosis markers and serves as an independent prognostic factor in gastric cancer. Tumour. Biol. 2010, 31, 1–7. [Google Scholar]
- Denkert, C.; Winzer, K.J.; Muller, B.M.; Weichert, W.; Pest, S.; Kobel, M.; Kristiansen, G.; Reles, A.; Siegert, A.; Guski, H.; Hauptmann, S. Elevated expression of cyclooxygenase-2 is a negative prognostic factor for disease free survival and overall survival in patients with breast carcinoma. Cancer 2003, 97, 2978–2987. [Google Scholar]
- Denkert, C.; Winzer, K.J.; Hauptmann, S. Prognostic impact of cyclooxygenase-2 in breast cancer. Clin. Breast Cancer 2004, 4, 428–433. [Google Scholar] [CrossRef]
- Wang, D.; DuBois, R.N. The role of COX-2 in intestinal inflammation and colorectal cancer. Oncogene 2010, 29, 781–788. [Google Scholar] [CrossRef]
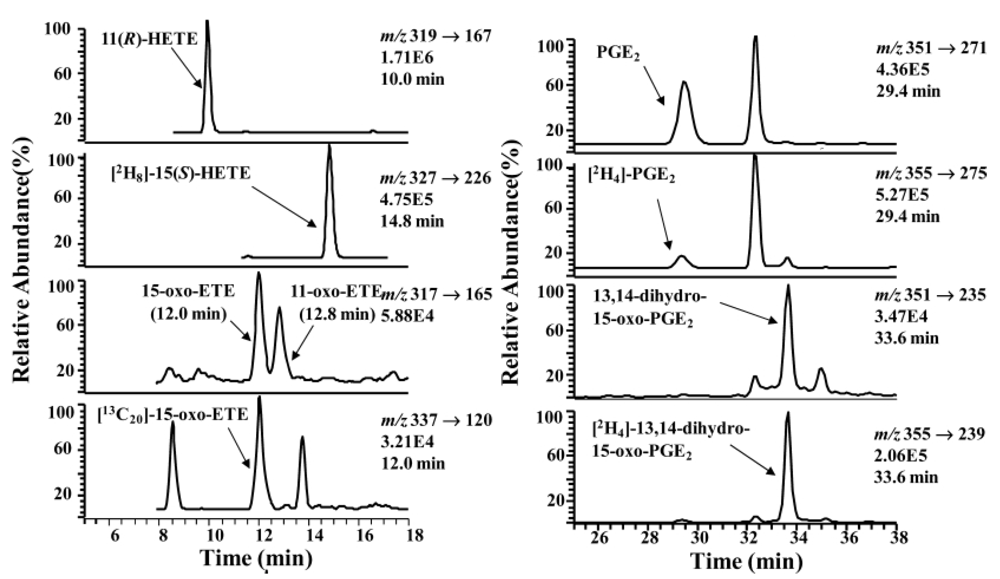
- Lee, S.H.; Rangiah, K.; Williams, M.V.; Wehr, A.Y.; DuBois, R.N.; Blair, I.A. Cyclooxygenase-2-mediated metabolism of arachidonic acid to 15-oxo-eicosatetraenoic acid by rat intestinal epithelial cells. Chem. Res. Toxicol. 2007, 20, 1665–1675. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, S.; Arora, J.S.; Snyder, N.W.; Shah, S.J.; Blair, I.A. 11-Oxoeicosatetraenoic Acid Is a Cyclooxygenase-2/15-Hydroxyprostaglandin Dehydrogenase-Derived Antiproliferative Eicosanoid. Chem. Res. Toxicol. 2011.
- Xin, X.; Yang, S.; Kowalski, J.; Gerritsen, M.E. Peroxisome proliferator-activated receptor gamma ligands are potent inhibitors of angiogenesis in vitro and in vivo. J. Biol. Chem. 1999, 274, 9116–9121. [Google Scholar]
- Backlund, M.G.; Mann, J.R.; Holla, V.R.; Shi, Q.; Daikoku, T.; Dey, S.K.; DuBois, R.N. Repression of 15-hydroxyprostaglandin dehydrogenase involves histone deacetylase 2 and snail in colorectal cancer. Cancer Res. 2008, 68, 9331–9337. [Google Scholar] [CrossRef]
- Lotzer, K.; Funk, C.D.; Habenicht, A.J. The 5-lipoxygenase pathway in arterial wall biology and atherosclerosis. Biochim. Biophys. Acta 2005, 1736, 30–37. [Google Scholar]
- Woods, J.W.; Evans, J.F.; Ethier, D.; Scott, S.; Vickers, P.J.; Hearn, L.; Heibein, J.A.; Charleson, S.; Singer, I.I. 5-lipoxygenase and 5-lipoxygenase-activating protein are localized in the nuclear envelope of activated human leukocytes. J. Exp. Med. 1993, 178, 1935–1946. [Google Scholar]
- Zhao, L.; Funk, C.D. Lipoxygenase pathways in atherogenesis. Trends Cardiovasc. Med. 2004, 14, 191–195. [Google Scholar] [CrossRef]
- Murphy, R.C.; Gijon, M.A. Biosynthesis and metabolism of leukotrienes. Biochem. J. 2007, 405, 379–395. [Google Scholar]
- Werz, O. 5-lipoxygenase: cellular biology and molecular pharmacology. Curr. Drug Targets Inflamm. Allergy 2002, 1, 23–44. [Google Scholar] [CrossRef]
- Sharma, J.N.; Mohammed, L.A. The role of leukotrienes in the pathophysiology of inflammatory disorders: is there a case for revisiting leukotrienes as therapeutic targets? Inflammopharmacology 2006, 14, 10–16. [Google Scholar] [CrossRef]
- Hicks, A.; Monkarsh, S.P.; Hoffman, A.F.; Goodnow, R., Jr. Leukotriene B4 receptor antagonists as therapeutics for inflammatory disease: preclinical and clinical developments. Expert. Opin. Investig. Drugs 2007, 16, 1909–1920. [Google Scholar] [CrossRef]
- Wymann, M.P.; Schneiter, R. Lipid signalling in disease. Nat. Rev. Mol. Cell Biol. 2008, 9, 162–176. [Google Scholar]
- Peters-Golden, M. Expanding roles for leukotrienes in airway inflammation. Curr. Allergy Asthma Rep. 2008, 8, 367–373. [Google Scholar] [CrossRef]
