Current Understanding of the Formation and Adaptation of Metabolic Systems Based on Network Theory

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Representation of Metabolic Networks
2.1. Substrate–Product Networks
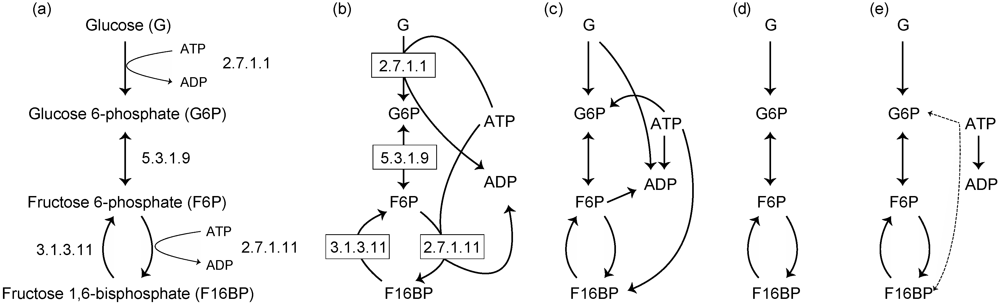
2.2. Reaction Networks
3. Networks Provide an Extended View of Metabolic Evolution
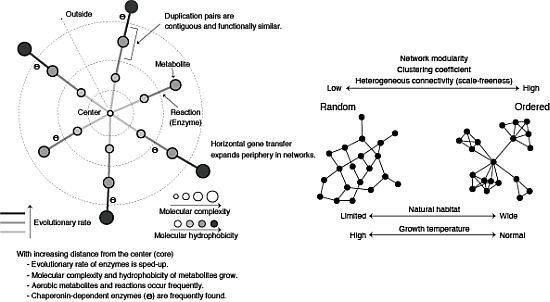
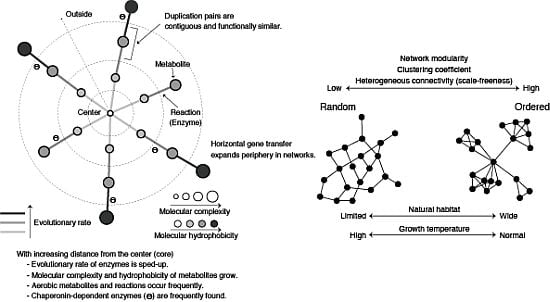
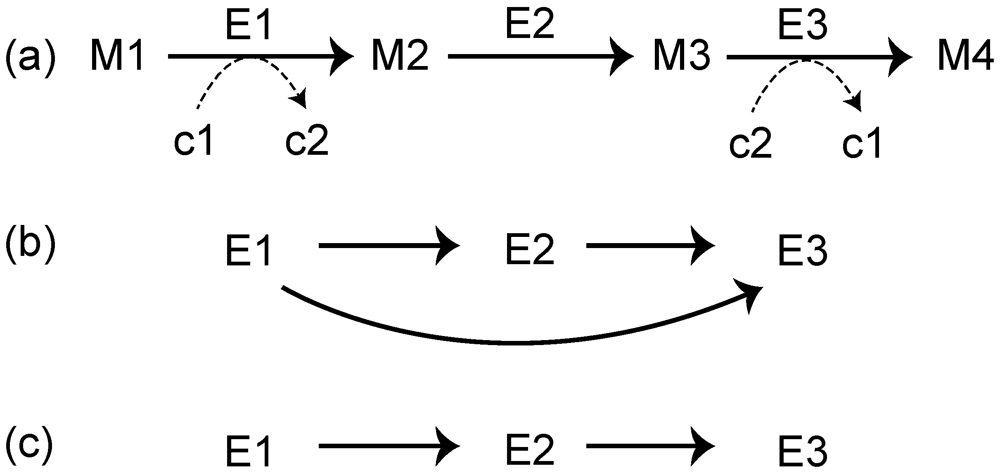
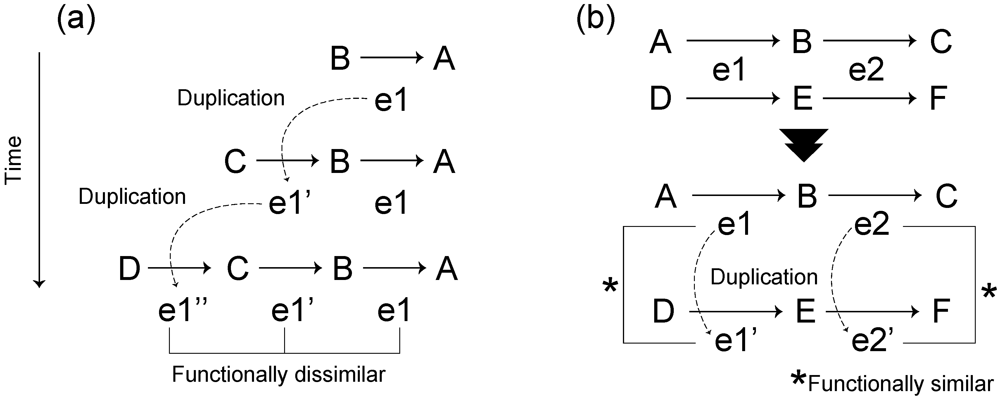
