
Low Complexity Induces Structure in Protein Regions Predicted as Intrinsically Disordered

 ,
,  , , ,
, , , 

Abstract
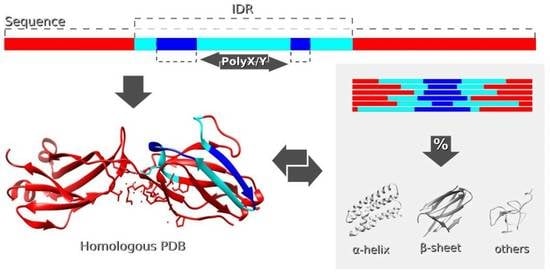
:
1. Introduction
2. Materials and Methods
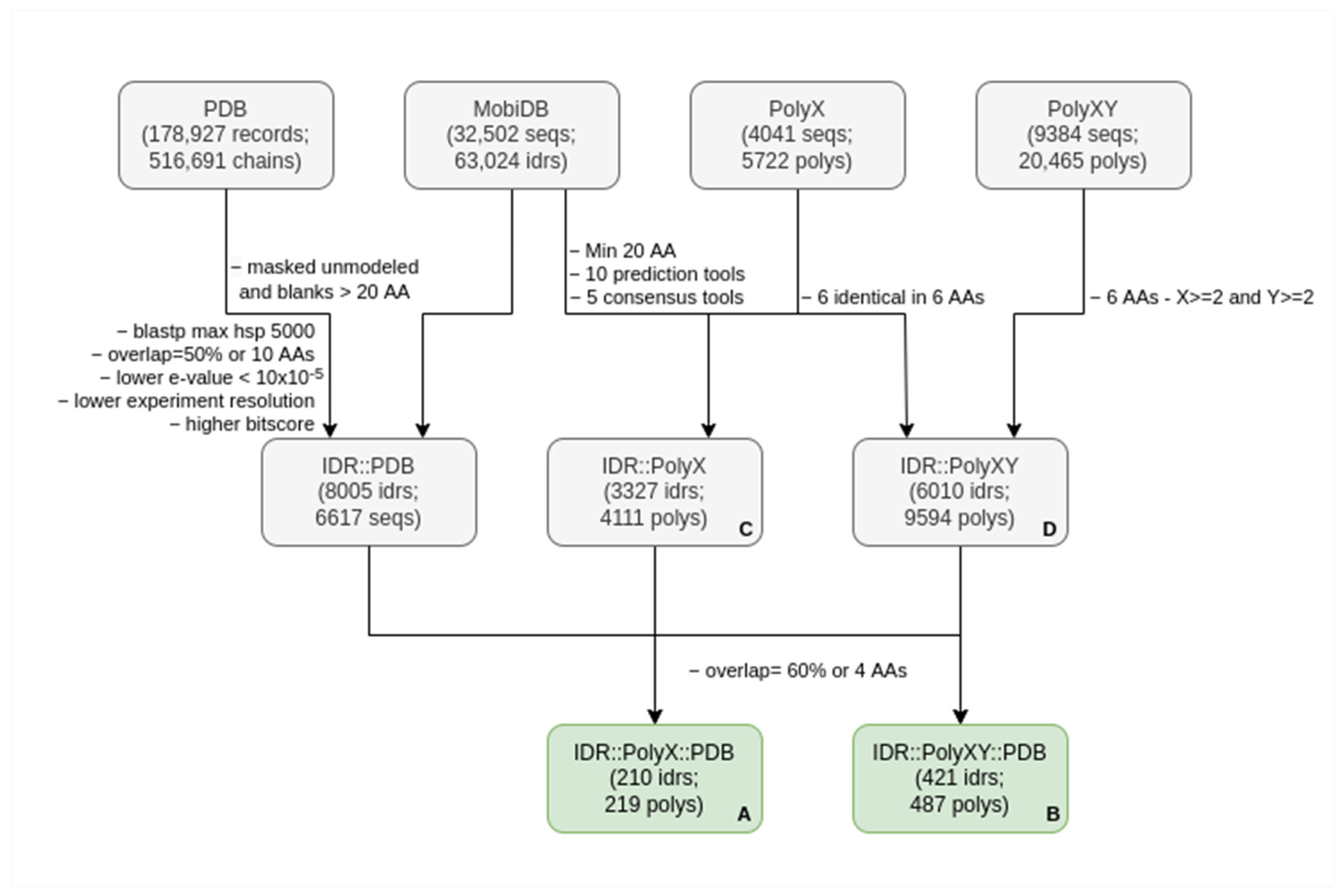
2.1. Dataset Construction
2.2. Finding Sequences of Proteins