- Fairweather, D.; Frisancho-Kiss, S. Mast cells and inflammatory heart disease: potential drug targets. Cardiovasc. Hematol. Disord. Drug Targets 2008, 8, 80–90. [Google Scholar] [CrossRef]
- Gupta, S.; Srivastava, M.; Ahmad, N.; Sakamoto, K.; Bostwick, D.G.; Mukhtar, H. Lipoxygenase-5 is overexpressed in prostate adenocarcinoma. Cancer 2001, 91, 737–743. [Google Scholar]
- Hennig, R.; Ding, X.Z.; Tong, W.G.; Schneider, M.B.; Standop, J.; Friess, H.; Buchler, M.W.; Pour, P.M.; Adrian, T.E. 5-Lipoxygenase and leukotriene B(4) receptor are expressed in human pancreatic cancers but not in pancreatic ducts in normal tissue. Am. J. Pathol. 2002, 161, 421–428. [Google Scholar] [CrossRef]
- Hennig, R.; Grippo, P.; Ding, X.Z.; Rao, S.M.; Buchler, M.W.; Friess, H.; Talamonti, M.S.; Bell, R.H.; Adrian, T.E. 5-Lipoxygenase, a marker for early pancreatic intraepithelial neoplastic lesions. Cancer Res. 2005, 65, 6011–6016. [Google Scholar]
- Chen, X.; Sood, S.; Yang, C.S.; Li, N.; Sun, Z. Five-lipoxygenase pathway of arachidonic acid metabolism in carcinogenesis and cancer chemoprevention. Curr. Cancer Drug Targets 2006, 6, 613–622. [Google Scholar] [CrossRef]
- Powell, W.S.; Gravelle, F.; Gravel, S. Metabolism of 5(S)-hydroxy-6,8,11,14-eicosatetraenoic acid and other 5(S)-hydroxyeicosanoids by a specific dehydrogenase in human polymorphonuclear leukocytes. J. Biol. Chem. 1992, 267, 19233–19241. [Google Scholar]
- Grant, G.E.; Rokach, J.; Powell, W.S. 5-Oxo-ETE and the OXE receptor. Prostaglandins Other Lipid Mediat. 2009, 89, 98–104. [Google Scholar] [CrossRef]
- Bowers, R.C.; Hevko, J.; Henson, P.M.; Murphy, R.C. A novel glutathione containing eicosanoid (FOG7) chemotactic for human granulocytes. J. Biol. Chem. 2000, 275, 29931–29934. [Google Scholar]
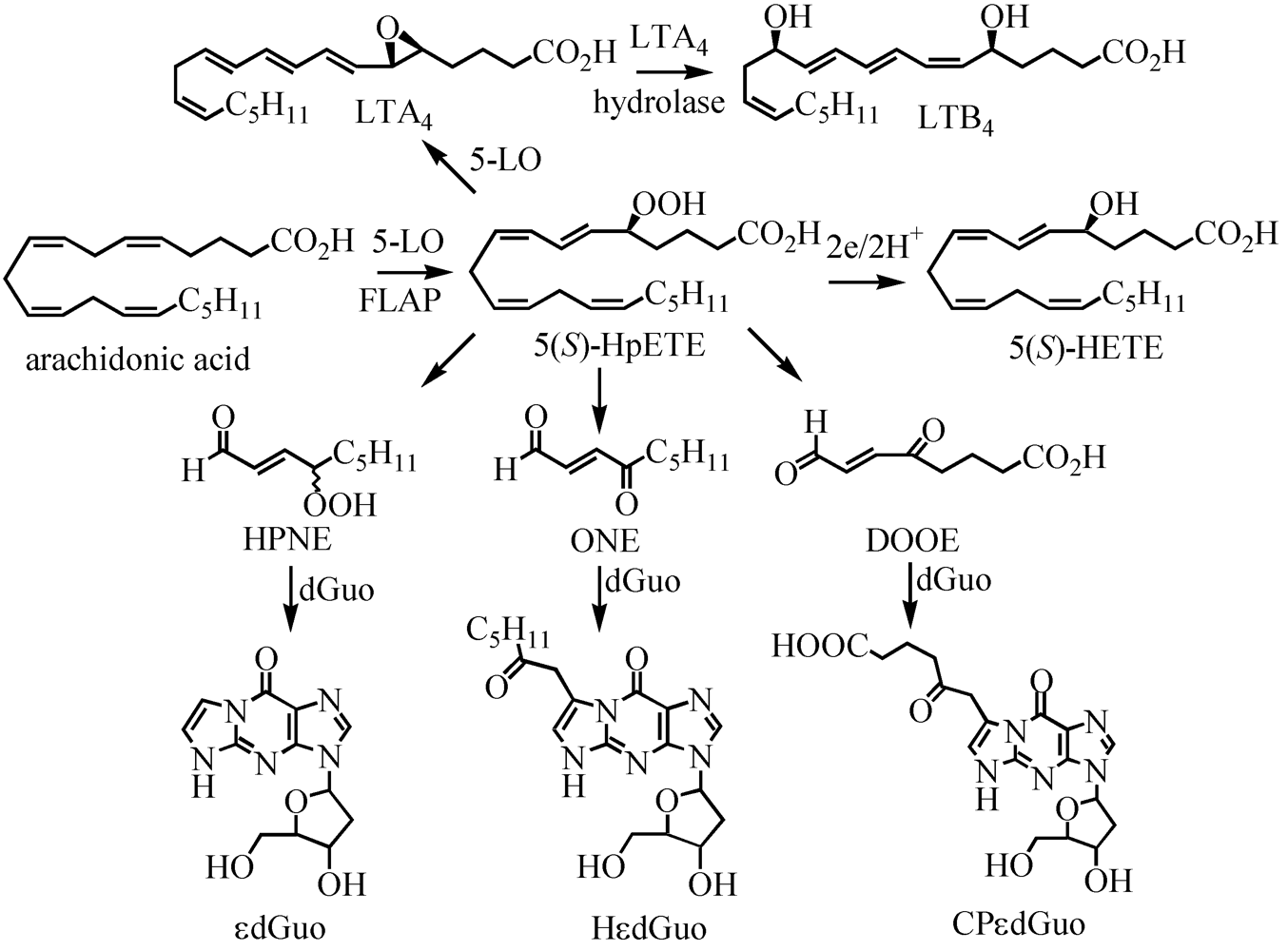
- Jian, W.; Lee, S.H.; Williams, M.V.; Blair, I.A. 5-Lipoxygenase-mediated endogenous DNA damage. J. Biol. Chem. 2009, 284, 16799–16807. [Google Scholar]
- Kuhn, H.; O'Donnell, V.B. Inflammation and immune regulation by 12/15-lipoxygenases. Prog. Lipid Res. 2006, 45, 334–356. [Google Scholar] [CrossRef]
- Kuhn, H.; Borchert, A. Regulation of enzymatic lipid peroxidation: the interplay of peroxidizing and peroxide reducing enzymes. Free Radic. Biol. Med. 2002, 33, 154–172. [Google Scholar]
- Brinckmann, R.; Schnurr, K.; Heydeck, D.; Rosenbach, T.; Kolde, G.; Kuhn, H. Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood 1998, 91, 64–74. [Google Scholar]
- Bryant, R.W.; Bailey, J.M.; Schewe, T.; Rapoport, S.M. Positional specificity of a reticulocyte lipoxygenase. Conversion of arachidonic acid to 15-S-hydroperoxy-eicosatetraenoic acid. J. Biol. Chem. 1982, 257, 6050–6055. [Google Scholar]
- Kuhn, H.; Chan, L. The role of 15-lipoxygenase in atherogenesis: pro- and antiatherogenic actions. Curr. Opin. Lipidol. 1997, 8, 111–117. [Google Scholar] [CrossRef]
- Walther, M.; Wiesner, R.; Kuhn, H. Investigations into calcium-dependent membrane association of 15-lipoxygenase-1. Mechanistic roles of surface-exposed hydrophobic amino acids and calcium. J. Biol. Chem. 2004, 279, 3717–3725. [Google Scholar]
