No Country for Old Worms: A Systematic Review of the Application of C. elegans to Investigate a Bacterial Source of Environmental Neurotoxicity in Parkinson’s Disease



Abstract
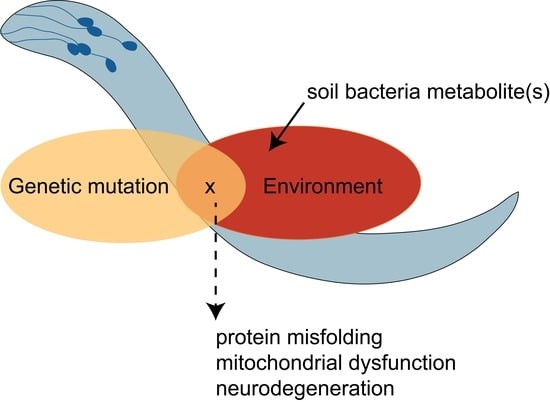
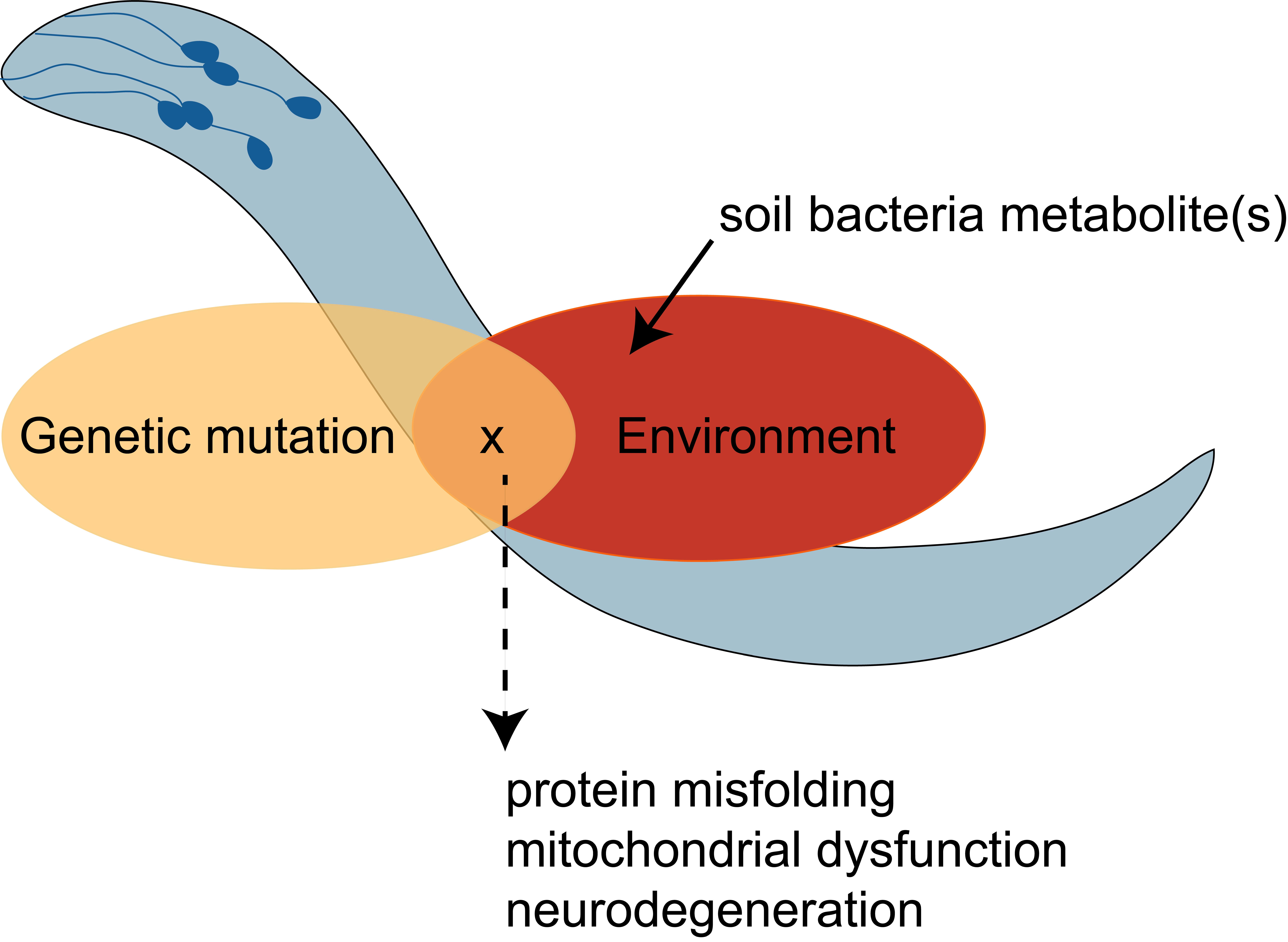
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Genetics of Parkinson’s Disease
2. Environmental Factors Impacting Neurodegeneration
3. Soil Bacteria and Neurodegeneration
4. Using C. elegans to Model Neurodegenerative Phenotypes
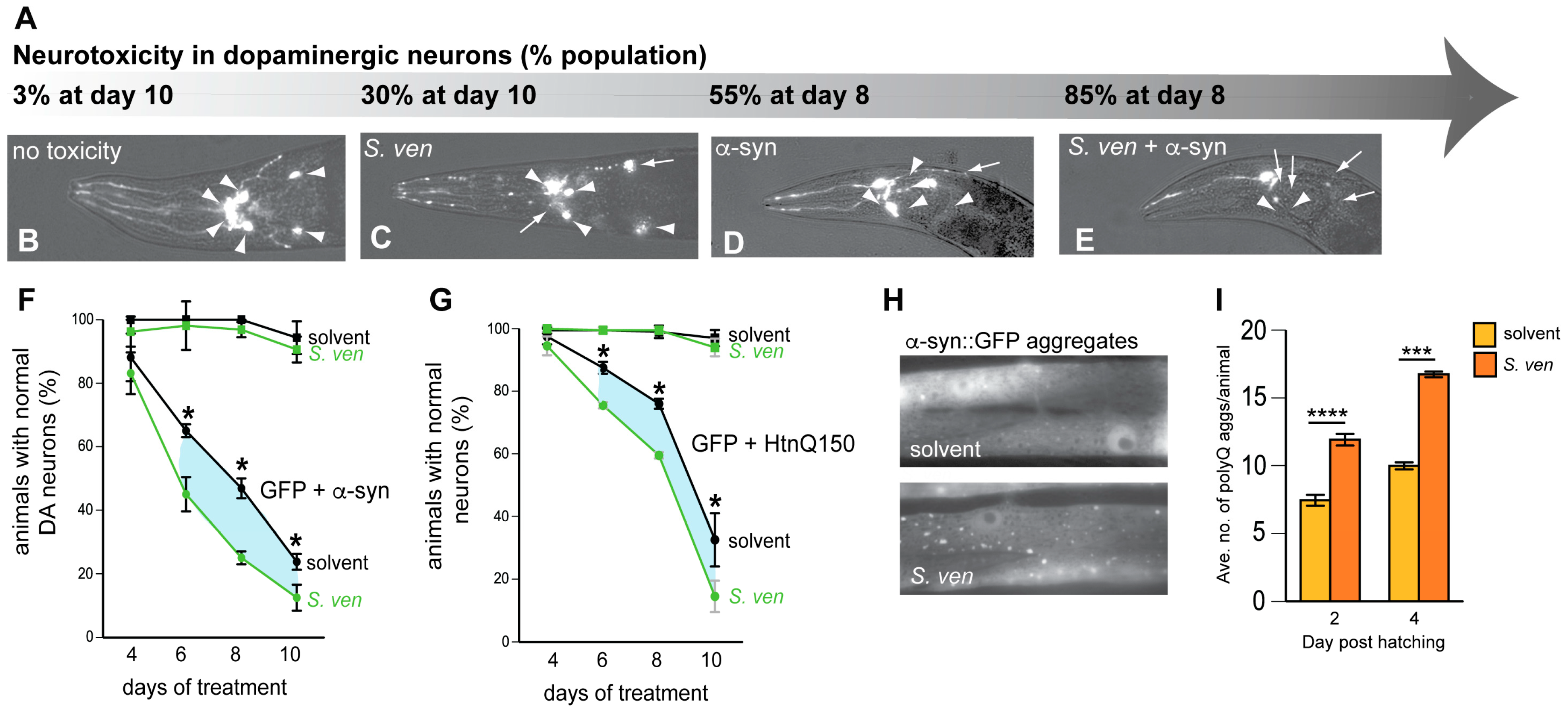
5. Gene-by-Environment (GxE) Interactions Modeled in C. elegans
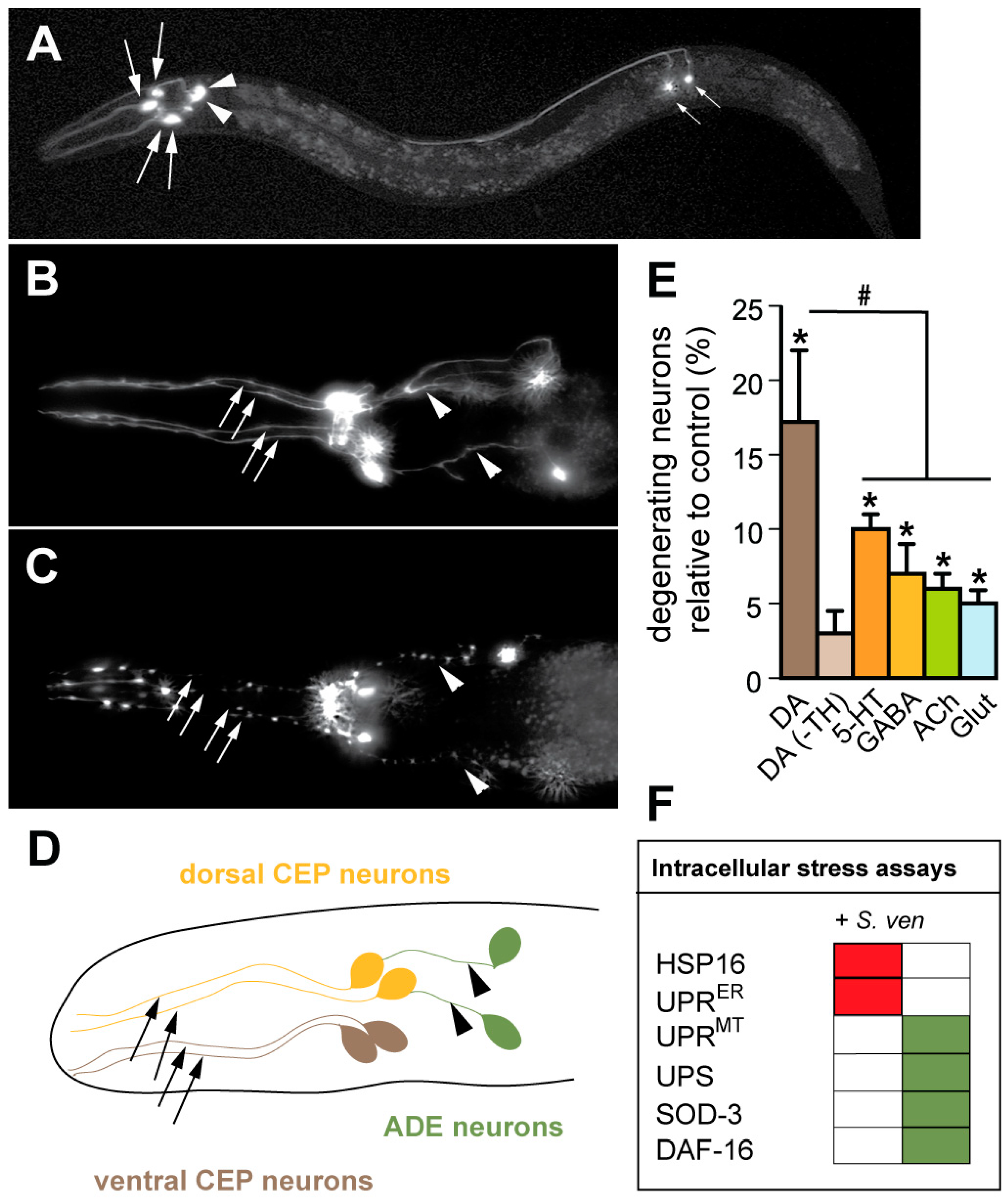
6. A Metabolic Fingerprint is Revealed in Response to the S. ven Metabolite
7. The S. ven Metabolite Causes Mitochondrial Dysfunction
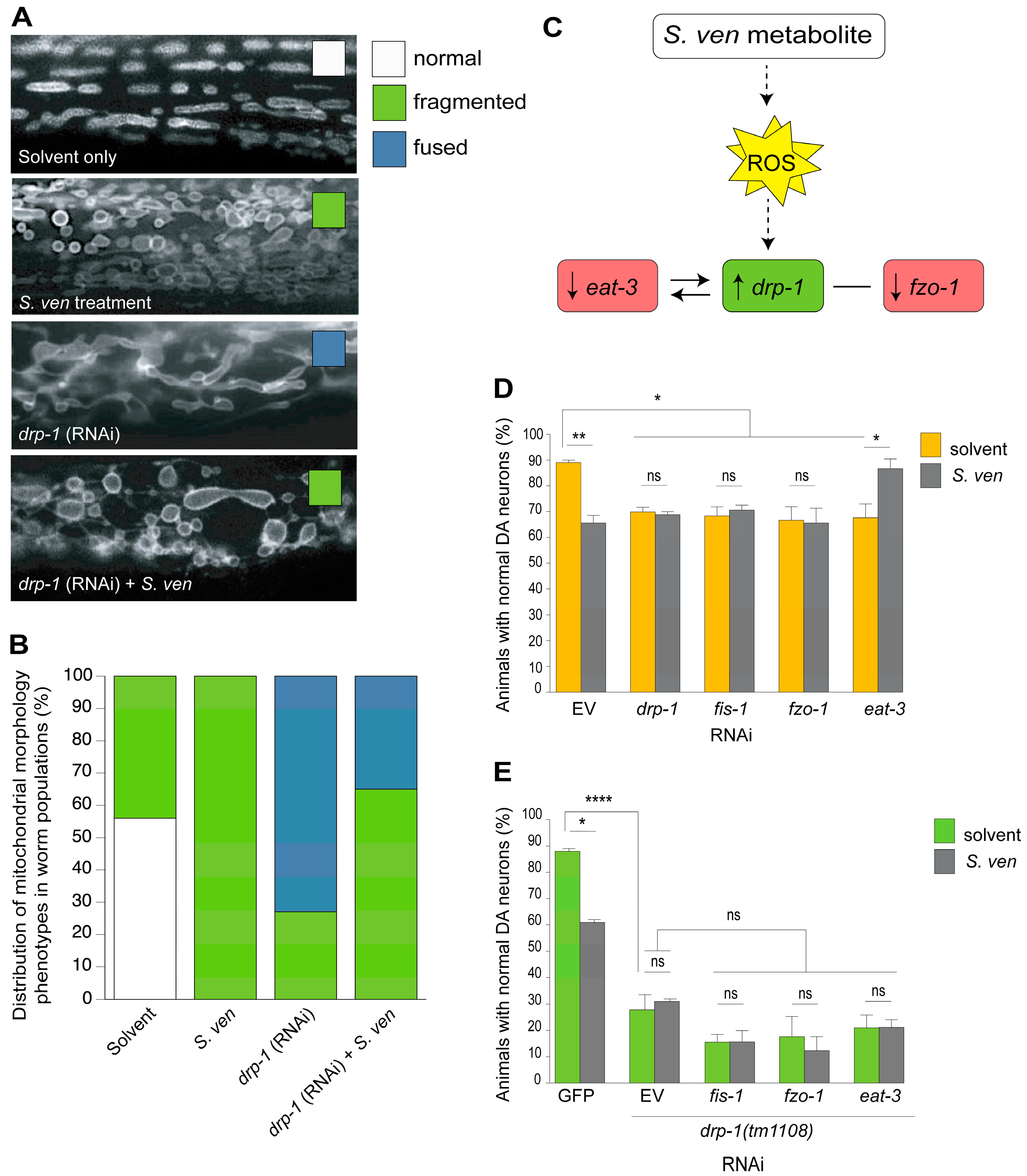
8. The S. ven Metabolite Disrupts Mitochondrial Homeostasis
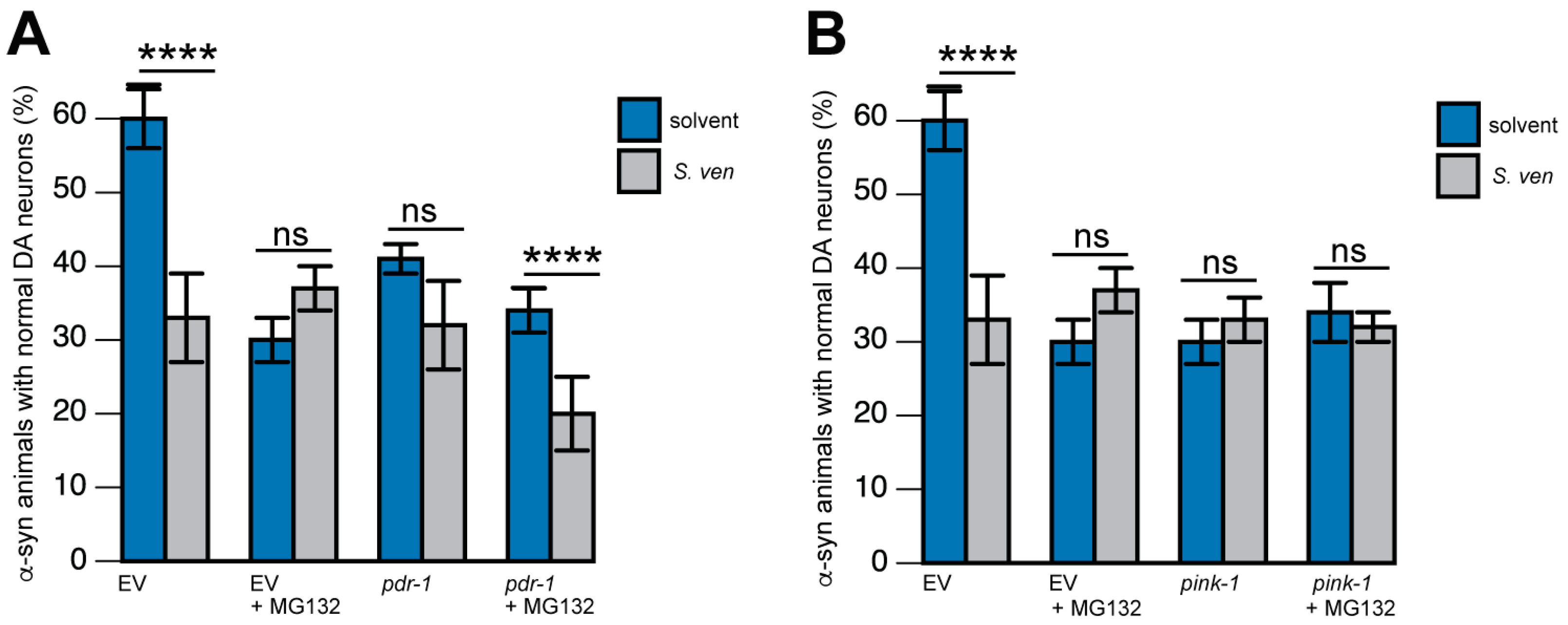
9. PINK-1/DRP-1-Dependent Fission Induced by S. ven Metabolite
10. Toward the Identification of the Neurotoxic Metabolite
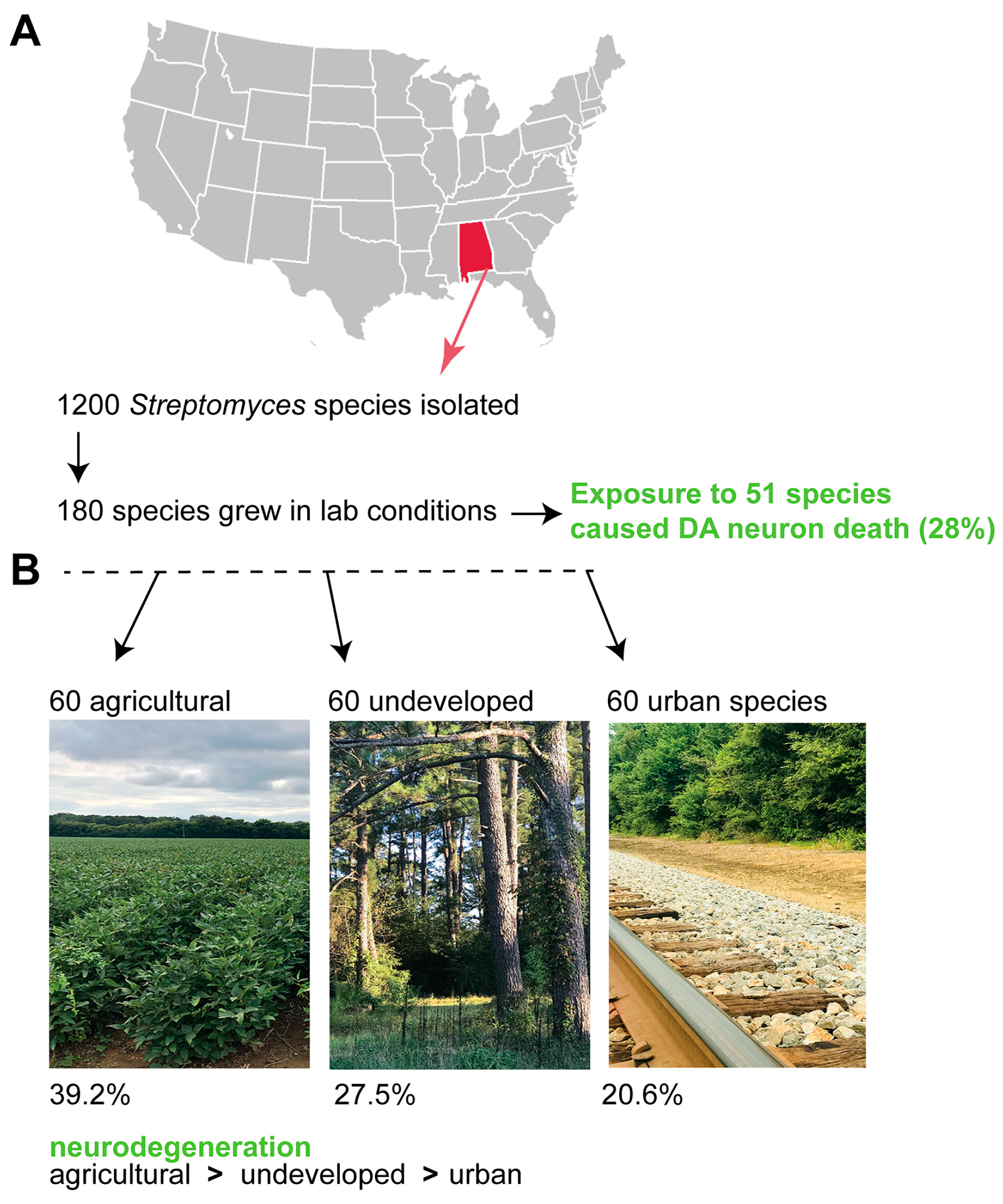
11. Exposure to Other Soil Streptomyces Species also Causes Neurodegeneration
12. Summary and Future Studies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goldwurm, S.; Di Fonzo, A.; Simons, E.J.; Rohé, C.F.; Zini, M.; Canesi, M.; Tesei, S.; Zecchinelli, A.; Antonini, A.; Mariani, C.; et al. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson’s disease and originates from a common ancestor. J. Med. Genet. 2005, 42, e65. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M. Occupational and environmental causes of parkinsonism. Occup. Med. 1992, 7, 503–513. [Google Scholar] [PubMed]
- Liou, H.H.; Tsai, M.C.; Chen, C.J.; Jeng, J.S.; Chang, Y.C.; Chen, S.Y.; Chen, R.C. Environmental risk factors and Parkinson’s disease: A case-control study in Taiwan. Neurology 1997, 48, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshi, A.; Khuder, S.A.; Schaub, E.A.; Priyadarshi, S.S. Environmental risk factors and Parkinson’s disease: A metaanalysis. Environ. Res. 2001, 86, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.; Cockburn, M.; Bronstein, J.; Zhang, X.; Ritz, B. Parkinson’s disease and residential exposure to maneb and paraquat from applications in the central valley of California. Am. J. Epidemiol. 2009, 169, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Gatto, N.M.; Cockburn, M.; Bronstein, J.; Manthripragada, A.D.; Ritz, B. Well-water consumption and Parkinson’s disease in rural California. Environ. Health Perspect. 2009, 117, 1912–1918. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.M.; Ross, G.W.; Jewell, S.A.; Hauser, R.A.; Jankovic, J.; Factor, S.A.; Bressman, S.; Deligtisch, A.; Marras, C.; Lyons, K.E.; et al. Occupation and risk of Parkinsonism: A multicenter case-control study. Arch. Neurol. 2009, 66, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Freire, C.; Koifman, S. Pesticide exposure and Parkinson’s disease: Epidemiological evidence of association. Neurotoxicology 2012, 22, 947–971. [Google Scholar] [CrossRef] [PubMed]
