Potential Metabolomic Linkage in Blood between Parkinson’s Disease and Traumatic Brain Injury

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Study Population Differences
2.2. Subacute mTBI Plasma Metabolomic Biomarkers–MetaboAnalyst 4.0 Method
2.3. Subacute Plasma mTBI Metabolomic Biomarkers–mixOmics, sPLS-DA Method
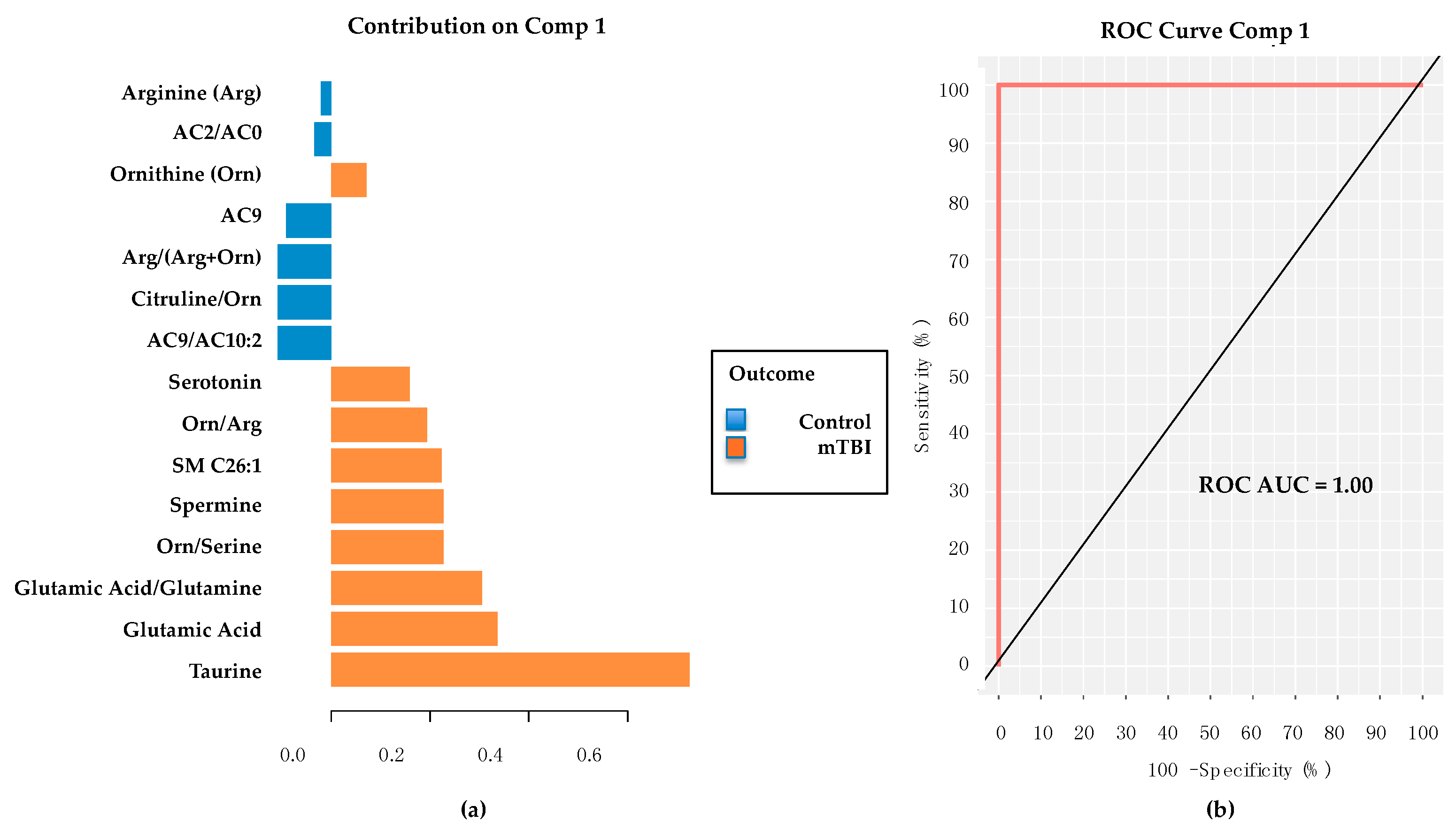
2.4. Subacute mTBI Plasma Metabolomic Biomarkers–Targeted Analysis via mixOmics
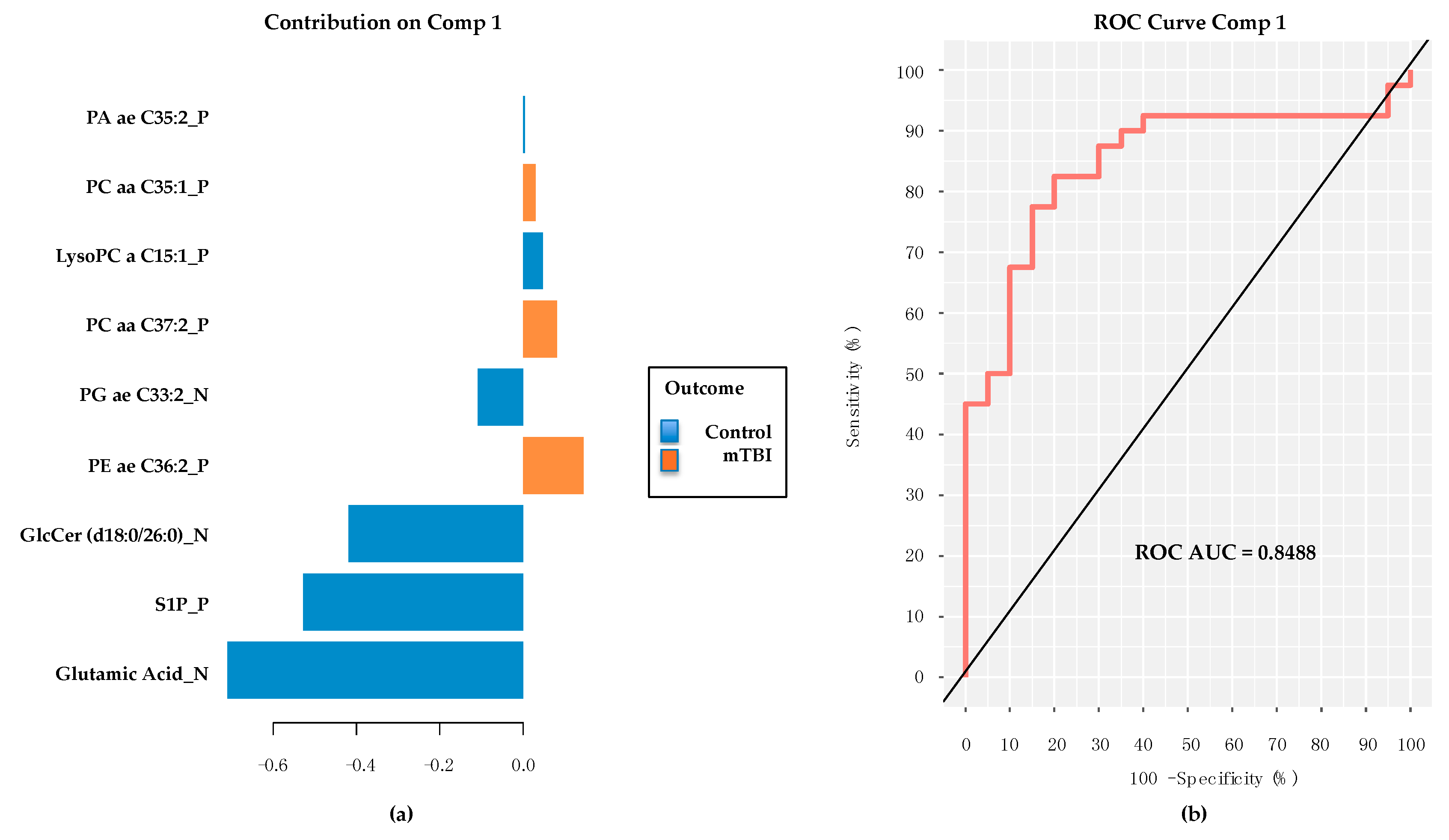
2.5. PD/PDD Serum Metabolomic Biomarkers–Utilizing the mixOmics-Derived sPLS-DA Top 20 Metabolites from Subacute mTBI Analysis
2.6. PD/PDD Serum Metabolomic Biomarkers–New Discovery Using mixOmics sPLS-DA
2.7. Evaluation of Glutamic Acid’s Performance as Sole Metabolite in mixOmics PLS-DA Classifier Models for Subacute mTBI and PD Cohorts
3. Discussion
4. Materials and Methods
4.1. Study Populations
4.2. Metabolomic Analyses and Data
4.3. Metabolomic Biomarker Development
4.4. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jafari, S.; Etminan, M.; Aminzadeh, F.; Samii, A. Head injury and risk of parkinson disease: A systematic review and meta-analysis. Mov. Disord. 2013, 28, 1222–1229. [Google Scholar] [CrossRef] [PubMed]
- Godbolt, A.K.; Cancelliere, C.; Hincapie, C.A.; Marras, C.; Boyle, E.; Kristman, V.L.; Coronado, V.G.; Cassidy, J.D. Systematic review of the risk of dementia and chronic cognitive impairment after mild traumatic brain injury: Results of the international collaboration on mild traumatic brain injury prognosis. Arch. Phys. Med. Rehabil. 2014, 95, S245–S256. [Google Scholar] [CrossRef] [PubMed]
- Kristman, V.L.; Borg, J.; Godbolt, A.K.; Salmi, L.R.; Cancelliere, C.; Carroll, L.J.; Holm, L.W.; Nygren-de Boussard, C.; Hartvigsen, J.; Abara, U.; et al. Methodological issues and research recommendations for prognosis after mild traumatic brain injury: Results of the international collaboration on mild traumatic brain injury prognosis. Arch. Phys. Med. Rehabil. 2014, 95, S265–S277. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Hincapie, C.A.; Kristman, V.L.; Cancelliere, C.; Soklaridis, S.; Li, A.; Borg, J.; af Geijerstam, J.-L.; Cassidy, J.D. Systematic review of the risk of parkinson’s disease after mild traumatic brain injury: Results of the international collaboration on mild traumatic brain injury prognosis. Arch. Phys. Med. Rehabil. 2014, 95, S238–S244. [Google Scholar] [CrossRef] [PubMed]
- Wirdefeldt, K.; Adami, H.O.; Cole, P.; Trichopoulos, D.; Mandel, J. Epidemiology and etiology of parkinson’s disease: A review of the evidence. Eur. J. Epidemiol. 2011, 26 (Suppl. 1), S1–S58. [Google Scholar] [CrossRef]