- Viita, H.; Markkanen, J.; Eriksson, E.; Nurminen, M.; Kinnunen, K.; Babu, M.; Heikura, T.; Turpeinen, S.; Laidinen, S.; Takalo, T.; Yla-Herttuala, S. 15-lipoxygenase-1 prevents vascular endothelial growth factor A- and placental growth factor-induced angiogenic effects in rabbit skeletal muscles via reduction in growth factor mRNA levels, NO bioactivity, and downregulation of VEGF receptor 2 expression. Circ. Res. 2008, 102, 177–184. [Google Scholar] [CrossRef]
- Harats, D.; Ben-Shushan, D.; Cohen, H.; Gonen, A.; Barshack, I.; Goldberg, I.; Greenberger, S.; Hodish, I.; Harari, A.; Varda-Bloom, N.; Levanon, K.; Grossman, E.; Chaitidis, P.; Kuhn, H.; Shaish, A. Inhibition of carcinogenesis in transgenic mouse models over-expressing 15-lipoxygenase in the vascular wall under the control of murine preproendothelin-1 promoter. Cancer Lett. 2005, 229, 127–134. [Google Scholar] [CrossRef]
- Wittwer, J.; Hersberger, M. The two faces of the 15-lipoxygenase in atherosclerosis. Prostaglandins Leukot. Essent. Fatty Acids 2007, 77, 67–77. [Google Scholar] [CrossRef]
- Fierro, I.M.; Colgan, S.P.; Bernasconi, G.; Petasis, N.A.; Clish, C.B.; Arita, M.; Serhan, C.N. Lipoxin A4 and aspirin-triggered 15-epi-lipoxin A4 inhibit human neutrophil migration: comparisons between synthetic 15 epimers in chemotaxis and transmigration with microvessel endothelial cells and epithelial cells. J. Immunol. 2003, 170, 2688–2694. [Google Scholar]
- Bannenberg, G.L.; Chiang, N.; Ariel, A.; Arita, M.; Tjonahen, E.; Gotlinger, K.H.; Hong, S.; Serhan, C.N. Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 2005, 174, 4345–4355. [Google Scholar]
- Brash, A.R.; Boeglin, W.E.; Chang, M.S. Discovery of a second 15S-lipoxygenase in humans. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 6148–6152. [Google Scholar]
- Shappell, S.B.; Boeglin, W.E.; Olson, S.J.; Kasper, S.; Brash, A.R. 15-lipoxygenase-2 (15-LOX-2) is expressed in benign prostatic epithelium and reduced in prostate adenocarcinoma. Am. J. Pathol. 1999, 155, 235–245. [Google Scholar] [CrossRef]
- Daurkin, I.; Eruslanov, E.; Stoffs, T.; Perrin, G.Q.; Algood, C.; Gilbert, S.M.; Rosser, C.J.; Su, L.M.; Vieweg, J.; Kusmartsev, S. Tumor-associated macrophages mediate immunosuppression in the renal cancer microenvironment by activating the 15-lipoxygenase-2 pathway. Cancer Res. 2011, 71, 6400–6409. [Google Scholar]
- Yeung, J.; Holinstat, M. 12-lipoxygenase: a potential target for novel anti-platelet therapeutics. Cardiovasc. Hematol. Agents Med. Chem. 2011, 9, 154–164. [Google Scholar] [CrossRef]
- Boeglin, W.E.; Kim, R.B.; Brash, A.R. A 12R-lipoxygenase in human skin: mechanistic evidence, molecular cloning, and expression. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 6744–6749. [Google Scholar] [CrossRef]
- Chen, Y.Q.; Duniec, Z.M.; Liu, B.; Hagmann, W.; Gao, X.; Shimoji, K.; Marnett, L.J.; Johnson, C.R.; Honn, K.V. Endogenous 12(S)-HETE production by tumor cells and its role in metastasis. Cancer Res. 1994, 54, 1574–1579. [Google Scholar]
- Guo, Y.; Zhang, W.; Giroux, C.; Cai, Y.; Ekambaram, P.; Dilly, A.K.; Hsu, A.; Zhou, S.; Maddipati, K.R.; Liu, J.; Joshi, S.; Tucker, S.C.; Lee, M.J.; Honn, K.V. Identification of the orphan G protein-coupled receptor GPR31 as a receptor for 12-(S)-hydroxyeicosatetraenoic acid. J. Biol. Chem. 2011, 286, 33832–33840. [Google Scholar]
- Schneider, C.; Brash, A.R. Lipoxygenase-catalyzed formation of R-configuration hydroperoxides. Prostaglandins Other Lipid Mediat. 2002, 68-69, 291–301. [Google Scholar] [CrossRef]