- Blesa, J.; Phani, S.; Jackson-Lewis, V.; Przedborski, S. Classic and new animal models of Parkinson’s disease. J. Biomed. Biotechnol. 2012, 2012, 845618. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Walker, J.E.; Youle, R. Mitochondrial quality control mediated by PINK1 and Parkin: Link to Parkinsonism. Cold Spring Harb. Perspect. Biol. 2012, 4, a011338. [Google Scholar] [CrossRef] [PubMed]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Richardson, R.J. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology 1998, 50, 1346–1350. [Google Scholar] [CrossRef] [PubMed]
- Roesch, L.F.W.; Fulthorpe, R.R.; Riva, A.; Casella, G.; Hadwin, A.K.M.; Kent, A.D.; Daroub, S.H.; Camargo, F.A.O.; Farmerie, W.G.; Triplett, E.W. Pyrosequencing enumerates and contrasts soil microbial diversity. ISME J. 2007, 1, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Gans, J.; Wolinsky, M.; Dunbar, J. Computational improvements reveal great bacterial diversity and high metal toxicity in soil. Science 2005, 309, 1387–1390. [Google Scholar] [CrossRef] [PubMed]
- Janssen, P.H. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl. Environ. Microbiol. 2006, 72, 1719–1728. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Omura, S. Metabolism and products of actinomycetes: An introduction. Actinomycetologica 1990, 4, 13–14. [Google Scholar] [CrossRef]
- Fenteany, G.; Standaert, R.F.; Lane, W.S.; Choi, S.; Corey, E.J.; Schreiber, S.L. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by lactacystin. Science 1995, 268, 726–731. [Google Scholar] [CrossRef] [PubMed]
- McNaught, K.S.P.; Perl, D.P.; Brownell, A.L.; Olanow, C.W. Systemic exposure to proteosome inhibitors causes a progressive model of Parkinson’s disease. Ann. Neurol. 2004, 56, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Bove, J.; Zhou, C.; Jackson-Lewis, V.; Taylor, J.; Chu, Y.; Rideout, H.J.; Wu, D.-C.; Kordower, J.H.; Petrucelli, L.; Przedborski, S. Proteasome inhibition and Parkinson’s disease modeling. Ann. Neurol. 2006, 60, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Kordower, J.H.; Kanann, N.M.; Chu, Y.; Babu, R.S.; Stansell, R.; Terpstra, B.T.; Sortwell, C.E.; Steece-Colllier, K.; Collier, T.J. Failure of proteasome inhibitor administration to provide a model of Parkinson’s disease in rats and monkeys. Ann. Neurol. 2006, 60, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Landau, A.M.; Kouassi, E.; Siegrist-Johnstone, R.; Desbarats, J. Proteasome inhibitor model of Parkinson’s disease in mice is confounded by neurotoxicity of the ethanol vehicle. Mov. Disord. 2007, 22, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Du, Y.; Fan, X.; Yang, D.; Luo, G.; Le, W. c-Jun N-terminal kinase mediates lactacystin-induced dopamine neuron degeneration. J. Neuropathol. Exp. Neurol. 2008, 67, 933–944. [Google Scholar] [CrossRef] [PubMed]