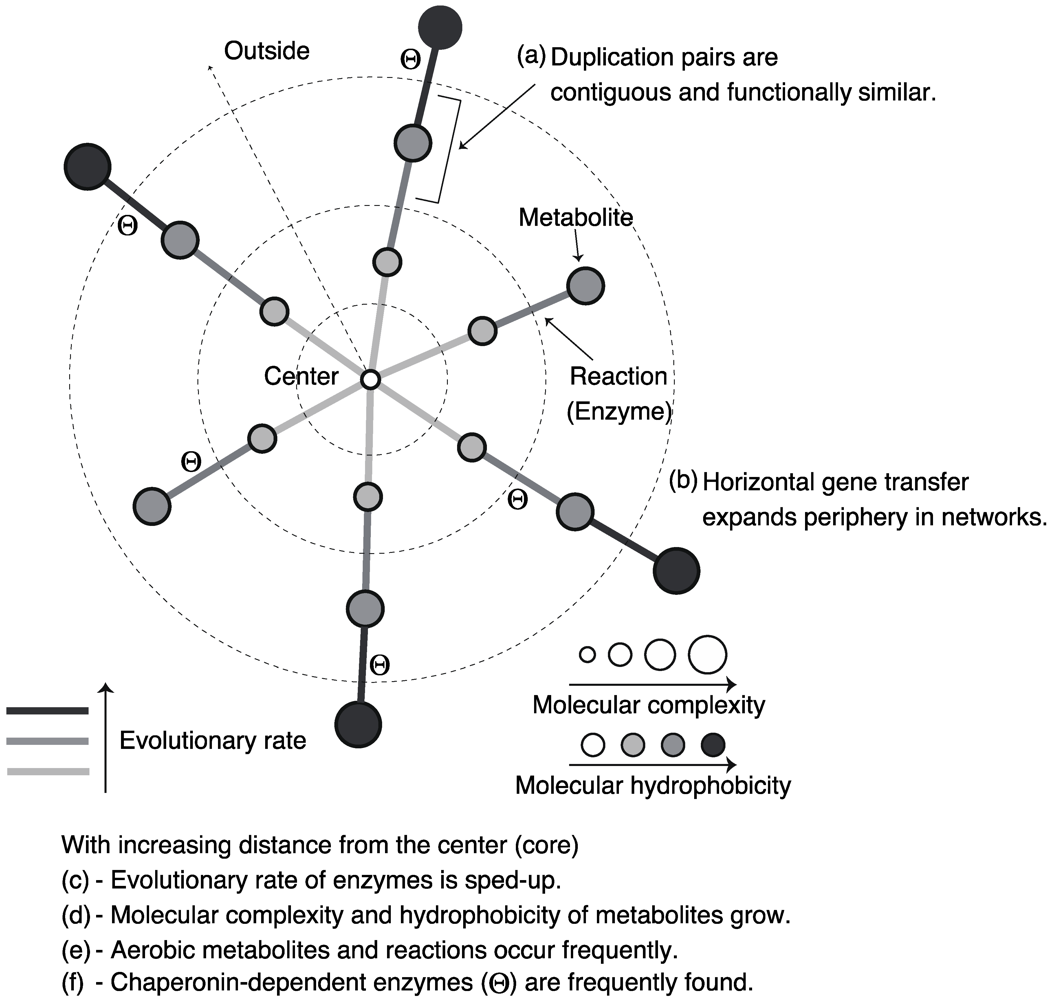
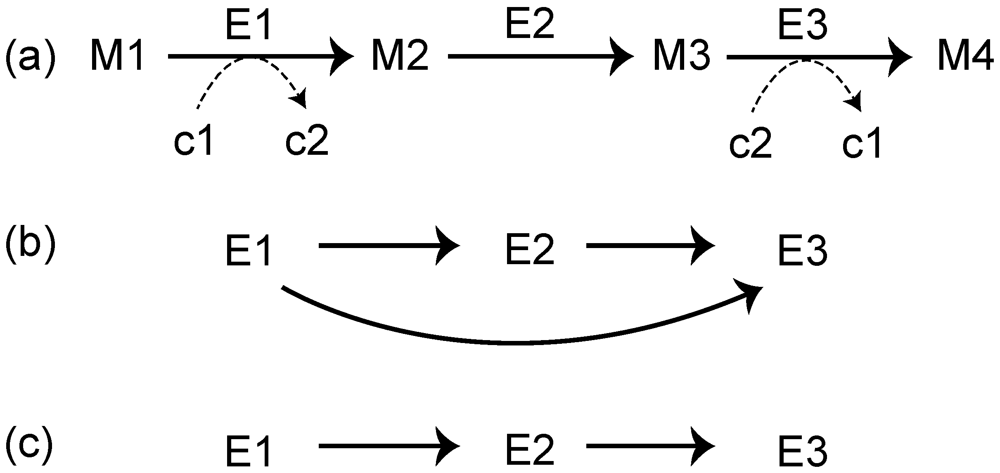
3.1. Roles of Gene Duplication
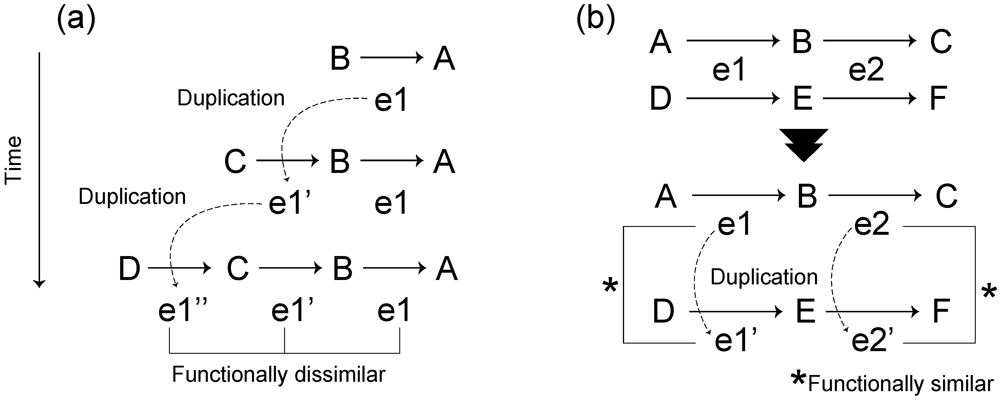
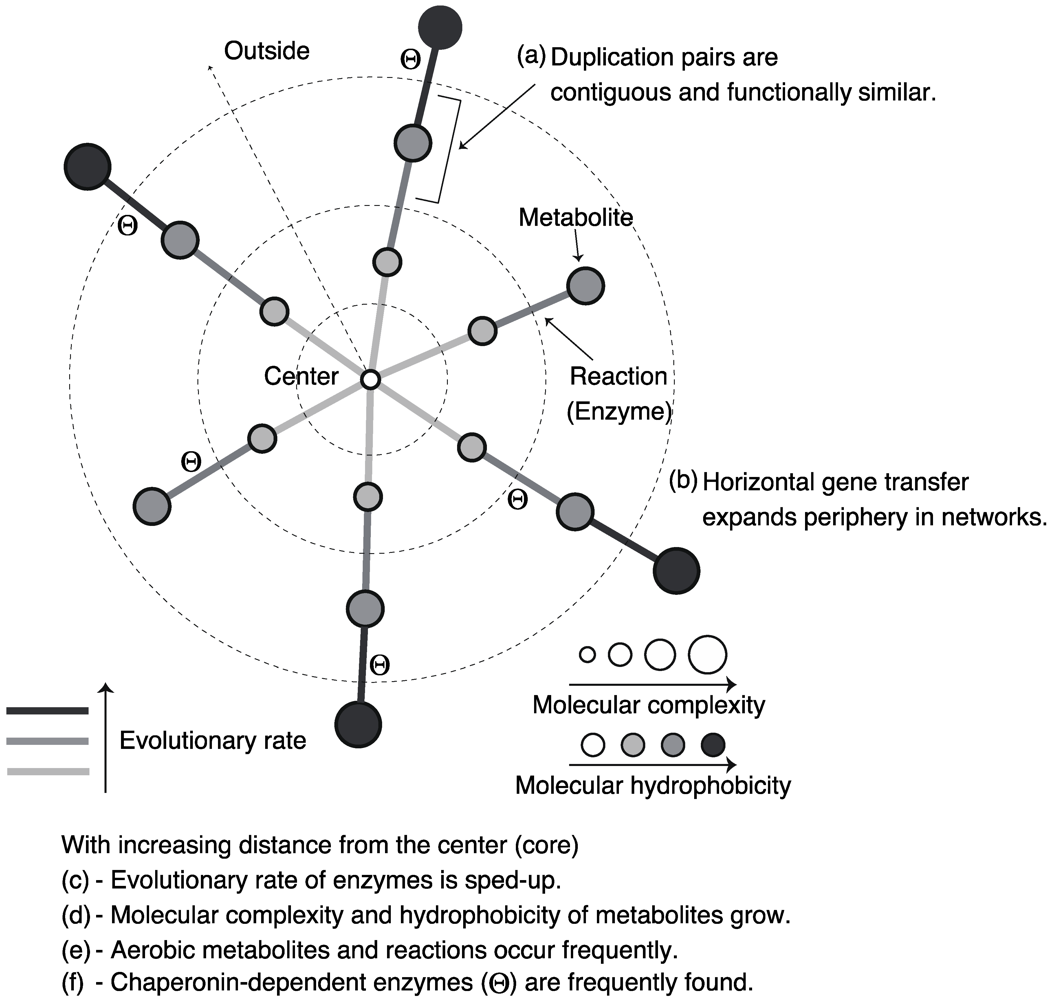
3.2. Role of Horizontal Gene Transfer
3.3. Evolutionary Rate in Metabolic Networks
3.4. From a Viewpoint of Chemical Properties of Metabolites
3.5. Roles of Chaperonin
4. Understanding Formation and Adaptation through Structural Properties
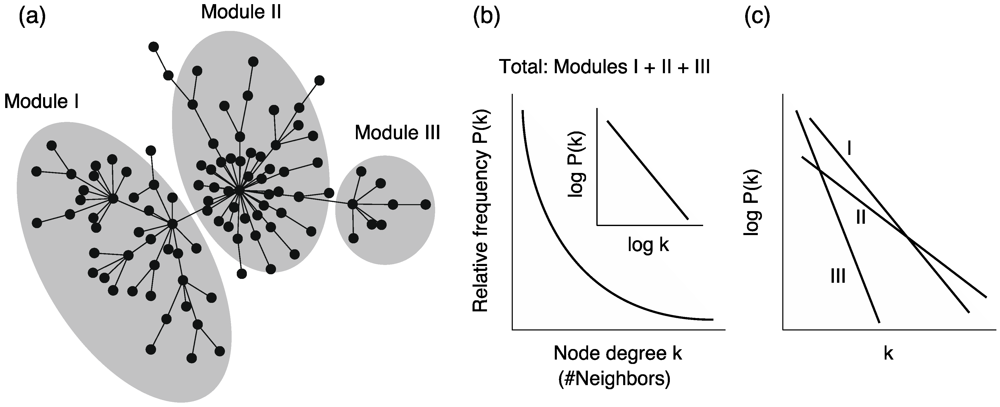
4.1. Scele-Free Connectivity