- Pace-Asciak, C.R. Hepoxilins in cancer and inflammation--use of hepoxilin antagonists. Cancer Metastasis Rev. 2011, 30, 493–506. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450: what have we learned and what are the future issues? Drug Metab Rev. 2004, 36, 159–197. [Google Scholar] [CrossRef]
- Capdevila, J.H.; Falck, J.R.; Imig, J.D. Roles of the cytochrome P450 arachidonic acid monooxygenases in the control of systemic blood pressure and experimental hypertension. Kidney Int. 2007, 72, 683–689. [Google Scholar] [CrossRef]
- Bylund, J.; Kunz, T.; Valmsen, K.; Oliw, E.H. Cytochromes P450 with bisallylic hydroxylation activity on arachidonic and linoleic acids studied with human recombinant enzymes and with human and rat liver microsomes. J. Pharmacol. Exp. Ther. 1998, 284, 51–60. [Google Scholar]
- Hsu, M.H.; Savas, U.; Griffin, K.J.; Johnson, E.F. Human cytochrome p450 family 4 enzymes: function, genetic variation and regulation. Drug Metab Rev. 2007, 39, 515–538. [Google Scholar] [CrossRef]
- Prakash, C.; Zhang, J.Y.; Falck, J.R.; Chauhan, K.; Blair, I.A. 20-Hydroxyeicosatetraenoic acid is excreted as a glucuronide conjugate in human urine. Biochem. Biophys. Res. Commun. 1992, 185, 728–733. [Google Scholar]
- Capdevila, J.H.; Falck, J.R.; Estabrook, R.W. Cytochrome P450 and the arachidonate cascade. FASEB J. 1992, 6, 731–736. [Google Scholar]
- Harder, D.R.; Gebremedhin, D.; Narayanan, J.; Jefcoat, C.; Falck, J.R.; Campbell, W.B.; Roman, R. Formation and action of a P-450 4A metabolite of arachidonic acid in cat cerebral microvessels. Am. J. Physiol 1994, 266, H2098–H2107. [Google Scholar]
- Schwartzman, M.L.; da Silva, J.L.; Lin, F.; Nishimura, M.; Abraham, N.G. Cytochrome P450 4A expression and arachidonic acid omega-hydroxylation in the kidney of the spontaneously hypertensive rat. Nephron 1996, 73, 652–663. [Google Scholar] [CrossRef]
- Fulton, D.; Falck, J.R.; McGiff, J.C.; Carroll, M.A.; Quilley, J. A method for the determination of 5,6-EET using the lactone as an intermediate in the formation of the diol. J. Lipid Res. 1998, 39, 1713–1721. [Google Scholar]
- Chacos, N.; Capdevila, J.; Falck, J.R.; Manna, S.; Martin-Wixtrom, C.; Gill, S.S.; Hammock, B.D.; Estabrook, R.W. The reaction of arachidonic acid epoxides (epoxyeicosatrienoic acids) with a cytosolic epoxide hydrolase. Arch. Biochem. Biophys. 1983, 223, 639–648. [Google Scholar] [CrossRef]
- Zeldin, D.C.; Plitman, J.D.; Kobayashi, J.; Miller, R.F.; Snapper, J.R.; Falck, J.R.; Szarek, J.L.; Philpot, R.M.; Capdevila, J.H. The rabbit pulmonary cytochrome P450 arachidonic acid metabolic pathway: characterization and significance. J. Clin. Invest 1995, 95, 2150–2160. [Google Scholar] [CrossRef]
- Spearman, M.E.; Prough, R.A.; Estabrook, R.W.; Falck, J.R.; Manna, S.; Leibman, K.C.; Murphy, R.C.; Capdevila, J. Novel glutathione conjugates formed from epoxyeicosatrienoic acids (EETs). Arch. Biochem. Biophys. 1985, 242, 225–230. [Google Scholar] [CrossRef]
- Smith, H.E.; Jones, J.P., III; Kalhorn, T.F.; Farin, F.M.; Stapleton, P.L.; Davis, C.L.; Perkins, J.D.; Blough, D.K.; Hebert, M.F.; Thummel, K.E.; Totah, R.A. Role of cytochrome P450 2C8 and 2J2 genotypes in calcineurin inhibitor-induced chronic kidney disease. Pharmacogenet. Genomics 2008, 18, 943–953. [Google Scholar] [CrossRef]
- Kaspera, R.; Totah, R.A. Epoxyeicosatrienoic acids: formation, metabolism and potential role in tissue physiology and pathophysiology. Expert. Opin. Drug Metab Toxicol. 2009.