- Kohbata, S.; Beaman, B.L. l-Dopa-responsive movement disorder caused by Nocardia asteroides localized in the brains of mice. Infect. Immun. 1991, 59, 181–191. [Google Scholar] [PubMed]
- Tam, S.; Barry, D.P.; Beaman, L.; Beaman, B.L. Neuroinvasive Nocardia asteroides GUH-2 induces apoptosis in the substantia nigra in vivo and dopaminergic cells in vitro. Exp. Neurol. 2002, 107, 453–460. [Google Scholar] [CrossRef]
- Ogata, S.A.; Beaman, B.L. Adherence of Nocardia asteroides within the murine brain. Infect. Immun. 1992, 60, 1800–1805. [Google Scholar] [PubMed]
- Ogata, S.A.; Beaman, B.L. Site-specific growth of Nocardia asteroides in the murine brain. Infect. Immun. 1992, 60, 3262–3267. [Google Scholar] [PubMed]
- Chapman, G.; Beaman, B.L.; Loeffler, D.A.; Camp, D.M.; Domino, E.F.; Dickson, D.W.; Ellis, W.G.; Chen, I.; Bachus, S.E.; LeWitt, P.A. In situ hybridization for detection of nocardial 16S rRNA: Reactivity within intracellular inclusions in experimentally infected cynomolgus monkeys--and in Lewy body-containing human brain specimens. Exp. Neurol. 2003, 184, 715–725. [Google Scholar] [CrossRef]
- Barry, D.P.; Beaman, B.L. Modulation of eukaryotic cells apoptosis by members of the bacterial order Actinomycetales. Apoptosis 2006, 11, 1695–1707. [Google Scholar] [CrossRef] [PubMed]
- Martinez, B.A.; Caldwell, K.A.; Caldwell, G.A. C. C. elegans as a model system to accelerate discovery for Parkinson disease. Curr. Opin. Genet. Dev. 2017, 44, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 2008 105, 145–150. [Google Scholar] [CrossRef]
- Gitler, A.D.; Chesi, A.; Geddie, M.L.; Strathearn, K.E.; Hamamichi, S.; Hill, K.J.; Caldwell, K.A.; Caldwell, G.A.; Cooper, A.A.; Rochet, J.C.; et al. Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat. Genet. 2009, 41, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Wilson, S.; Yacoubian, T.A.; Xie, Z.-L.; Speake, L.D.; Parks, R.; Crabtree, D.; et al. A neuroprotective role of lysosomal enzyme cathepsin D against alpha-synuclein pathogenesis. Mol. Brain 2008, 1, 17. [Google Scholar] [CrossRef] [PubMed]
- Yacoubian, T.A.; Slone, S.R.; Harrington, A.J.; Hamamichi, S.; Schieltz, J.M.; Caldwell, K.A.; Caldwell, G.A.; Standaert, D.G. Differential neuroprotective effects of 14-3-3 proteins in models of Parkinson’s disease. Cell Death Dis. 2010, 1, e2. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Harrington, A.J.; Caldwell, K.A.; Caldwell, G.A.; Standaert, D.G. VPS41, a protein involved in lysosomal trafficking, is protective in Caenorhabditis elegans and mammalian cellular models of Parkinson’s disease. Neurobiol. Dis. 2010, 37, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Chalfie, M.; White, J. The nervous system. In The Nematode Caenorhabditis Elegans; Wood, W.B., Ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1988; pp. 337–391. ISBN 978-087969433-3. [Google Scholar]