- Prince, C.; Bruhns, M.E. Evaluation and treatment of mild traumatic brain injury: The role of neuropsychology. Brain Sci. 2017, 7, 105. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Liu, R.; Zhao, E.; Huang, X.; Nalls, M.A.; Singleton, A.B.; Chen, H. Head injury, potential interaction with genes, and risk for parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.C.; Burke, J.F.; Nettiksimmons, J.; Goldman, S.; Tanner, C.M.; Yaffe, K. Traumatic brain injury in later life increases risk for parkinson disease. Ann. Neurol. 2015, 77, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.C.; Yaffe, K. Epidemiology of mild traumatic brain injury and neurodegenerative disease. Mol. Cell Neurosci. 2015, 66, 75–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.C.; Hazrati, L.N. Parkinson’s disease, parkinsonism, and traumatic brain injury. Crit. Rev. Clin. Lab. Sci. 2013, 50, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Loane, D.J. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics 2015, 12, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Mapstone, M.; Mahmoodi, A.; Gross, T.; Macciardi, F.; Cheema, A.K.; Merchant-Borna, K.; Bazarian, J.; Federoff, H.J. Plasma metabolomic biomarkers accurately classify acute mild traumatic brain injury from controls. PLoS ONE 2018, 13, e0195318. [Google Scholar] [CrossRef] [PubMed]
- Shutter, L.; Tong, K.A.; Holshouser, B.A. Proton mrs in acute traumatic brain injury: Role for glutamate/glutamine and choline for outcome prediction. J. Neurotrauma 2004, 21, 1693–1705. [Google Scholar] [CrossRef] [PubMed]
- Ashwal, S.; Holshouser, B.; Tong, K.; Serna, T.; Osterdock, R.; Gross, M.; Kido, D. Proton mr spectroscopy detected glutamate/glutamine is increased in children with traumatic brain injury. J. Neurotrauma 2004, 21, 1539–1552. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.; Takahashi, K.; Azuma, J. Role of osmoregulation in the actions of taurine. Amino Acids 2000, 19, 527–546. [Google Scholar] [CrossRef] [PubMed]
- El Idrissi, A. Taurine increases mitochondrial buffering of calcium: Role in neuroprotection. Amino Acids 2008, 34, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.A.; Dawson, R., Jr. Attenuation of oxidative damage to DNA by taurine and taurine analogs. Adv. Exp. Med. Biol. 2000, 483, 355–367. [Google Scholar] [PubMed]
- Huxtable, R.J. Taurine in the central nervous system and the mammalian actions of taurine. Prog. Neurobiol. 1989, 32, 471–533. [Google Scholar] [CrossRef]
- Hawkins, R.A. The blood-brain barrier and glutamate. Am. J. Clin. Nutr. 2009, 90, 867S–874S. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, S.; Nakajima, Y.; Masu, M.; Ueda, Y.; Nakahara, K.; Watanabe, D.; Yamaguchi, S.; Kawabata, S.; Okada, M. Glutamate receptors: Brain function and signal transduction. Brain Res. Rev. 1998, 26, 230–235. [Google Scholar] [CrossRef]
- Yudkoff, M. Interactions in the metabolism of glutamate and the branched-chain amino acids and ketoacids in the CNS. Neurochem. Res. 2017, 42, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, R.J.; Ryan, R.M. Mechanisms of glutamate transport. Physiol. Rev. 2013, 93, 1621–1657. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, M.; Wang, Y.; Teichberg, V.I. Blood-mediated scavenging of cerebrospinal fluid glutamate. J. Neurochem. 2003, 87, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helms, H.C.; Madelung, R.; Waagepetersen, H.S.; Nielsen, C.U.; Brodin, B. In vitro evidence for the brain glutamate efflux hypothesis: Brain endothelial cells cocultured with astrocytes display a polarized brain-to-blood transport of glutamate. Glia 2012, 60, 882–893. [Google Scholar] [CrossRef] [PubMed]