- Spector, A.A. Arachidonic acid cytochrome P450 epoxygenase pathway. J. Lipid Res. 2009, 50 Suppl, S52–S56. [Google Scholar]
- Capdevila, J.H.; Wei, S.; Yan, J.; Karara, A.; Jacobson, H.R.; Falck, J.R.; Guengerich, F.P.; DuBois, R.N. Cytochrome P-450 arachidonic acid epoxygenase. Regulatory control of the renal epoxygenase by dietary salt loading. J. Biol. Chem. 1992, 267, 21720–21726. [Google Scholar]
- Karara, A.; Dishman, E.; Blair, I.; Falck, J.R.; Capdevila, J.H. Endogenous epoxyeicosatrienoic acids. Cytochrome P-450 controlled stereoselectivity of the hepatic arachidonic acid epoxygenase. J. Biol. Chem. 1989, 264, 19822–19827. [Google Scholar]
- Karara, A.; Dishman, E.; Jacobson, H.; Falck, J.R.; Capdevila, J.H. Arachidonic acid epoxygenase. Stereochemical analysis of the endogenous epoxyeicosatrienoic acids of human kidney cortex. FEBS Lett. 1990, 268, 227–230. [Google Scholar] [CrossRef]
- Wu, S.; Moomaw, C.R.; Tomer, K.B.; Falck, J.R.; Zeldin, D.C. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. J. Biol. Chem. 1996, 271, 3460–3468. [Google Scholar]
- Capdevila, J.H.; Dishman, E.; Karara, A.; Falck, J.R. Cytochrome P450 arachidonic acid epoxygenase: stereochemical characterization of epoxyeicosatrienoic acids. Methods Enzymol. 1991, 206, 441–453. [Google Scholar]
- Roman, R.J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev. 2002, 82, 131–185. [Google Scholar]
- Harder, D.R.; Campbell, W.B.; Roman, R.J. Role of cytochrome P-450 enzymes and metabolites of arachidonic acid in the control of vascular tone. J. Vasc. Res. 1995, 32, 79–92. [Google Scholar] [CrossRef]
- Campbell, W.B. New role for epoxyeicosatrienoic acids as anti-inflammatory mediators. Trends Pharmacol. Sci. 2000, 21, 125–127. [Google Scholar] [CrossRef]
- Fleming, I. DiscrEET regulators of homeostasis: epoxyeicosatrienoic acids, cytochrome P450 epoxygenases and vascular inflammation. Trends Pharmacol. Sci. 2007, 28, 448–452. [Google Scholar] [CrossRef]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar]
- Spector, A.A.; Norris, A.W. Action of epoxyeicosatrienoic acids on cellular function. Am. J. Physiol Cell Physiol 2007, 292, C996–1012. [Google Scholar] [CrossRef]
- Fitzpatrick, F.A.; Ennis, M.D.; Baze, M.E.; Wynalda, M.A.; McGee, J.E.; Liggett, W.F. Inhibition of cyclooxygenase activity and platelet aggregation by epoxyeicosatrienoic acids. Influence of stereochemistry. J. Biol. Chem. 1986, 261, 15334–15338. [Google Scholar]
- Panigrahy, D.; Edin, M.L.; Lee, C.R.; Huang, S.; Bielenberg, D.R.; Butterfield, C.E.; Barnes, C.M.; Mammoto, A.; Mammoto, T.; Luria, A.; Benny, O.; Chaponis, D.M.; Dudley, A.C.; Greene, E.R.; Vergilio, J.A.; Pietramaggiori, G.; Scherer-Pietramaggiori, S.S.; Short, S.M.; Seth, M.; Lih, F.B.; Tomer, K.B.; Yang, J.; Schwendener, R.A.; Hammock, B.D.; Falck, J.R.; Manthati, V.L.; Ingber, D.E.; Kaipainen, A.; D'Amore, P.A.; Kieran, M.W.; Zeldin, D.C. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Invest 2012, 122, 178–191. [Google Scholar] [Green Version]
- Wang, D.; DuBois, R.N. Epoxyeicosatrienoic acids: a double-edged sword in cardiovascular diseases and cancer. J. Clin. Invest 2012, 122, 19–22. [Google Scholar] [CrossRef]
- Mesaros, C.; Lee, S.H.; Blair, I.A. Targeted quantitative analysis of eicosanoid lipids in biological samples using liquid chromatography-tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2009, 877, 2736–2745. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Lopez, R.; Tamimi, T.A.; Yerian, L.; Chung, Y.M.; Berk, M.; Zhang, R.; McIntyre, T.M.; Hazen, S.L. Mass spectrometric profiling of oxidized lipid products in human nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. J. Lipid Res. 2010, 51, 3046–3054. [Google Scholar] [CrossRef]
- Yang, R.; Chiang, N.; Oh, S.F.; Serhan, C.N. Metabolomics-lipidomics of eicosanoids and docosanoids generated by phagocytes. Curr. Protoc. Immunol. 2011, Chapter 14. [Google Scholar]
- Oh, S.F.; Vickery, T.W.; Serhan, C.N. Chiral lipidomics of E-series resolvins: aspirin and the biosynthesis of novel mediators. Biochim. Biophys. Acta 2011, 1811, 737–747. [Google Scholar] [CrossRef]
- Blaho, V.A.; Buczynski, M.W.; Brown, C.R.; Dennis, E.A. Lipidomic analysis of dynamic eicosanoid responses during the induction and resolution of Lyme arthritis. J. Biol. Chem. 2009, 284, 21599–21612. [Google Scholar] [CrossRef]
- Sanak, M.; Gielicz, A.; Nagraba, K.; Kaszuba, M.; Kumik, J.; Szczeklik, A. Targeted eicosanoids lipidomics of exhaled breath condensate in healthy subjects. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2010, 878, 1796–1800. [Google Scholar] [CrossRef]
- Altmaier, E.; Kastenmuller, G.; Romisch-Margl, W.; Thorand, B.; Weinberger, K.M.; Illig, T.; Adamski, J.; Doring, A.; Suhre, K. Questionnaire-based self-reported nutrition habits associate with serum metabolism as revealed by quantitative targeted metabolomics. Eur. J. Epidemiol. 2011, 26, 145–156. [Google Scholar] [CrossRef]
- Mal, M.; Koh, P.K.; Cheah, P.Y.; Chan, E.C. Ultra-pressure liquid chromatography/tandem mass spectrometry targeted profiling of arachidonic acid and eicosanoids in human colorectal cancer. Rapid Commun. Mass Spectrom. 2011, 25, 755–764. [Google Scholar] [CrossRef]
- Manna, J.D.; Reyzer, M.L.; Latham, J.C.; Weaver, C.D.; Marnett, L.J.; Caprioli, R.M. High-throughput quantification of bioactive lipids by MALDI mass spectrometry: application to prostaglandins. Anal. Chem. 2011, 83, 6683–6688. [Google Scholar]
- Clugston, R.D.; Jiang, H.; Lee, M.X.; Piantedosi, R.; Yuen, J.J.; Ramakrishnan, R.; Lewis, M.J.; Gottesman, M.E.; Huang, L.S.; Goldberg, I.J.; Berk, P.D.; Blaner, W.S. Altered hepatic lipid metabolism in C57BL/6 mice fed alcohol: a targeted lipidomic and gene expression study. J. Lipid Res. 2011, 52, 2021–2031. [Google Scholar] [CrossRef]
- Boger, M.S.; Bian, A.; Shintani, A.; Milne, G.L.; Morrow, J.D.; Erdem, H.; Mitchell, V.; Haas, D.W.; Hulgan, T. Sex differences in urinary biomarkers of vascular and endothelial function in HIV-infected persons receiving antiretroviral therapy. Antivir. Ther. 2011.