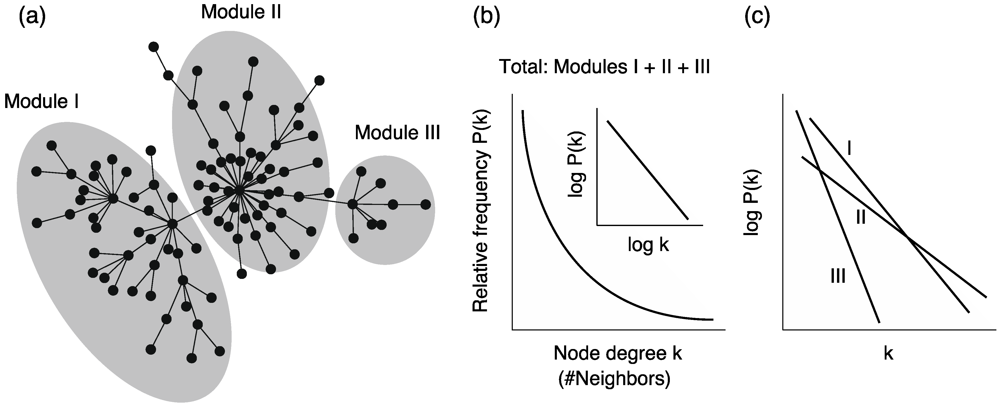
 , where P(k) is the relative frequency of nodes with degree k (Figure 5b).
, where P(k) is the relative frequency of nodes with degree k (Figure 5b). is satisfied, where C and α are constants, when considering a power-law function
is satisfied, where C and α are constants, when considering a power-law function  .
.
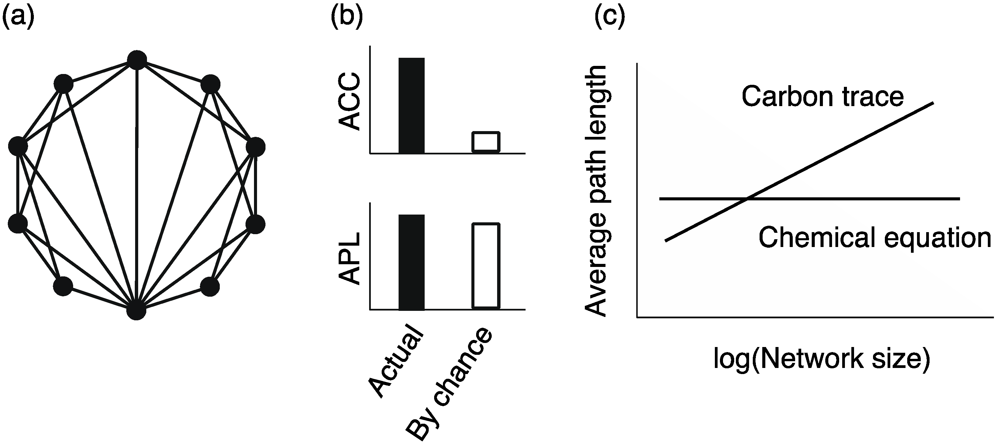
4.2. Small Worldness
 , where mi and ki indicate the number of edges among neighbors of node i and the degree of node i, respectively.