- al-Sarraf, H.; Preston, J.E.; Segal, M.B. Changes in the kinetics of the acidic amino acid brain and csf uptake during development in the rat. Dev. Brain Res. 1997, 102, 127–134. [Google Scholar] [CrossRef]
- al-Sarraf, H.; Preston, J.E.; Segal, M.B. Acidic amino acid accumulation by rat choroid plexus during development. Dev. Brain Res. 1997, 102, 47–52. [Google Scholar] [CrossRef]
- Teichberg, V.I.; Cohen-Kashi-Malina, K.; Cooper, I.; Zlotnik, A. Homeostasis of glutamate in brain fluids: An accelerated brain-to-blood efflux of excess glutamate is produced by blood glutamate scavenging and offers protection from neuropathologies. Neuroscience 2009, 158, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.H.; Hazell, A.S. Excitotoxic mechanisms and the role of astrocytic glutamate transporters in traumatic brain injury. Neurochem. Int. 2006, 48, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and nmda receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Zauner, A.; Bullock, R. The role of excitatory amino acids in severe brain trauma: Opportunities for therapy: A review. J. Neurotrauma 1995, 12, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Ho, O.L.; Rhee, V. Cytotoxic effects of acidic and sulphur containing amino acids on the infant mouse central nervous system. Exp. Brain Res. 1971, 14, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, H.S.; Donaldson, D.; Dempsey, R.J.; Prasad, M.R. Regional levels of free fatty acids and evans blue extravasation after experimental brain injury. J. Neurotrauma 1994, 11, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Pilitsis, J.G.; Coplin, W.M.; O’Regan, M.H.; Wellwood, J.M.; Diaz, F.G.; Fairfax, M.R.; Michael, D.B.; Phillis, J.W. Free fatty acids in cerebrospinal fluids from patients with traumatic brain injury. Neurosci. Lett. 2003, 349, 136–138. [Google Scholar] [CrossRef]
- Scheff, S.W.; Dhillon, H.S. Creatine-enhanced diet alters levels of lactate and free fatty acids after experimental brain injury. Neurochem. Res. 2004, 29, 469–479. [Google Scholar] [CrossRef] [PubMed]
- Contreras, M.A.; Chang, M.C.; Kirkby, D.; Bell, J.M.; Rapoport, S.I. Reduced palmitate turnover in brain phospholipids of pentobarbital-anesthetized rats. Neurochem. Res. 1999, 24, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Newkirk, J.D.; Waite, M. Identification of a phospholipase a1 in plasma membranes of rat liver. Biochim. Biophys. Acta 1971, 225, 224–233. [Google Scholar] [CrossRef]
- Su, X.; Chu, Y.; Kordower, J.H.; Li, B.; Cao, H.; Huang, L.; Nishida, M.; Song, L.; Wang, D.; Federoff, H.J. Pgc-1α promoter methylation in parkinson’s disease. PLoS ONE 2015, 10, e0134087. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. Pgc-1α, a potential therapeutic target for early intervention in parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [PubMed]