- Jonker, M.J.; Piskiewicz, A.M.; Ivorra i Castella, N.; Kammenga, J.E. Toxicity of binary mixtures of cadmium-copper and carbendazim-copper to the nematode Caenorhabditis elegans. Environ. Toxicol. Chem. 2004, 23, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Peres, T.V.; Schettinger, M.R.; Chen, P.; Carvalho, F.; Avila, D.S.; Bowman, A.B.; Aschner, M. Manganese-induced neurotoxicity: A review of its behavioral consequences and neuroprotective strategies. BMC Pharmacol. Toxicol. 2016, 17, 57. [Google Scholar] [CrossRef] [PubMed]
- Locke, C.J.; Fox, S.A.; Caldwell, G.A.; Caldwell, K.A. Acetaminophen attenuates dopamine neuron degeneration in animal models of Parkinson’s disease. Neurosci. Lett. 2008, 439, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Hewett, J.W.; Yokoi, F.; Lu, J.; Buckley, A.C.; Burdette, A.J.; Chen, P.; Nery, F.C.; Li, Y.; Breakefield, X.O.; et al. Chemical enhancement of torsinA function in cell and animal models of torsion dystonia. Dis. Model. Mech. 2010, 3, 386–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardiff, D.F.; Jui, N.T.; Khurana, V.; Tambe, M.A.; Thompson, M.L.; Chung, C.Y.; Kamadurai, H.B.; Kim, H.T.; Lancaster, A.K.; Caldwell, K.A.; et al. Yeast reveal a “druggable” Rsp5/Nedd4 network that ameliorates α-synuclein toxicity in neurons. Science 2013, 342, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Nass, R.; Hall, D.H.; Miller, D.M., 3rd; Blakely, R.D. Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 2002, 99, 3264–3269. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Martinez, B.A.; Berkowitz, L.A.; Caldwell, G.A.; Caldwell, K.A. Mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson’s model. Cell Death Dis. 2014, 5, e984. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lu, H.; Bargmann, C.I. Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans. Nature 2005, 438, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, K.A.; Tucci, M.L.; Armagost, J.; Hodges, T.W.; Chen, J.; Memon, S.B.; Blalock, J.E.; DeLeon, S.M.; Findlay, R.H.; Ruan, Q.; et al. Investigating Bacterial Sources of Toxicity as an Environmental Contributor to Dopaminergic Neurodegeneration. PLoS ONE 2009, 4, e7227. [Google Scholar] [CrossRef] [PubMed]
- Sulston, J.; Dew, M.; Brenner, S. Dopaminergic neurons in the nematode Caenorhabditis elegans. J. Comp. Neurol. 1975, 163, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, S.; Wintle, R.F.; Kindt, K.S.; Nuttley, W.M.; Arvan, R.; Fitzmaurice, P.; Bigras, E.; Merz, D.C.; Hébert, T.E.; van der Kooy, D.; et al. Dopamine modulates the plasticity of mechanosensory responses in Caenorhabditis elegans. EMBO J. 2003, 23, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. Alpha-synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed]
- Manning-Bog, A.B.; McCormack, A.L.; Li, J.; Uversky, V.N.; Fink, A.L.; Di Monte, D.A. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: Paraquat and alpha-synuclein. J. Biol. Chem. 2002, 277, 1641–1644. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.B.; Li, J.; Fink, A.L. Pesticides directly accelerate the rate of alpha-synclein fibril formation: A possible factor in Parkinson’s Disease. FEBS Lett. 2001, 500, 105–108. [Google Scholar] [CrossRef]