, where mi and ki indicate the number of edges among neighbors of node i and the degree of node i, respectively.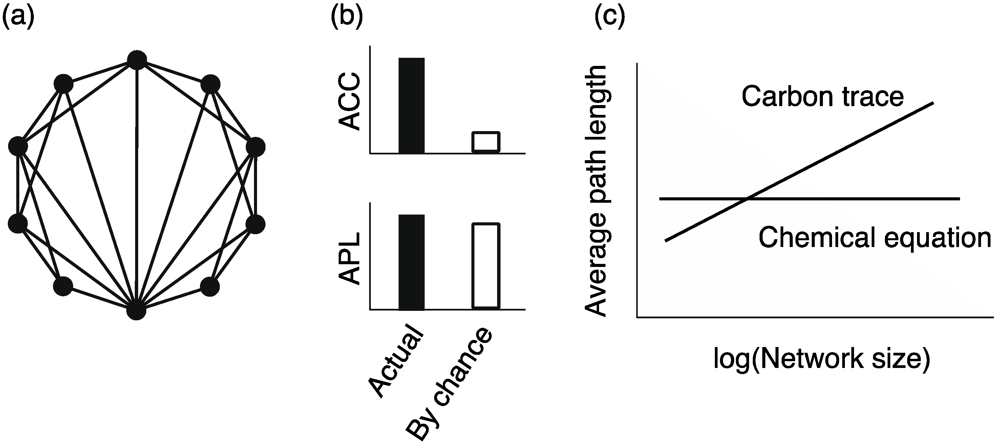
4.3. Preferential Attachment Mechanism
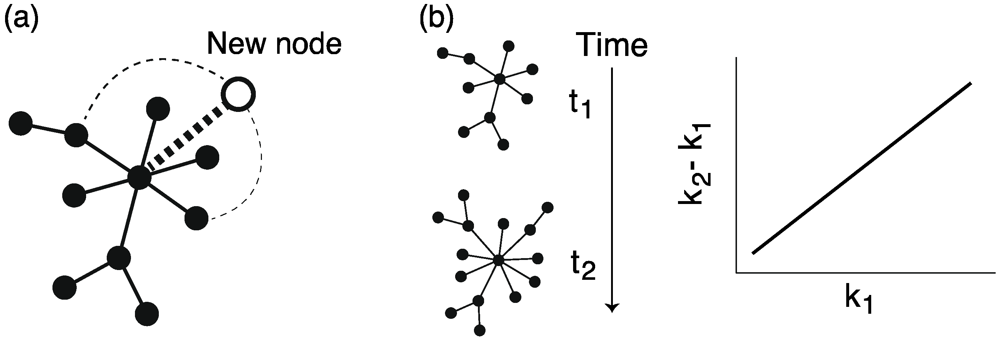
4.4. Network Modularity

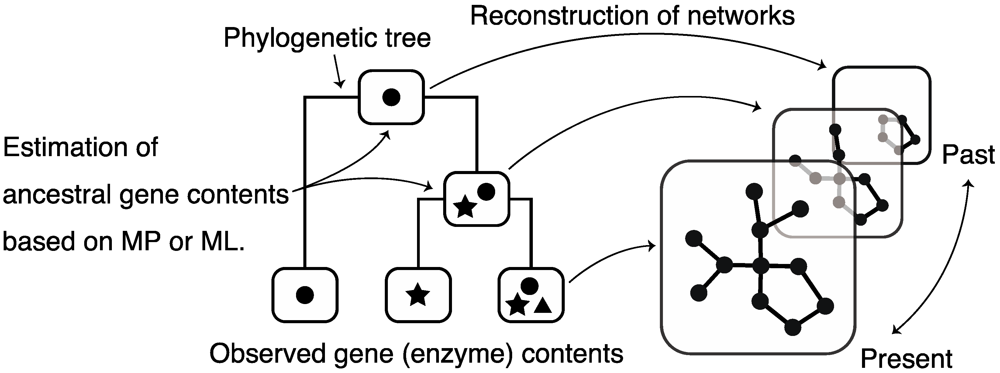
5. Reconstruction of Ancestral Metabolic Networks
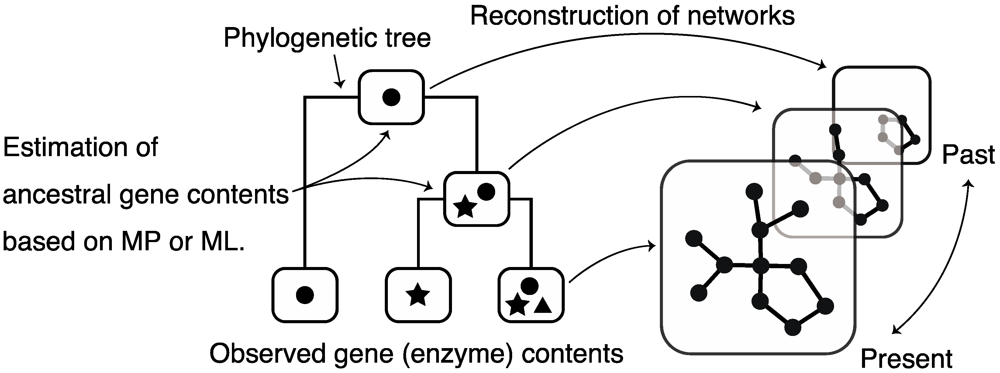
6. Measuring Metabolic Network Robustness
7. Mathematical Models for Metabolic Network Formation
8. Metabolite Distribution across Species
9. Metabolism in Ecosystems
10. Large-Scale Mutational Analyses and Laboratory Evolution Experiments
11. Conclusions
Acknowledgments
Conflict of Interest
References
- Kanehisa, M.; Goto, S.; Sato, Y.; Furumichi, M.; Tanabe, M. KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 2011, 40, 109–114. [Google Scholar]
- Caspi, R.; Altman, T.; Dreher, K.; Fulcher, C.A.; Subhraveti, P.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Latendresse, M.; Mueller, L.A.; Ong, Q.; Paley, S.; Pujar, A.; Shearer, A.G.; Travers, M.; Weerasinghe, D.; Zhang, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2011, 40, 742–753. [Google Scholar]
- Chassagnole, C.; Noisommit-Rizzi, N.; Schmid, J.W.; Mauch, K.; Reuss, M. Dynamic modeling of the central carbon metabolism of Escherichia coli. Biotechnol. Bioeng. 2002, 79, 53–73. [Google Scholar] [CrossRef]
- Lewis, N.E.; Nagarajan, H.; Palsson, B.O. Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 2012, 10, 291–305. [Google Scholar]
- Jeong, H.; Tombor, B.; Albert, R.; Oltvai, Z.N.; Barabási, A.-L. The large-scale organization of metabolic networks. Nature 2000, 407, 651–654. [Google Scholar]
- Ravasz, E.; Somera, A.L.; Mongru, D.A.; Oltvai, Z.N.; Barabási, A.-L. Hierarchical organization of modularity in metabolic networks. Science 2002, 297, 1551–1555. [Google Scholar]
- Barabási, A.-L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef]
- Lazcano, A.; Miller, S.L. On the origin of metabolic pathways. J. Mol. Evol. 1999, 49, 424–431. [Google Scholar] [CrossRef]
- Rison, S.C.G.; Thornton, J.M. Pathway evolution, structurally speaking. Curr. Opin. Struct. Biol. 2002, 12, 374–382. [Google Scholar] [CrossRef]
- Fani, R.; Fondi, M. Origin and evolution of metabolic pathways. Phys. Life Rev. 2009, 6, 23–52. [Google Scholar] [CrossRef]
- Fondi, M.; Emiliani, G.; Fani, R. Origin and evolution of operons and metabolic pathways. Res. Microbiol. 2009, 160, 502–512. [Google Scholar] [CrossRef]
- Yamada, T.; Bork, P. Evolution of biomolecular networks: lessons from metabolic and protein interactions. Nat. Rev. Mol. Cell Biol. 2009, 10, 791–803. [Google Scholar] [CrossRef]
- Schmidt, S.; Sunyaev, S.; Bork, P.; Dandekar, T. Metabolites: a helping hand for pathway evolution? Trends Biochem. Sci. 2003, 28, 336–341. [Google Scholar] [CrossRef]
- Papp, B.; Teusink, B.; Notebaart, R.A. A critical view of metabolic network adaptations. HFSP J. 2009, 3, 24–35. [Google Scholar] [CrossRef]
- Nam, H.; Conrad, T.M.; Lewis, N.E. The role of cellular objectives and selective pressures in metabolic pathway evolution. Curr. Opin. Biotechnol. 2011, 22, 595–600. [Google Scholar] [CrossRef]
- Hickey, D.A.; Singer, G.A.C. Genomic and proteomic adaptations to growth at high temperature. Genome Biol. 2004, 5, 117. [Google Scholar] [CrossRef]
- Papp, B.; Notebaart, R.A.; Pál, C. Systems-biology approaches for predicting genomic evolution. Nat. Rev. Genet. 2011, 12, 591–602. [Google Scholar] [CrossRef]
- Conrad, T.M.; Lewis, N.E.; Palsson, B.O. Microbial laboratory evolution in the era of genome-scale science. Mol. Syst. Biol. 2011, 7, 509:1–509:11. [Google Scholar]
- Keating, S.M.; Bornstein, B.J.; Finney, A.; Hucka, M. SBMLToolbox: an SBML toolbox for MATLAB users. Bioinformatics 2006, 22, 1275–1277. [Google Scholar] [CrossRef]
- Demir, E.; Cary, M.P.; Paley, S.; Fukuda, K.; Lemer, C.; Vastrik, I.; Wu, G.; DEustachio, P.; Schaefer, C.; Luciano, J.; Schacherer, F.; Martinez-Flores, I.; Hu, Z.; Jimenez-Jacinto, V.; Joshi-Tope, G.; Kandasamy, K.; Lopez-Fuentes, A.C.; Mi, H.; Pichler, E.; Rodchenkov, I.; Splendiani, A.; Tkachev, S.; Zucker, J.; Gopinath, G.; Rajasimha, H.; Ramakrishnan, R.; Shah, I.; Syed, M.; Anwar, N.; Babur, O.; Blinov, M.; Brauner, E.; Corwin, D.; Donaldson, S.; Gibbons, F.; Goldberg, R.; Hornbeck, P.; Luna, A.; Murray-Rust, P.; Neumann, E.; Reubenacker, O.; Samwald, M.; van Iersel, M.; Wimalaratne, S.; Allen, K.; Braun, B.; Whirl-Carrillo, M.; Cheung, K.-H.; Dahlquist, K.; Finney, A.; Gillespie, M.; Glass, E.; Gong, L.; Haw, R.; Honig, M.; Hubaut, O.; Kane, D.; Krupa, S.; Kutmon, M.; Leonard, J.; Marks, D.; Merberg, D.; Petri, V.; Pico, A.; Ravenscroft, D.; Ren, L.; Shah, N.; Sunshine, M.; Tang, R.; Whaley, R.; Letovksy, S.; Buetow, K.H.; Rzhetsky, A.; Schachter, V.; Sobral, B.S.; Dogrusoz, U.; McWeeney, S.; Aladjem, M.; Birney, E.; Collado-Vides, J.; Goto, S.; Hucka, M.; Le Novere, N.; Maltsev, N.; Pandey, A.; Thomas, P.; Wingender, E.; Karp, P.D.; Sander, C.; Bader, G.D. The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 2010, 28, 935–942. [Google Scholar]