- Murphy, R.C.; Barkley, R.M.; Zemski, B.K.; Hankin, J.; Harrison, K.; Johnson, C.; Krank, J.; McAnoy, A.; Uhlson, C.; Zarini, S. Electrospray ionization and tandem mass spectrometry of eicosanoids. Anal. Biochem. 2005, 346, 1–42. [Google Scholar] [CrossRef]
- Singh, G.; Gutierrez, A.; Xu, K.; Blair, I.A. Liquid chromatography/electron capture atmospheric pressure chemical ionization/mass spectrometry: analysis of pentafluorobenzyl derivatives of biomolecules and drugs in the attomole range. Anal. Chem. 2000, 72, 3007–3013. [Google Scholar]
- Lee, S.H.; Williams, M.V.; DuBois, R.N.; Blair, I.A. Targeted lipidomics using electron capture atmospheric pressure chemical ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 2168–2176. [Google Scholar] [CrossRef]
- Lee, S.H.; Williams, M.V.; Blair, I.A. Targeted chiral lipidomics analysis. Prostaglandins Other Lipid Mediat. 2005, 77, 141–157. [Google Scholar] [CrossRef]
- Lee, S.H.; Blair, I.A. Targeted chiral lipidomics analysis by liquid chromatography electron capture atmospheric pressure chemical ionization mass spectrometry (LC-ECAPCI/MS). Methods Enzymol. 2007, 433, 159–174. [Google Scholar]
- Blair, I.A.; Barrow, S.E.; Waddell, K.A.; Lewis, P.J.; Dollery, C.T. Prostacyclin is not a circulating hormone in man. Prostaglandins 1982, 23, 579–589. [Google Scholar] [CrossRef]
- Blair, I.A. Electron-capture negative-ion chemical ionization mass-spectrometry of lipid mediators. Methods Enzymol. 1990, 187, 13–23. [Google Scholar] [CrossRef]
- Jian, W.; Lee, S.H.; Williams, M.V.; Blair, I.A. 5-Lipoxygenase-mediated endogenous DNA damage. J. Biol. Chem. 2009, 284, 16799–16807. [Google Scholar]
- Wei, S.; Brittin, J.J.; Falck, J.R.; Anjaiah, S.; Nithipatikom, K.; Cui, L.; Campbell, W.B.; Capdevila, J.H. Chiral resolution of the epoxyeicosatrienoic acids, arachidonic acid epoxygenase metabolites. Anal. Biochem. 2006, 352, 129–134. [Google Scholar]
- Liu, X.; Zhang, S.; Arora, J.S.; Snyder, N.W.; Shah, S.J.; Blair, I.A. 11-Oxoeicosatetraenoic acid is a cyclooxygenase-2/15-hydroxyprostaglandin dehydrogenase-derived antiproliferative eicosanoid. Chem. Res. Toxicol. 2011, 24, 2227–2236. [Google Scholar] [CrossRef]
- Matsumura, F. The significance of the nongenomic pathway in mediating inflammatory signaling of the dioxin-activated Ah receptor to cause toxic effects. Biochem. Pharmacol. 2009, 77, 608–626. [Google Scholar] [CrossRef]
- Ouyang, W.; Ma, Q.; Li, J.; Zhang, D.; Ding, J.; Huang, Y.; Xing, M.M.; Huang, C. Benzo[a]pyrene diol-epoxide (B[a]PDE) upregulates COX-2 expression through MAPKs/AP-1 and IKKbeta/NF-kappaB in mouse epidermal Cl41 cells. Mol. Carcinog. 2007, 46, 32–41. [Google Scholar]
- Huang, R.Y.; Chen, G.G. Cigarette smoking, cyclooxygenase-2 pathway and cancer. Biochim. Biophys. Acta 2011, 1815, 158–169. [Google Scholar]
- Backlund, M.G.; Mann, J.R.; Holla, V.R.; Buchanan, F.G.; Tai, H.H.; Musiek, E.S.; Milne, G.L.; Katkuri, S.; DuBois, R.N. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J. Biol. Chem. 2005, 280, 3217–3223. [Google Scholar]
- Chou, W.L.; Chuang, L.M.; Chou, C.C.; Wang, A.H.; Lawson, J.A.; FitzGerald, G.A.; Chang, Z.F. Identification of a novel prostaglandin reductase reveals the involvement of prostaglandin E2 catabolism in regulation of peroxisome proliferator-activated receptor gamma activation. J. Biol. Chem. 2007, 282, 18162–18172. [Google Scholar]
- Hughes, D.; Otani, T.; Yang, P.; Newman, R.A.; Yantiss, R.K.; Altorki, N.K.; Port, J.L.; Yan, M.; Markowitz, S.D.; Mazumdar, M.; Tai, H.H.; Subbaramaiah, K.; Dannenberg, A.J. NAD+-dependent 15-hydroxyprostaglandin dehydrogenase regulates levels of bioactive lipids in non-small cell lung cancer. Cancer Prev. Res. (Phila) 2008, 1, 241–249. [Google Scholar] [CrossRef]
- Tai, H.H.; Tong, M.; Ding, Y. 15-hydroxyprostaglandin dehydrogenase (15-PGDH) and lung cancer. Prostaglandins Other Lipid Mediat. 2007, 83, 203–208. [Google Scholar] [CrossRef]
- Lee, S.H.; Williams, M.V.; DuBois, R.N.; Blair, I.A. Cyclooxygenase-2-mediated DNA damage. J. Biol. Chem. 2005, 280, 28337–28346. [Google Scholar] [CrossRef]
- Wei, C.; Zhu, P.; Shah, S.J.; Blair, I.A. 15-oxo-Eicosatetraenoic acid, a metabolite of macrophage 15-hydroxyprostaglandin dehydrogenase that inhibits endothelial cell proliferation. Mol. Pharmacol. 2009, 76, 516–525. [Google Scholar] [CrossRef]