- Sheng, B.; Wang, X.; Su, B.; Lee, H.G.; Casadesus, G.; Perry, G.; Zhu, X. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in alzheimer’s disease. J. Neurochem. 2012, 120, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, R.; Tyagi, E.; Vergnes, L.; Reue, K.; Gomez-Pinilla, F. Coupling energy homeostasis with a mechanism to support plasticity in brain trauma. Biochim. Biophys. Acta 2014, 1842, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Y.; Jankovic, J.; Wu, Y.C. Epigenetic mechanisms in parkinson’s disease. J. Neurol. Sci. 2015, 349, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Haapasalo, A.; Kauppinen, A.; Kaarniranta, K.; Soininen, H.; Hiltunen, M. Impaired mitochondrial energy metabolism in alzheimer’s disease: Impact on pathogenesis via disturbed epigenetic regulation of chromatin landscape. Prog. Neurobiol. 2015, 131, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Benveniste, H.; Drejer, J.; Schousboe, A.; Diemer, N.H. Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J. Neurochem. 1984, 43, 1369–1374. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Olney, J.W. Glutamate and the pathophysiology of hypoxic-ischemic brain damage. Ann. Neurol. 1986, 19, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Blandini, F.; Greenamyre, J.T.; Nappi, G. The role of glutamate in the pathophysiology of parkinson’s disease. Funct. Neurol. 1996, 11, 3–15. [Google Scholar] [PubMed]
- Erecinska, M.; Dagani, F. Relationships between the neuronal sodium/potassium pump and energy metabolism. Effects of k+, na+, and adenosine triphosphate in isolated brain synaptosomes. J. Gen. Physiol. 1990, 95, 591–616. [Google Scholar] [CrossRef] [PubMed]
- Albin, R.L.; Greenamyre, J.T. Alternative excitotoxic hypotheses. Neurology 1992, 42, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Beal, M.F.; Hyman, B.T.; Koroshetz, W. Do defects in mitochondrial energy-metabolism underlie the pathology of neurodegenerative diseases. Trends Neurosci. 1993, 16, 125–131. [Google Scholar] [CrossRef]
- Blandini, F.; Porter, R.H.; Greenamyre, J.T. Glutamate and parkinson’s disease. Mol. Neurobiol. 1996, 12, 73–94. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.G.; Greenamyre, J.T. Exacerbation of nmda, ampa, and l-glutamate excitotoxicity by the succinate dehydrogenase inhibitor malonate. J. Neurochem. 1995, 64, 2332–2338. [Google Scholar] [CrossRef] [PubMed]
- Havelund, J.F.; Heegaard, N.H.H.; Faergeman, N.J.K.; Gramsbergen, J.B. Biomarker research in parkinson’s disease using metabolite profiling. Metabolites 2017, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Lei, S.; Zavala-Flores, L.; Garcia-Garcia, A.; Nandakumar, R.; Huang, Y.; Madayiputhiya, N.; Stanton, R.C.; Dodds, E.D.; Powers, R.; Franco, R. Alterations in energy/redox metabolism induced by mitochondrial and environmental toxins: A specific role for glucose-6-phosphate-dehydrogenase and the pentose phosphate pathway in paraquat toxicity. ACS Chem. Biol. 2014, 9, 2032–2048. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.S.; Santosh, W.; Kumar, S.; Christlet, H.T. Metabolic profiling of parkinson’s disease: Evidence of biomarker from gene expression analysis and rapid neural network detection. J. Biomed. Sci. 2009, 16, 63. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Zhou, Y.; Huang, F.J.; Tang, H.D.; Xu, X.H.; Liu, J.J.; Wang, Y.; Deng, Y.L.; Ren, R.J.; Xu, W.; et al. Plasma metabolite profiles of alzheimer’s disease and mild cognitive impairment. J. Proteome Res. 2014, 13, 2649–2658. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.; Ikeda, K.; Shiojima, T.; Kinoshita, M. Increased plasma concentrations of aspartate, glutamate and glycine in parkinson’s disease. Neurosci. Lett. 1992, 145, 175–177. [Google Scholar] [CrossRef]
- Mulder, C.; Wahlund, L.O.; Teerlink, T.; Blomberg, M.; Veerhuis, R.; van Kamp, G.J.; Scheltens, P.; Scheffer, P.G. Decreased lysophosphatidylcholine/phosphatidylcholine ratio in cerebrospinal fluid in alzheimer’s disease. J. Neural Transm. 2003, 110, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Klavins, K.; Koal, T.; Dallmann, G.; Marksteiner, J.; Kemmler, G.; Humpel, C. The ratio of phosphatidylcholines to lysophosphatidylcholines in plasma differentiates healthy controls from patients with alzheimer’s disease and mild cognitive impairment. Alzheimers Dement. 2015, 1, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Adachi, J.; Asano, M.; Yoshioka, N.; Nushida, H.; Ueno, Y. Analysis of phosphatidylcholine oxidation products in human plasma using quadrupole time-of-flight mass spectrometry. Kobe J. Med. Sci. 2006, 52, 127–140. [Google Scholar] [PubMed]
- Mapstone, M.; Cheema, A.; Zhong, X.; Fiandaca, M.; Federoff, H. Biomarker validation: Methods and matrix matter (letter to the editor). Alzheimers Dement. 2017, 13, 608–609. [Google Scholar] [CrossRef] [PubMed]
- Oresic, M.; Posti, J.P.; Kamstrup-Nielsen, M.H.; Takala, R.S.; Lingsma, H.F.; Mattila, I.; Jantti, S.; Katila, A.J.; Carpenter, K.L.; Ala-Seppala, H.; et al. Human serum metabolites associate with severity and patient outcomes in traumatic brain injury. EBioMedicine 2016, 12, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Gagnon, J.F.; Vendette, M.; Fantini, M.L.; Massicotte-Marquez, J.; Montplaisir, J. Quantifying the risk of neurodegenerative disease in idiopathic rem sleep behavior disorder. Neurology 2009, 72, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Bertrand, J.A.; Montplaisir, J.; Desjardins, C.; Vendette, M.; Rios Romenets, S.; Panisset, M.; Gagnon, J.F. Rapid eye movement sleep behavior disorder and risk of dementia in parkinson’s disease: A prospective study. Mov. Disord. 2012, 27, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Holtbernd, F.; Gagnon, J.F.; Postuma, R.B.; Ma, Y.; Tang, C.C.; Feigin, A.; Dhawan, V.; Vendette, M.; Soucy, J.P.; Eidelberg, D.; et al. Abnormal metabolic network activity in rem sleep behavior disorder. Neurology 2014, 82, 620–627. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Aa, J.; Wang, G.; Yan, B.; Zhang, Y.; Wang, X.; Zhao, C.; Cao, B.; Shi, J.; Li, M.; et al. Differences in metabolite profile between blood plasma and serum. Anal. Biochem. 