- Arita, M. The metabolic world of Escherichia coli is not small. Proc. Natl. Acad. Sci. USA 2004, 101, 1543–1547. [Google Scholar] [CrossRef]
- Wagner, A.; Fell, D.A. The small world inside large metabolic networks. Proc. R. Soc. B 2001, 268, 1803–1810. [Google Scholar] [CrossRef]
- Huss, M.; Holme, P. Currency and commodity metabolites: Their identification and relation to the modularity of metabolic networks. IET Syst. Biol. 2007, 1, 280–285. [Google Scholar] [CrossRef]
- Ma, H.; Zeng, A.-P. Reconstruction of metabolic networks from genome data and analysis of their global structure for various organisms. Bioinformatics 2003, 19, 270–277. [Google Scholar] [CrossRef]
- Arita, M. In silico atomic tracing by substrate-product relationships in Escherichia coli intermediary metabolism. Genome Res. 2003, 13, 2455–2466. [Google Scholar] [CrossRef]
- Kotera, M.; Okuno, Y.; Hattori, M.; Goto, S.; Kanehisa, M. Computational assignment of the EC numbers for genomic-scale analysis of enzymatic reactions. J. Am. Chem. Soc. 2004, 126, 16487–16498. [Google Scholar]
- Arita, M.; Suwa, K. Search extension transforms Wiki into a relational system: a case for flavonoid metabolite database. BioData Min. 2008, 1, 7:1–7:8. [Google Scholar]
- Arita, M. A pitfall of wiki solution for biological databases. Brief. Bioinform. 2009, 10, 295–296. [Google Scholar] [CrossRef]
- Díaz-Mejía, J.J.; Pérez-Rueda, E.; Segovia, L. A network perspective on the evolution of metabolism by gene duplication. Genome Biol. 2007, 8, R26:1–R26:10. [Google Scholar]
- Light, S.; Kraulis, P. Network analysis of metabolic enzyme evolution in Escherichia coli. BMC Bioinformatics 2004, 5, 15:1–15:16. [Google Scholar]
- Horowitz, N.H. On the evolution of biochemical syntheses. Proc. Natl. Acad. Sci. USA 1945, 31, 153–157. [Google Scholar] [CrossRef]
- Zhang, Y.; Thiele, I.; Weekes, D.; Li, Z.; Jaroszewski, L.; Ginalski, K.; Deacon, A.M.; Wooley, J.; Lesley, S.A.; Wilson, I.A.; Palsson, B.; Osterman, A.; Godzik, A. Three-dimensional structural view of the central metabolic network of Thermotoga maritima. Science 2009, 325, 1544–1549. [Google Scholar]
- Lazcano, A.; Miller, S.L. The origin and early evolution of life: prebiotic chemistry, the pre-RNA world, and time. Cell 1996, 85, 793–798. [Google Scholar] [CrossRef]
- Jensen, R.A. Enzyme recruitment in evolution of new function. Annu. Rev. Microbiol. 1976, 30, 409–425. [Google Scholar] [CrossRef]
- Wilmanns, M.; Hyde, C.C.; Davies, D.R.; Kirschner, K.; Jansonius, J.N. Structural conservation in parallel beta/alpha-barrel enzymes that catalyze three sequential reactions in the pathway of tryptophan biosynthesis. Biochemistry 1991, 30, 9161–9169. [Google Scholar]
- Copley, R.R.; Bork, P. Homology among (βα)(8) barrels: Implications for the evolution of metabolic pathways. J. Mol. Biol. 2000, 303, 627–641. [Google Scholar] [CrossRef]
- Pál, C.; Papp, B.; Lercher, M.J. Adaptive evolution of bacterial metabolic networks by horizontal gene transfer. Nat. Genet. 2005, 37, 1372–1375. [Google Scholar]
- Rausher, M.D.; Miller, R.E.; Tiffin, P. Patterns of evolutionary rate variation among genes of the anthocyanin biosynthetic pathway. Mol. Biol. Evol. 1999, 16, 266–274. [Google Scholar] [CrossRef]
- Ramsay, H.; Rieseberg, L.H.; Ritland, K. The correlation of evolutionary rate with pathway position in plant terpenoid biosynthesis. Mol. Biol. Evol. 2009, 26, 1045–1053. [Google Scholar] [CrossRef]
- Vitkup, D.; Kharchenko, P.; Wagner, A. Influence of metabolic network structure and function on enzyme evolution. Genome Biol. 2006, 7, R39:1–R39:9. [Google Scholar]
- Freeman, L.C. Centrality in social networks conceptual clarification. Soc. Networks 1978, 1, 215–239. [Google Scholar] [CrossRef]
- Holme, P. Model validation of simple-graph representations of metabolism. J. Roy. Soc. Interface 2009, 6, 1027–1034. [Google Scholar] [CrossRef]
- Zhu, Q.; Qin, T.; Jiang, Y.-Y.; Ji, C.; Kong, D.-X.; Ma, B.-G.; Zhang, H.-Y. Chemical basis of metabolic network organization. PLoS Comput. Biol. 2011, 7, e1002214:1–e1002214:8. [Google Scholar]