- Shao, J.; Sheng, H.; Inoue, H.; Morrow, J.D.; DuBois, R.N. Regulation of constitutive cyclooxygenase-2 expression in colon carcinoma cells. J. Biol. Chem. 2000, 275, 33951–33956. [Google Scholar]
- Waddington, E.; Sienuarine, K.; Puddey, I.; Croft, K. Identification and quantitation of unique fatty acid oxidation products in human atherosclerotic plaque using high-performance liquid chromatography. Anal. Biochem. 2001, 292, 234–244. [Google Scholar]
- Yan, M.; Mehta, J.L.; Zhang, W.; Hu, C. LOX-1, Oxidative Stress and Inflammation: A Novel Mechanism for Diabetic Cardiovascular Complications. Cardiovasc. Drugs Ther. 2011, 25, 451–459. [Google Scholar]
- Greene, E.R.; Huang, S.; Serhan, C.N.; Panigrahy, D. Regulation of inflammation in cancer by eicosanoids. Prostaglandins Other Lipid Mediat. 2011, 96, 27–36. [Google Scholar] [CrossRef]
- Williams, M.V.; Lee, S.H.; Blair, I.A. Liquid chromatography/mass spectrometry analysis of bifunctional electrophiles and DNA adducts from vitamin C mediated decomposition of 15-hydroperoxyeicosatetraenoic acid. Rapid Commun. Mass Spectrom. 2005, 19, 849–858. [Google Scholar] [CrossRef]
- Lee, S.H.; Oe, T.; Blair, I.A. Vitamin C-induced decomposition of lipid hydroperoxides to endogenous genotoxins. Science 2001, 292, 2083–2086. [Google Scholar] [CrossRef]
- Lee, S.H.; Arora, J.A.; Oe, T.; Blair, I.A. 4-Hydroperoxy-2-nonenal-induced formation of 1,N2-etheno-2'-deoxyguanosine adducts. Chem. Res. Toxicol. 2005, 18, 780–786. [Google Scholar] [CrossRef]
- Lee, S.H.; Silva Elipe, M.V.; Arora, J.S.; Blair, I.A. Dioxododecenoic acid: a lipid hydroperoxide-derived bifunctional electrophile responsible for etheno DNA adduct formation. Chem. Res. Toxicol. 2005, 18, 566–578. [Google Scholar] [CrossRef]
- Williams, M.V.; Lee, S.H.; Pollack, M.; Blair, I.A. Endogenous lipid hydroperoxide-mediated DNA-adduct formation in min mice. J. Biol. Chem. 2006, 281, 10127–10133. [Google Scholar] [CrossRef]
- Berry, C.N.; Hoult, J.R.; Peers, S.H.; Agback, H. Inhibition of prostaglandin 15-hydroxydehydrogenase by sulphasalazine and a novel series of potent analogues. Biochem. Pharmacol. 1983, 32, 2863–2871. [Google Scholar] [CrossRef]
- Quidville, V.; Segond, N.; Lausson, S.; Frenkian, M.; Cohen, R.; Jullienne, A. 15-Hydroxyprostaglandin-dehydrogenase is involved in anti-proliferative effect of non-steroidal anti-inflammatory drugs COX-1 inhibitors on a human medullary thyroid carcinoma cell line. Prostaglandins Other Lipid Mediat. 2006, 81, 14–30. [Google Scholar] [CrossRef]
- Gulliksson, M.; Brunnstrom, A.; Johannesson, M.; Backman, L.; Nilsson, G.; Harvima, I.; Dahlen, B.; Kumlin, M.; Claesson, H.E. Expression of 15-lipoxygenase type-1 in human mast cells. Biochim. Biophys. Acta 2007, 1771, 1156–1165. [Google Scholar]
- Murphy, R.C.; Zarini, S. Glutathione adducts of oxyeicosanoids. Glutathione adducts of oxyeicosanoids. 2002, 68-69, 471–482. [Google Scholar]
- Blair, I.A. Endogenous glutathione adducts. Cur. Drug Metab. 2006, 7, 853–872. [Google Scholar] [CrossRef]
- Blair, I.A. Analysis of endogenous glutathione-adducts and their metabolites. Biomed. Chromatogr. 2010, 24, 29–38. [Google Scholar] [CrossRef]
- Fierro, I.M.; Kutok, J.L.; Serhan, C.N. Novel lipid mediator regulators of endothelial cell proliferation and migration: aspirin-triggered-15R-lipoxin A(4) and lipoxin A(4). J. Pharmacol. Exp. Ther. 2002, 300, 385–392. [Google Scholar] [CrossRef]
- Merched, A.J.; Ko, K.; Gotlinger, K.H.; Serhan, C.N.; Chan, L. Atherosclerosis: evidence for impairment of resolution of vascular inflammation governed by specific lipid mediators. FASEB J. 2008, 22, 3595–3606. [Google Scholar] [CrossRef]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar]
- Mesaros, C.; Lee, S.H.; Blair, I.A. Analysis of epoxyeicosatrienoic acids by chiral liquid chromatography/electron capture atmospheric pressure chemical ionization mass spectrometry using [13C]-analog internal standards. Rapid Commun. Mass Spectrom. 2010, 24, 3237–3247. [Google Scholar] [CrossRef]
- Capdevila, J.H.; Kishore, V.; Dishman, E.; Blair, I.A.; Falck, J.R. A novel pool of rat liver inositol and ethanolamine phospholipids contains epoxyeicosatrienoic acids (EETs). Biochem. Biophys. Res. Commun. 1987, 146, 638–644. [Google Scholar]