2010, 406, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Wedge, D.C.; Allwood, J.W.; Dunn, W.; Vaughan, A.A.; Simpson, K.; Brown, M.; Priest, L.; Blackhall, F.H.; Whetton, A.D.; Dive, C.; et al. Is serum or plasma more appropriate for intersubject comparisons in metabolomic studies? An assessment in patients with small-cell lung cancer. Anal. Chem. 2011, 83, 6689–6697. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.; Merchant-Borna, K.; Jeromin, A.; Livingston, W.; Bazarian, J. Acute plasma tau relates to prolonged return to play after concussion. Neurology 2017, 88, 595–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawton, M.; Baig, F.; Rolinski, M.; Ruffman, C.; Nithi, K.; May, M.T.; Ben-Shlomo, Y.; Hu, M.T. Parkinson’s disease subtypes in the oxford parkinson disease centre (OPDC) discovery cohort. J. Parkinsons Dis. 2015, 5, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Burn, D.; Goetz, C.; Aarsland, D.; Brown, R.G.; Broe, G.A.; Dickson, D.; Duyckaerts, C.; Cummings, J.; Gauthier, S.; et al. Diagnostic procedures for parkinson’s disease dementia: Recommendations from the movement disorder society task force. Mov. Disord. 2007, 22, 2314–2324. [Google Scholar] [CrossRef] [PubMed]
- Mapstone, M.; Cheema, A.K.; Fiandaca, M.S.; Zhong, X.; Mhyre, T.R.; MacArthur, L.H.; Hall, W.J.; Fisher, S.G.; Peterson, D.R.; Haley, J.M.; et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat. Med. 2014, 20, 415–418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dromerick, A.W.; Edwardson, M.A.; Edwards, D.F.; Giannetti, M.L.; Barth, J.; Brady, K.P.; Chan, E.; Tan, M.T.; Tamboli, I.; Chia, R.; et al. Critical periods after stroke study: Translating animal stroke recovery experiments into a clinical trial. Front. Hum. Neurosci. 2015, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Zhong, X.; Cheema, A.K.; Orquiza, M.H.; Chidambaram, S.; Tan, M.T.; Gresenz, C.R.; FitzGerald, K.T.; Nalls, M.A.; Singleton, A.B.; et al. Plasma 24-metabolite panel predicts preclinical transition to clinical stages of alzheimer’s disease. Front. Neurol. 2015, 6, 237. [Google Scholar] [CrossRef] [PubMed]
- Mapstone, M.; Lin, F.; Nalls, M.A.; Cheema, A.K.; Singleton, A.B.; Fiandaca, M.S.; Federoff, H.J. What success can teach us about failure: The plasma metabolome of older adults with superior memory and lessons for alzheimer’s disease. Neurobiol. Aging 2017, 51, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Xu, Y. An extremely simple method for extraction of lysophospholipids and phospholipids from blood samples. J. Lipid Res. 2010, 51, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Tautenhahn, R.; Patti, G.J.; Rinehart, D.; Siuzdak, G. Xcms online: A web-based platform to process untargeted metabolomic data. Anal. Chem. 2012, 84, 5035–5039. [Google Scholar] [CrossRef] [PubMed]
- Huan, T.; Forsberg, E.M.; Rinehart, D.; Johnson, C.H.; Ivanisevic, J.; Benton, H.P.; Fang, M.; Aisporna, A.; Hilmers, B.; Poole, F.L.; et al. Systems biology guided by xcms online metabolomics. Nat. Methods 2017, 14, 461–462. [Google Scholar] [CrossRef] [PubMed]
- MSFmetabolomics. Available online: https://www.mathworks.com/matlabcentral/fileexchange/60607-msfmetabolomics?s_tid=prof_contriblnk (accessed on 7 September 2018).