- Bar-Even, A.; Noor, E.; Flamholz, A.; Buescher, J.M.; Milo, R. Hydrophobicity and charge shape cellular metabolite concentrations. PLoS Comput. Biol. 2011, 7, e1002166:1–e1002166:7. [Google Scholar]
- Jiang, Y.-Y.; Kong, D.-X.; Qin, T.; Li, X.; Caetano-Anolles, G.; Zhang, H.-Y. The impact of oxygen on metabolic evolution: a chemoinformatic investigation. PLoS Comput. Biol. 2012, 8, e1002426:1–e1002426:8. [Google Scholar]
- Raymond, J.; Segré, D. The effect of oxygen on biochemical networks and the evolution of complex life. Science 2006, 311, 1764–1767. [Google Scholar] [CrossRef]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular chaperones and protein quality control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef]
- Kerner, M.J.; Naylor, D.J.; Ishihama, Y.; Maier, T.; Chang, H.-C.; Stines, A.P.; Georgopoulos, C.; Frishman, D.; Hayer-Hartl, M.; Mann, M.; Hartl, F.U. Proteome-wide analysis of chaperonin-dependent protein folding in Escherichia coli. Cell 2005, 122, 209–220. [Google Scholar] [CrossRef]
- Fujiwara, K.; Ishihama, Y.; Nakahigashi, K.; Soga, T.; Taguchi, H. A systematic survey of in vivo obligate chaperonin-dependent substrates. EMBO J. 2010, 29, 1552–1564. [Google Scholar] [CrossRef]
- Takemoto, K.; Niwa, T.; Taguchi, H. Difference in the distribution pattern of substrate enzymes in the metabolic network of Escherichia coli, according to chaperonin requirement. BMC Syst. Biol. 2011, 5, 98. [Google Scholar] [CrossRef]
- Rutherford, S.L.; Lindquist, S. Hsp90 as a capacitor for morphological evolution. Nature 1998, 396, 336–342. [Google Scholar] [CrossRef]
- Queitsch, C.; Sangster, T.A.; Lindquist, S. Hsp90 as a capacitor of phenotypic variation. Nature 2002, 417, 618–624. [Google Scholar] [CrossRef]
- Tokuriki, N.; Tawfik, D.S. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature 2009, 459, 668–673. [Google Scholar] [CrossRef]
- Bogumil, D.; Dagan, T. Chaperonin-dependent accelerated substitution rates in prokaryotes. Genome Biol. Evol. 2010, 2, 602–608. [Google Scholar] [CrossRef]
- Williams, T.A.; Fares, M.A. The effect of chaperonin buffering on protein evolution. Genome Biol. Evol. 2010, 2, 609–619. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Albert, R. Emergence of scaling in random networks. Science 1999, 286, 509–512. [Google Scholar] [CrossRef]
- Albert, R. Scale-free networks in cell biology. J. Cell Sci. 2005, 118, 4947–4957. [Google Scholar] [CrossRef]
- Albert, R.; Barabási, A.-L. Statistical mechanics of complex networks. Rev. Mod. Phys. 2002, 74, 47–97. [Google Scholar] [CrossRef]
- Song, C.; Havlin, S.; Makse, H.A. Self-similarity of complex networks. Nature 2005, 433, 392–395. [Google Scholar] [CrossRef]
- Arita, M. Scale-freeness and biological networks. J. Biochem. 2005, 138, 1–4. [Google Scholar] [CrossRef]
- Li, L.; Alderson, D.; Doyle, J.C.; Willinger, W. Towards a theory of scale-free graphs: Definition, properties, and implication. Internet Mathematics 2005, 2, 431–523. [Google Scholar] [CrossRef]
- Tanaka, R. Scale-rich metabolic networks. Phys. Rev. Lett. 2005, 94, 168101:1–168101:4. [Google Scholar]
- Watts, D.J.; Strogatz, S.H. Collective dynamics of “small-world” networks. Nature 1998, 393, 440–442. [Google Scholar] [CrossRef]
- Light, S.; Kraulis, P.; Elofsson, A. Preferential attachment in the evolution of metabolic networks. BMC Genomics 2005, 6, 159:1–159:11. [Google Scholar]
- Pastor-Satorras, R.; Smith, E.; Solé, R.V. Evolving protein interaction networks through gene duplication. J. Theor. Biol. 2003, 222, 199–210. [Google Scholar] [CrossRef]
- Qian, J.; Luscombe, N.M.; Gerstein, M. Protein family and fold occurrence in genomes: power-law behaviour and evolutionary model. J. Mol. Biol. 2001, 313, 673–681. [Google Scholar] [CrossRef]
- Eisenberg, E.; Levanon, E. Preferential attachment in the protein network evolution. Phys. Rev. Lett. 2003, 91, 1–4. [Google Scholar]
- Teichmann, S.A.; Babu, M.M. Gene regulatory network growth by duplication. Nat. Genet. 2004, 36, 492–496. [Google Scholar] [CrossRef]
- Pfeiffer, T.; Soyer, O.S.; Bonhoeffer, S. The evolution of connectivity in metabolic networks. PLoS Biol. 2005, 3, e228:1–e228:8. [Google Scholar]
- Hartwell, L.H.; Hopfield, J.J.; Leibler, S.; Murray, A.W. From molecular to modular cell biology. Nature 1999, 402, C47–C52. [Google Scholar] [CrossRef]
- Fortunato, S. Community detection in graphs. Phys. Rep. 2010, 486, 75–174. [Google Scholar] [CrossRef]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef]
- Parter, M.; Kashtan, N.; Alon, U. Environmental variability and modularity of bacterial metabolic networks. BMC Evol. Biol. 2007, 7, 169:1–169:8. [Google Scholar]