- Martinez, B.A.; Kim, H.; Ray, A.; Caldwell, G.A.; Caldwell, K.A. A bacterial metabolite induces glutathione-tractable proteostatic damage, proteasomal disturbances, and PINK1-dependent autophagy in C. elegans. Cell Death Dis. 2015, 6, e1908–e1913. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. Genetic analysis of pathways to Parkinson’s disease. Neuron 2010, 68, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Caroni, P. Selective neuronal vulnerability in neurodegenerative diseases: From stressor thresholds to degeneration. Neuron 2011, 71, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Saha, S.; Guillily, M.D.; Ferree, A.; Lanceta, J.; Chan, D.; Ghosh, J.; Hsu, C.H.; Segal, L.; Raghavan, K.; Matsumoto, K.; et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in C. elegans. J. Neurosci. 2009, 29, 9210–9218. [Google Scholar] [CrossRef] [PubMed]
- Mortiboys, H.; Johansen, K.K.; Aasly, J.O.; Bandmann, O. Mitochondrial impairment in patients with Parkinson disease with the G2019S mutation in LRRK2. Neurology 2010, 75, 2017–2020. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.H.; Mok, S.Z.; Koh, C.; Ouyang, X.; Fivaz, M.L.; Tan, E.K.; Dawson, V.L.; Dawson, T.M.; Yu, F.; Lim, K.L. Parkin Protects against LRRK2 G2019S Mutant-Induced Dopaminergic Neurodegeneration in Drosophila. J. Neurosci. 2009, 29, 11257–11262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jha, N.; Kumar, J.; Bonplueang, R.; Andersen, J.K. Glutathione decreases in dopaminergic PC12 cells interfere with the ubiquitin protein degradation pathway: Relevance for Parkinson’s disease? J. Neurochem. 2002, 80, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Romero-Aristizabal, C.; Marks, D.S.; Fontana, W.; Apfeld, J. Regulated spatial organization and sensitivity of cytosolic protein oxidation in Caenorhabditis elegans. Nat. Commun. 2014, 5, 5020. [Google Scholar] [CrossRef] [PubMed]
- Back, P.; Braeckma, B.P.; Matthijssens, F. ROS in Aging Caenorhabditis elegans: Damage or signaling. Oxidative Med. Cell. Longev. 2012, 2012, 608478. [Google Scholar] [CrossRef] [PubMed]
- Springer, W.; Hoppe, T.; Schmidt, E.; Baumeister, R. A Caenorhabditis elegans Parkin mutant with altered solubility couples α-synuclein aggregation to proteotoxic stress. Hum. Mol. Genet. 2005, 14, 3407–3423. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ouyang, Y.; Yang, L.; Beal, M.F.; McQulbban, A.; Vogel, H.; Lu, B. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc. Natl. Acad. Sci. USA 2008, 105, 7070–7075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.A.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Pargalija, D.; Klinkenberg, M.; Dominiguez-Bautista, J.; Hetzel, M.; Gispert, S.; Chimi, M.A.; Dröse, S.; Mai, S.; Brandt, U.; Auburger, G.; et al. Loss of PINK1 impairs stress-induced autophagy and cell survival. PLoS ONE 2014, 9, e95288. [Google Scholar]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef] [PubMed]