- Xie, W.; Zhang, H.; Zeng, J.; Chen, H.; Zhao, Z.; Liang, Z. Tissues-based chemical profiling and semi-quantitative analysis of bioactive components in the root of salvia miltiorrhiza bunge by using laser microdissection system combined with uplc-q-tof-ms. Chem. Cent. J. 2016, 10, 42. [Google Scholar] [CrossRef] [PubMed]
- Evans, A.M.; DeHaven, C.D.; Barrett, T.; Mitchell, M.; Milgram, E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 2009, 81, 6656–6667. [Google Scholar] [CrossRef] [PubMed]
- Illig, T.; Gieger, C.; Zhai, G.; Romisch-Margl, W.; Wang-Sattler, R.; Prehn, C.; Altmaier, E.; Kastenmuller, G.; Kato, B.S.; Mewes, H.W.; et al. A genome-wide perspective of genetic variation in human metabolism. Nat. Genet. 2010, 42, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Romisch-Margl, W.; Prehn, C.; Bogumil, R.; Rohring, C.; Suhre, K.; Adamski, J. Procedure for tissue sample preparation and metabolite extraction for high-throughput targeted metabolomics. Metabolomics 2012, 8, 133–142. [Google Scholar] [CrossRef]
- Gross, T.J.; Mapstone, M.; Miramontes, R.; Padilla, R.; Cheema, A.K.; Macciardi, F.; Federoff, H.J.; Fiandaca, M.S. Toward reproducible results from targeted metabolomic studies: Perspectives for data pre-processing and a basis for analytic pipeline development. Curr. Top. Med. Chem. 2018, 18, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.; Soufan, O.; Li, C.; Caraus, I.; Li, S.; Bourque, G.; Wishart, D.S.; Xia, J. Metaboanalyst 4.0: Towards more transparent and integrative metabolomics analysis. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef] [Green Version]
- Worley, B.; Powers, R. Multivariate analysis in metabolomics. Curr. Metab. 2013, 1, 92–107. [Google Scholar] [PubMed]
- Chen, T.; Cao, Y.; Zhang, Y.; Liu, J.; Bao, Y.; Wang, C.; Jia, W.; Zhao, A. Random forest in clinical metabolomics for phenotypic discrimination and biomarker selection. Evid. Based Complement. Altern. Med. 2013, 2013, 298183. [Google Scholar] [CrossRef] [PubMed]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B Methodol. 1996, 58, 267–288. [Google Scholar]
- Rohart, F.; Gautier, B.; Singh, A.; Le Cao, K.A. Mixomics: An R package for ‘omics feature selection and multiple data integration. PLoS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