- Kashtan, N.; Alon, U. Spontaneous evolution of modularity and network motifs. Proc. Natl. Acad. Sci. USA 2005, 102, 13773–13778. [Google Scholar] [CrossRef]
- Takemoto, K.; Borjigin, S. Metabolic network modularity in archaea depends on growth conditions. PLoS ONE 2011, 6, e25874:1–e25874:6. [Google Scholar]
- Takemoto, K.; Nacher, J.C.; Akutsu, T. Correlation between structure and temperature in prokaryotic metabolic networks. BMC Bioinformatics 2007, 8, 303:1–303:11. [Google Scholar]
- Takemoto, K.; Akutsu, T. Origin of structural difference in metabolic networks with respect to temperature. BMC Syst. Biol. 2008, 2, 82:1–82:13. [Google Scholar]
- Wang, H.-C.; Hickey, D.A. Evidence for strong selective constraint acting on the nucleotide composition of 16S ribosomal RNA genes. Nucleic Acids Res. 2002, 30, 2501–2507. [Google Scholar] [CrossRef]
- Friedman, R.; Drake, J.W.; Hughes, A.L. Genome-wide patterns of nucleotide substitution reveal stringent functional constraints on the protein sequences of thermophiles. Genetics 2004, 167, 1507–1512. [Google Scholar] [CrossRef]
- Mirkin, B.G.; Fenner, T.I.; Galperin, M.Y.; Koonin, E.V. Algorithms for computing parsimonious evolutionary scenarios for genome evolution, the last universal common ancestor and dominance of horizontal gene transfer in the evolution of prokaryotes. BMC Evol. Biol. 2003, 3, 2:1–2:34. [Google Scholar]
- Csűrös, M.; Miklós, I. A probabilistic model for gene content evolution with duplication, loss, and horizontal transfer. Lecture Notes Comp. Sci. 2006, 3909, 206–220. [Google Scholar]
- Iwasaki, W.; Takagi, T. Reconstruction of highly heterogeneous gene-content evolution across the three domains of life. Bioinformatics 2007, 23, i230–i239. [Google Scholar] [CrossRef]
- Iwasaki, W.; Takagi, T. Rapid pathway evolution facilitated by horizontal gene transfers across prokaryotic lineages. PLoS Genet. 2009, 5, e1000402:1–e1000402:8. [Google Scholar]
- Bak, P.; Sneppen, K. Punctuated equilibrium and criticality in a simple model of evolution. Phys. Rev. Lett. 1993, 71, 4083–4086. [Google Scholar] [CrossRef]
- Kreimer, A.; Borenstein, E.; Gophna, U.; Ruppin, E. The evolution of modularity in bacterial metabolic networks. Proc. Natl. Acad. Sci. USA 2008, 105, 6976–6981. [Google Scholar]
- Caetano-Anollés, G.; Kim, H.S.; Mittenthal, J.E. The origin of modern metabolic networks inferred from phylogenomic analysis of protein architecture. Proc. Natl. Acad. Sci. USA 2007, 104, 9358–9363. [Google Scholar]
- Tanaka, M.; Yamada, T.; Itoh, M.; Okuda, S.; Goto, S.; Kanehisa, M. Analysis of the differences in metabolic network expansion between prokaryotes and eukaryotes. Genome Inform. 2006, 17, 230–239. [Google Scholar]
- Kitano, H. Biological robustness. Nat. Rev. Genet. 2004, 5, 826–837. [Google Scholar] [CrossRef]
- Burgard, A.P.; Pharkya, P.; Maranas, C.D. Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol. Bioeng. 2003, 84, 647–657. [Google Scholar] [CrossRef]
- Behre, J.; Wilhelm, T.; von Kamp, A.; Ruppin, E.; Schuster, S. Structural robustness of metabolic networks with respect to multiple knockouts. J. Theor. Biol. 2008, 252, 433–441. [Google Scholar] [CrossRef]
- Handorf, T.; Ebenhöh, O.; Heinrich, R. Expanding metabolic networks: scopes of compounds, robustness, and evolution. J. Mol. Evol. 2005, 61, 498–512. [Google Scholar] [CrossRef]
- Smart, A.G.; Amaral, L.A.N.; Ottino, J.M. Cascading failure and robustness in metabolic networks. Proc. Natl. Acad. Sci. USA 2008, 105, 13223–13228. [Google Scholar]
- Jiang, D.; Zhou, S.; Chen, Y.-P.P. Compensatory ability to null mutation in metabolic networks. Biotechnol. Bioeng. 2009, 103, 361–369. [Google Scholar] [CrossRef]
- Takemoto, K.; Tamura, T.; Cong, Y.; Ching, W.-K.; Vert, J.-P.; Akutsu, T. Analysis of the impact degree distribution in metabolic networks using branching process approximation. Physica A 2012, 391, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Maslov, S.; Krishna, S.; Pang, T.Y.; Sneppen, K. Toolbox model of evolution of prokaryotic metabolic networks and their regulation. Proc. Natl. Acad. Sci. USA 2009, 106, 9743–9748. [Google Scholar]
- Pang, T.Y.; Maslov, S. A toolbox model of evolution of metabolic pathways on networks of arbitrary topology. PLoS Comput. Biol. 2011, 7, e1001137:1–e1001137:11. [Google Scholar]
- Takemoto, K.; Arita, M. Heterogeneous distribution of metabolites across plant species. Physica A 2009, 388, 2771–2780. [Google Scholar] [CrossRef]