- Gosai, S.J.; Kwak, J.H.; Luke, C.J.; Long, O.S.; King, D.E.; Kovatch, K.J.; Johnston, P.A.; Shun, T.Y.; Lazo, J.S.; Perlmutter, D.H.; et al. Automated high-content live animal drug screening using C. elegans expressing the aggregation prone serpin α1-antitrypsin Z. PLoS ONE 2010, 5, e15460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spires, T.L.; Hannan, A.J. Nature, nurture and neurology: Gene-environment interactions in neurodegenerative disease. FASEB J. 2005, 272, 2347–2361. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Hyde, B.; Shirihai, O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: The bioenergetic view. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lämmermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of Parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef] [PubMed]
- Haynes, C.M.; Ron, D. The mitochondrial UPR–protecting organelle protein homeostasis. J. Cell Sci. 2010, 123, 3849–3855. [Google Scholar] [CrossRef] [PubMed]
- Murfitt, R.R.; Vogel, K.; Sanadi, D.R. Characterization of the mitochondria of the free-living nematode, Caenorhabditis elegans. Comp. Biochem. Physiol. 1976, 53, 423–430. [Google Scholar] [CrossRef]
- Ved, R.; Saha, S.; Westlund, B.; Perier, C.; Burnam, L.; Sluder, A.; Hoener, M.; Rodrigues, C.M.; Alfonso, A.; Steer, C.; et al. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J. Biol. Chem. 2005, 280, 42655–42668. [Google Scholar] [CrossRef] [PubMed]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Lemire, B.D. Mitochondrial complex I mutations in Caenorhabditis elegans produce cytochrome c oxidase stress and vitamin-responsive lactic acidosis. Hum. Mol. Genet. 2004, 13, 303–314. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, N.; Jofuku, A.; Eura, Y.; Mihara, K. Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochim Biophys. Acta 2003, 301, 891–898. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Koopman, W.J.H.; Verkaart, S.; Visch, H.-J.; van der Westhuizen, F.H.; Murphy, M.P.; van den Heuvel, L.W.P.J.; Smeitink, J.A.M.; Willems, P.H.G.M. Inhibition of complex I of the electron transport chain causes O2−-mediated mitochondrial outgrowth. Am. J. Physiol. Cell Physiol. 2005, 288, C1440–C1450. [Google Scholar] [CrossRef] [PubMed]
- Arnold, B.; Cassady, S.J.; VanLaar, V.S.; Berman, S.B. Integrating multiple aspects of mitochondrial dynamics in neurons: Age-related differences and dynamic changes in a chronic rotenone model. Neurobiol. Dis. 2011, 41, 189–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Su, B.; Liu, W.; He, X.; Gao, Y.; Castellani, R.J.; Perry, G.; Smith, M.A.; Zhu, X. DLP1-dependent mitochondrial fragmentation mediates 1-methyl-4-phenylpyridinium toxicity in neurons: Implications for Parkinson’s disease. Aging Cell 2011, 10, 807–823. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, P.; Sangar, M.C.; Singh, S.; Tang, W.; Bansal, S.; Chowdhury, G.; Cheng, Q.; Fang, J.-K.; Martin, M.V.; Guengerich, F.P.; et al. Metabolism of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine by mitochondrion-targeted cytochrome P450 2D6: Implications in Parkinson disease. J. Biol. Chem. 2013, 288, 4436–4451. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Yang, L.; Wang, J.; Ye, F.; Dan, G.; Zhao, Y.; Cai, Y.; Cui, Z.; Ao, L.; Liu, J.; et al. The Interaction of Mitochondrial Biogenesis and Fission/Fusion Mediated by PGC-1α Regulates Rotenone-Induced Dopaminergic Neurotoxicity. Mol. Neurobiol. 2017, 54, 3783–3797. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhou, F.; Zhang, Z.; Xing, D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission–fusion proteins. FEBS J. 2011, 278, 941–954. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Perentis, R.J.; Caldwell, G.A.; Caldwell, K.A. Gene-by-environment interactions that disrupt mitochondrial homeostasis cause neurodegeneration in C. elegans Parkinson’s models. Cell Death Dis. 2018, 9, 555. [Google Scholar] [CrossRef] [PubMed]
- Harrington, A.J.; Yacoubian, T.A.; Slone, S.R.; Caldwell, K.A.; Caldwell, G.A. Functional analysis of VPS41-mediated neuroprotection in Caenorhabditis elegans and mammalian models of Parkinson’s disease. J. Neurosci. 2012, 32, 2142–2153. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Zappaterra, M.D.; Hasegawa, A.; Wright, A.P.; Newman-Smith, E.D.; Buttle, K.F.; McDonald, K.; Mannella, C.A.; van der Bliek, A.M. The C. elegans Opa1 Homologue EAT-3 Is Essential for Resistance to Free Radicals. PLoS Genet. 2008, 4, e1000022. [Google Scholar] [CrossRef] [PubMed]
- Sandebring, A.; Thomas, K.J.; Beilina, A.; van der Brug, M.; Cleland, M.M.; Ahmad, R.; Miller, D.W.; Zambrano, I.; Cowburn, R.F.; Behbahani, H.; et al. Mitochondrial alterations in PINK1 deficient cells are influenced by calcineurin-dependent dephosphorylation of dynamin-related protein 1. PLoS ONE 2009, 4, e5701. [Google Scholar] [CrossRef] [PubMed]
- Buhlman, L.; Damiano, M.; Bertolin, G.; Ferrando-Miguel, R.; Lombès, A.; Brice, A.; Corti, O. Functional interplay between Parkin and Drp1 in mitochondrial fission and clearance. BBA Mol. Cell Res. 2014, 1843, 2012–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, L.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Youle, R. Cell biology: Form follows function for mitochondria. Lett. Nat. 2016, 530, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Watkins, A.L.; Ray, A.R.; Roberts, L.; Caldwell, K.A.; Olson, J.B. The Prevalence and Distribution of Neurodegenerative Compound-Producing Soil Streptomyces spp. Sci. Rep. 2016, 6, 22566. [Google Scholar] [CrossRef] [PubMed]
- Challis, G.L.; Hopwood, D.A. Synergy and contingency as driving forces for the evolution of multiple secondary metabolite production by Streptomyces species. Proc. Natl. Acad. Sci. USA 2003, 100, 14555–14561. [Google Scholar] [CrossRef] [PubMed]