- Hammonds, T.D.; Blair, I.A.; Falck, J.R.; Capdevila, J.H. Resolution of epoxyeicosatrienoate enantiomers by chiral phase chromatography. Anal. Biochem. 1989, 182, 300–303. [Google Scholar] [CrossRef]
- VanderNoot, V.A.; VanRollins, M. Capillary electrophoresis of cytochrome P-450 epoxygenase metabolites of arachidonic acid. 1. Resolution of regioisomers. Anal. Chem. 2002, 74, 5859–5865. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Blair, I.A. Direct resolution of epoxyeicosatrienoic acid enantiomers by chiral-phase high-performance liquid chromatography. J. Chromatogr. B Biomed. Appl. 1994, 657, 23–29. [Google Scholar] [CrossRef]
- Hammonds, T.D.; Blair, I.A.; Falck, J.R.; Capdevila, J.H. Resolution of epoxyeicosatrienoate enantiomers by chiral phase chromatography. Anal. Biochem. 1989, 182, 300–303. [Google Scholar] [CrossRef]
- Bylund, J.; Ericsson, J.; Oliw, E.H. Analysis of cytochrome P450 metabolites of arachidonic and linoleic acids by liquid chromatography-mass spectrometry with ion trap MS. Anal. Biochem. 1998, 265, 55–68. [Google Scholar]
- Wei, S.; Brittin, J.J.; Falck, J.R.; Anjaiah, S.; Nithipatikom, K.; Cui, L.; Campbell, W.B.; Capdevila, J.H. Chiral resolution of the epoxyeicosatrienoic acids, arachidonic acid epoxygenase metabolites. Anal. Biochem. 2006, 352, 129–134. [Google Scholar]
- Kiss, L.; Roder, Y.; Bier, J.; Weissmann, N.; Seeger, W.; Grimminger, F. Direct eicosanoid profiling of the hypoxic lung by comprehensive analysis via capillary liquid chromatography with dual online photodiode-array and tandem mass-spectrometric detection. Anal. Bioanal. Chem. 2008, 390, 697–714. [Google Scholar]
- Zhang, J.Y.; Prakash, C.; Yamashita, K.; Blair, I.A. Regiospecific and enantioselective metabolism of 8,9-epoxyeicosatrienoic acid by cyclooxygenase. Biochem. Biophys. Res. Commun. 1992, 183, 138–143. [Google Scholar] [CrossRef]
- Homma, T.; Zhang, J.Y.; Shimizu, T.; Prakash, C.; Blair, I.A.; Harris, R.C. Cyclooxygenase-derived metabolites of 8,9-epoxyeicosatrienoic acid are potent mitogens for cultured rat glomerular mesangial cells. Biochem. Biophys. Res. Commun. 1993, 191, 282–288. [Google Scholar] [CrossRef]
- Ciccimaro, E.; Blair, I.A. Stable-isotope dilution LC-MS for quantitative biomarker analysis. Bioanalysis. 2010, 2, 311–341. [Google Scholar] [CrossRef]
- Tan, B.; O'Dell, D.K.; Yu, Y.W.; Monn, M.F.; Hughes, H.V.; Burstein, S.; Walker, J.M. Identification of endogenous acyl amino acids based on a targeted lipidomics approach. J. Lipid Res. 2010, 51, 112–119. [Google Scholar] [CrossRef]
- Huang, S.M.; Bisogno, T.; Petros, T.J.; Chang, S.Y.; Zavitsanos, P.A.; Zipkin, R.E.; Sivakumar, R.; Coop, A.; Maeda, D.Y.; De, P.L.; Burstein, S.; Di, M., V; Walker, J.M. Identification of a new class of molecules, the arachidonyl amino acids, and characterization of one member that inhibits pain. J. Biol. Chem. 2001, 276, 42639–42644. [Google Scholar]
- Huang, S.M.; Walker, J.M. Enhancement of spontaneous and heat-evoked activity in spinal nociceptive neurons by the endovanilloid/endocannabinoid N-arachidonoyldopamine (NADA). J. Neurophysiol. 2006, 95, 1207–1212. [Google Scholar]
- Wehr, A.Y.; Hwang, W.T.; Blair, I.A.; Yu, K.H. Relative Quantification of Serum Proteins from Pancreatic Ductal Adenocarcinoma Patients by Stable Isotope Dilution Liquid Chromatography-Mass Spectrometry. J. Proteome Res. 2012, 11, 1749–1758. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mesaros, C.; Blair, I.A. Targeted Chiral Analysis of Bioactive Arachidonic Acid Metabolites Using Liquid-Chromatography-Mass Spectrometry. Metabolites 2012, 2, 337-365. https://doi.org/10.3390/metabo2020337
Mesaros C, Blair IA. Targeted Chiral Analysis of Bioactive Arachidonic Acid Metabolites Using Liquid-Chromatography-Mass Spectrometry. Metabolites. 2012; 2(2):337-365. https://doi.org/10.3390/metabo2020337
Chicago/Turabian StyleMesaros, Clementina, and Ian A. Blair. 2012. "Targeted Chiral Analysis of Bioactive Arachidonic Acid Metabolites Using Liquid-Chromatography-Mass Spectrometry" Metabolites 2, no. 2: 337-365. https://doi.org/10.3390/metabo2020337
APA StyleMesaros, C., & Blair, I. A. (2012). Targeted Chiral Analysis of Bioactive Arachidonic Acid Metabolites Using Liquid-Chromatography-Mass Spectrometry. Metabolites, 2(2), 337-365. https://doi.org/10.3390/metabo2020337