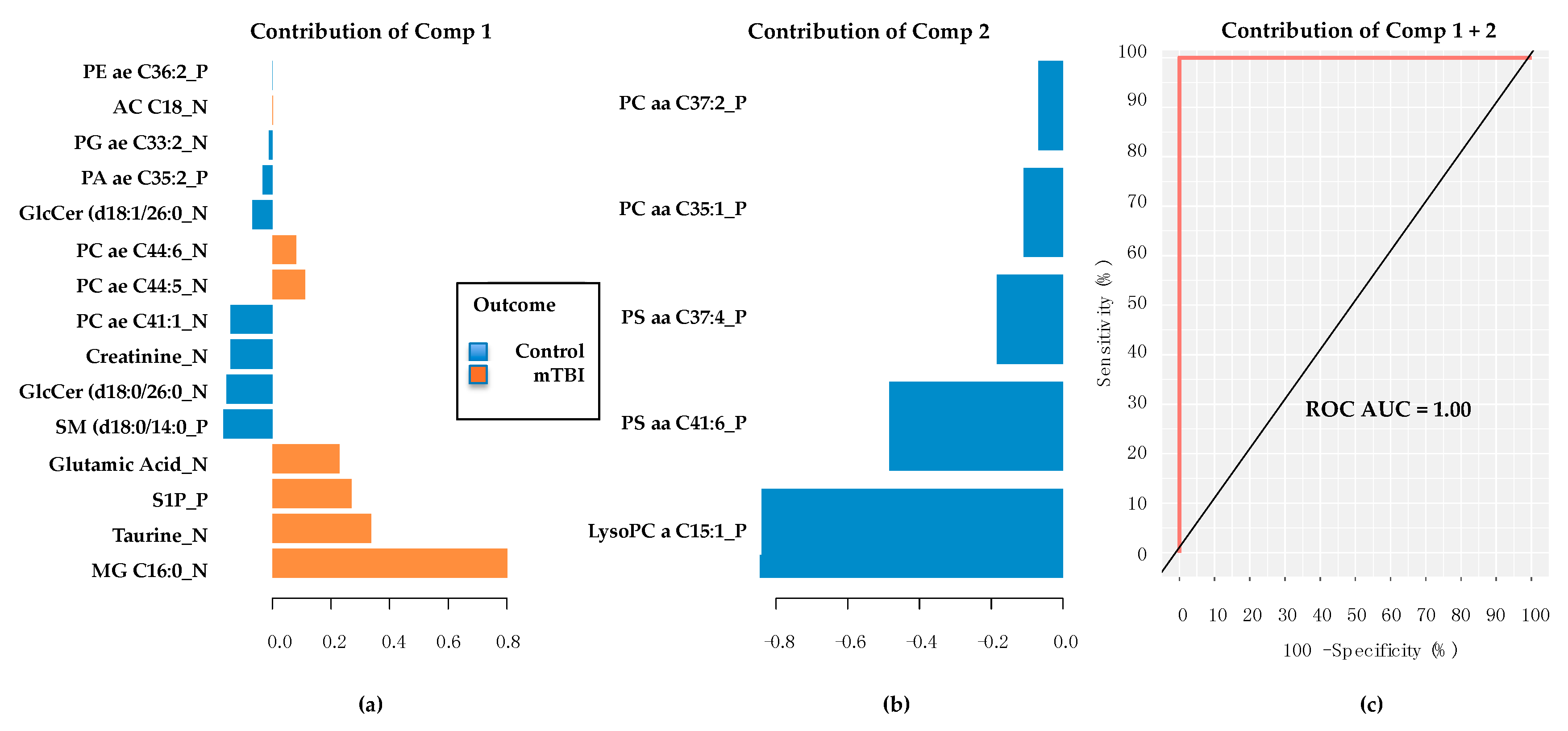{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population Characteristic | Subacute TBI Cases | TBI Controls | PD Cases (PD/PDD) | PD Controls |
|---|---|---|---|---|
| Number of subjects (n) | 75 | 20 | 40 | 20 |
| Age in years (mean ± S.D.) | 24.9 ± 5.2 * | 18.7 ± 0.8 * | 67.2 ± 11.4 NS | 65.9 ± 10.3 NS |
| Sex (n; M/F) | 71/4 ** | 8/12 ** | 22/18 NS | 11/9 NS |
| Preliminary Annotation | RVU in TBI Controls | RVU in Subacute mTBI Cases |
|---|---|---|
| * Monoacylglycerol (MG) C16:0_N | Low | High |
| Taurine_N | Low | High |
| Sphingosine 1 Phosphate_P (S1P_P) | Low | High |
| * Glutamic Acid_N | Low | High |
| Glucosylceramide (GlcCer) d18:1/26:0_N | High | Low |
| * Creatinine_N | High | Low |
| GlcCer d18:0/26:0_N | High | Low |
| Phosphatidylcholine (PC) ae C41:1_N | High | Low |
| PC ae C44:5_N | Low | High |
| Classification Algorithm for Model | ROC AUC | 95% CI | Sensitivity/Specificity |
|---|---|---|---|
| LinSVM | 0.968 | 0.945–0.992 | - |
| PLS-DA | 0.977 | 0.945–0.992 | - |
| RandFor | 0.965 | 0.882–1.00 | - |
| LR | 0.939 | 0.734–0.984 | - |
| LR + 10FCV Discovery | 0.993 | 0.984–1.00 | 0.981/0.939 |
| LR + 10FCV Internal Validation | 0.893 | 0.789–0.996 | 0.947/0.850 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiandaca, M.S.; Gross, T.J.; Johnson, T.M.; Hu, M.T.; Evetts, S.; Wade-Martins, R.; Merchant-Borna, K.; Bazarian, J.; Cheema, A.K.; Mapstone, M.; et al. Potential Metabolomic Linkage in Blood between Parkinson’s Disease and Traumatic Brain Injury. Metabolites 2018, 8, 50. https://doi.org/10.3390/metabo8030050
Fiandaca MS, Gross TJ, Johnson TM, Hu MT, Evetts S, Wade-Martins R, Merchant-Borna K, Bazarian J, Cheema AK, Mapstone M, et al. Potential Metabolomic Linkage in Blood between Parkinson’s Disease and Traumatic Brain Injury. Metabolites. 2018; 8(3):50. https://doi.org/10.3390/metabo8030050
Chicago/Turabian StyleFiandaca, Massimo S., Thomas J. Gross, Thomas M. Johnson, Michele T. Hu, Samuel Evetts, Richard Wade-Martins, Kian Merchant-Borna, Jeffrey Bazarian, Amrita K. Cheema, Mark Mapstone, and et al. 2018. "Potential Metabolomic Linkage in Blood between Parkinson’s Disease and Traumatic Brain Injury" Metabolites 8, no. 3: 50. https://doi.org/10.3390/metabo8030050