- Van Lanen, S.G.; Shen, B. Microbial genomics for the improvement of natural product discovery. Curr. Opin. Microbiol. 2006, 9, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef]
- Lee, V.M.; Trojanowski, J.Q. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: New targets for drug discovery. Neuron 2006, 52, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.P.; Youle, R.J. Targeting mitochondrial dysfunction: Role for PINK1 and Parkin in mitochondrial quality control. Antioxid. Redox Signal. 2011, 14, 1929–1938. [Google Scholar] [CrossRef] [PubMed]
- Vidal, M.; Cusick, M.E.; Barabasi, A.L. Interactome networks and human disease. Cell 2011, 144, 986–998. [Google Scholar] [CrossRef] [PubMed]
- Towlson, E.K.; Vértes, P.E.; Yan, G.; Chew, Y.L.; Walker, D.S.; Schafer, W.R.; Barabási, A.L. Caenorhabditis elegans and the network control framework-FAQs. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170372. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.W.; Smith, R.L.; van Weeghel, M.; Kamble, R.; Janssens, G. E.; Houtkooper, R.H. Identification of key pathways and metabolic fingerprints of longevity in C. elegans. Exp. Gerontol. 2018, 113, 128–140. [Google Scholar] [CrossRef] [PubMed]
- Van Assche, R.; Temmerman, L.; Dias, D.A.; Boughton, B.; Boonen, K.; Braeckman, B.P.; Schoofs, L.; Roessner, U. Metabolic profiling of a transgenic Caenorhabditis elegans Alzheimer model. Metabolomics 2015, 11, 477–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, A.W.; Sterken, M.G.; Uit de Bos, J.; van Creij, J.; Kamble, R.; Snoek, B.L.; Kammenga, J.E.; Houtkooper, R.H. Natural genetic variation in C. elegans identified genomic loci controlling metabolite levels. Genome Res. 2018, 28, 1296–1308. [Google Scholar] [CrossRef] [PubMed]
- Whitman, W.B.; Coleman, D.C.; Wiebe, W.J. Prokaryotes: The unseen majority. Proc. Natl. Acad. Sci. USA 1998, 95, 6578–6583. [Google Scholar] [CrossRef] [PubMed] [Green Version]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caldwell, K.A.; Thies, J.L.; Caldwell, G.A. No Country for Old Worms: A Systematic Review of the Application of C. elegans to Investigate a Bacterial Source of Environmental Neurotoxicity in Parkinson’s Disease. Metabolites 2018, 8, 70. https://doi.org/10.3390/metabo8040070
Caldwell KA, Thies JL, Caldwell GA. No Country for Old Worms: A Systematic Review of the Application of C. elegans to Investigate a Bacterial Source of Environmental Neurotoxicity in Parkinson’s Disease. Metabolites. 2018; 8(4):70. https://doi.org/10.3390/metabo8040070
Chicago/Turabian StyleCaldwell, Kim A., Jennifer L. Thies, and Guy A. Caldwell. 2018. "No Country for Old Worms: A Systematic Review of the Application of C. elegans to Investigate a Bacterial Source of Environmental Neurotoxicity in Parkinson’s Disease" Metabolites 8, no. 4: 70. https://doi.org/10.3390/metabo8040070