- Takemoto, K. Global architecture of metabolite distributions across species and its formation mechanisms. Biosystems 2010, 100, 8–13. [Google Scholar] [CrossRef]
- Klitgord, N.; Segrè, D. Ecosystems biology of microbial metabolism. Curr. Opin. Biotechnol. 2011, 22, 541–546. [Google Scholar] [CrossRef]
- Christian, N.; Handorf, T.; Ebenhöh, O. Metabolic synergy: Increasing biosynthetic capabilities by network cooperation. Genome Inform. 2007, 18, 320–329. [Google Scholar]
- Borenstein, E.; Feldman, M.W. Topological signatures of species interactions in metabolic networks. J. Comput. Biol. 2009, 16, 191–200. [Google Scholar] [CrossRef]
- Borenstein, E.; Kupiec, M.; Feldman, M.W.; Ruppin, E. Large-scale reconstruction and phylogenetic analysis of metabolic environments. Proc. Natl. Acad. Sci. USA 2008, 105, 14482–14487. [Google Scholar]
- Freilich, S.; Zarecki, R.; Eilam, O.; Segal, E.S.; Henry, C.S.; Kupiec, M.; Gophna, U.; Sharan, R.; Ruppin, E. Competitive and cooperative metabolic interactions in bacterial communities. Nat. Commun. 2011, 2, 589:1–589:7. [Google Scholar]
- Ibarra, R.U.; Edwards, J.S.; Palsson, B.O. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 2002, 420, 186–189. [Google Scholar] [CrossRef]
- Fong, S.S.; Palsson, B.O. Metabolic gene-deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat. Genet. 2004, 36, 1056–1058. [Google Scholar] [CrossRef]
- Lewis, N.E.; Hixson, K.K.; Conrad, T.M.; Lerman, J.A.; Charusanti, P.; Polpitiya, A.D.; Adkins, J.N.; Schramm, G.; Purvine, S.O.; Lopez-Ferrer, D.; Weitz, K.K.; Eils, R.; König, R.; Smith, R.D.; Palsson, B.O. Omic data from evolved E. coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 2010, 6, 390:1–390:13. [Google Scholar]
- Kishimoto, T.; Iijima, L.; Tatsumi, M.; Ono, N.; Oyake, A.; Hashimoto, T.; Matsuo, M.; Okubo, M.; Suzuki, S.; Mori, K.; Kashiwagi, A.; Furusawa, C.; Ying, B.-W.; Yomo, T. Transition from positive to neutral in mutation fixation along with continuing rising fitness in thermal adaptive evolution. PLoS Genet. 2010, 6, e1001164:1–e1001164:10. [Google Scholar]
- Horinouchi, T.; Tamaoka, K.; Furusawa, C.; Ono, N.; Suzuki, S.; Hirasawa, T.; Yomo, T.; Shimizu, H. Transcriptome analysis of parallel-evolved Escherichia coli strains under ethanol stress. BMC Genomics 2010, 11, 579:1–579:11. [Google Scholar]
- Lü, L.; Zhou, T. Link prediction in complex networks: A survey. Physica A 2011, 390, 1150–1170. [Google Scholar] [CrossRef]
- Khersonsky, O.; Tawfik, D.S. Enzyme promiscuity: a mechanistic and evolutionary perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [CrossRef]
- Patrick, W.M.; Quandt, E.M.; Swartzlander, D.B.; Matsumura, I. Multicopy suppression underpins metabolic evolvability. Mol. Biol. Evol. 2007, 24, 2716–2722. [Google Scholar] [CrossRef]
- Copley, S.D. Toward a systems biology perspective on enzyme evolution. J. Biol. Chem. 2012, 287, 3–10. [Google Scholar] [CrossRef]
- Carbonell, P.; Lecointre, G.; Faulon, J.-L. Origins of specificity and promiscuity in metabolic networks. J. Bio. Chem. 2011, 286, 43994–44004. [Google Scholar]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; Mende, D.R.; Li, J.; Xu, J.; Li, S.; Li, D.; Cao, J.; Wang, B.; Liang, H.; Zheng, H.; Xie, Y.; Tap, J.; Lepage, P.; Bertalan, M.; Batto, J.-M.; Hansen, T.; Le Paslier, D.; Linneberg, A.; Nielsen, H.B.; Pelletier, E.; Renault, P.; Sicheritz-Ponten, T.; Turner, K.; Zhu, H.; Yu, C.; Li, S.; Jian, M.; Zhou, Y.; Li, Y.; Zhang, X.; Li, S.; Qin, N.; Yang, H.; Wang, J.; Brunak, S.; Doré, J.; Guarner, F.; Kristiansen, K.; Pedersen, O.; Parkhill, J.; Weissenbach, J.; Bork, P.; Ehrlich, S.D.; Wang, J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [Green Version]
- Daniel, R. The metagenomics of soil. Nat. Rev. Microbiol. 2005, 3, 470–478. [Google Scholar] [CrossRef]
- Kennedy, J.; Marchesi, J.R.; Dobson, A.D. Marine metagenomics: strategies for the discovery of novel enzymes with biotechnological applications from marine environments. Microb. Cell Fact. 2008, 7, 27:1–27:8. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Takemoto, K. Current Understanding of the Formation and Adaptation of Metabolic Systems Based on Network Theory. Metabolites 2012, 2, 429-457. https://doi.org/10.3390/metabo2030429
Takemoto K. Current Understanding of the Formation and Adaptation of Metabolic Systems Based on Network Theory. Metabolites. 2012; 2(3):429-457. https://doi.org/10.3390/metabo2030429
Chicago/Turabian StyleTakemoto, Kazuhiro. 2012. "Current Understanding of the Formation and Adaptation of Metabolic Systems Based on Network Theory" Metabolites 2, no. 3: 429-457. https://doi.org/10.3390/metabo2030429