in the PDB with Homology to IDRs
2.3. Secondary Structure Annotation
2.4. Filtering PolyXs and PolyXYs Related to IDRs
2.5. Additional Extractions and Analyses
3. Results and Discussions
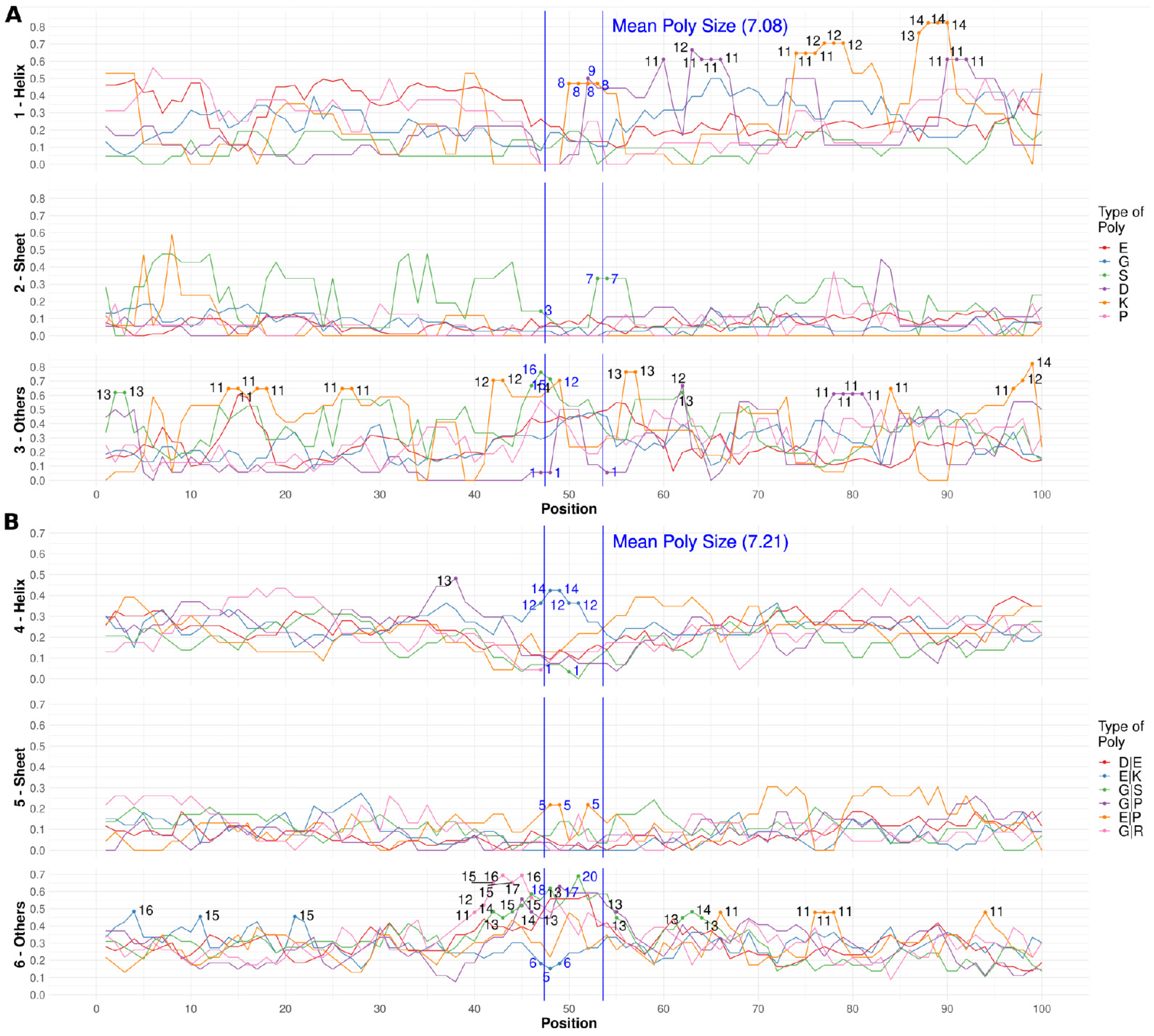
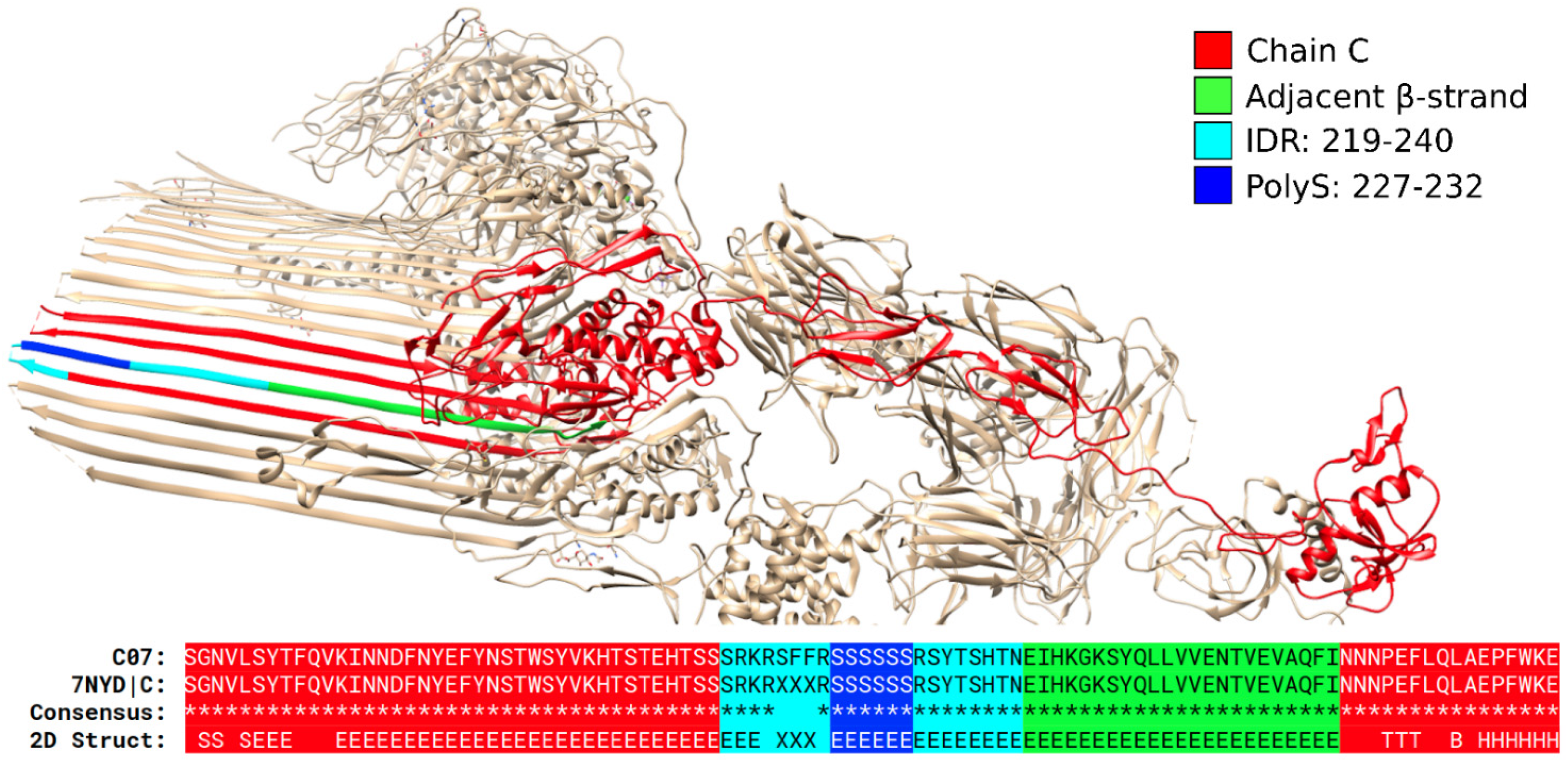
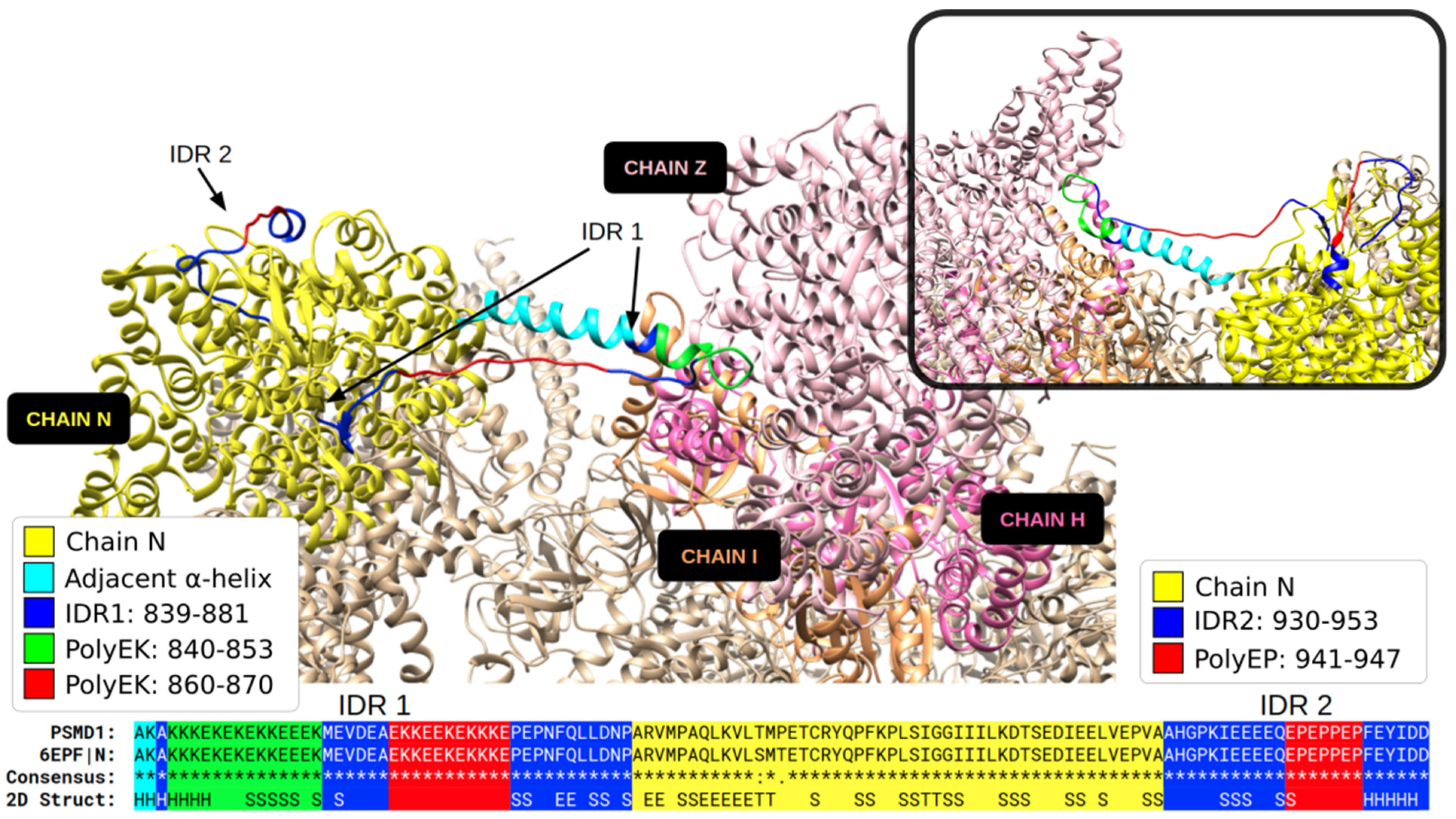
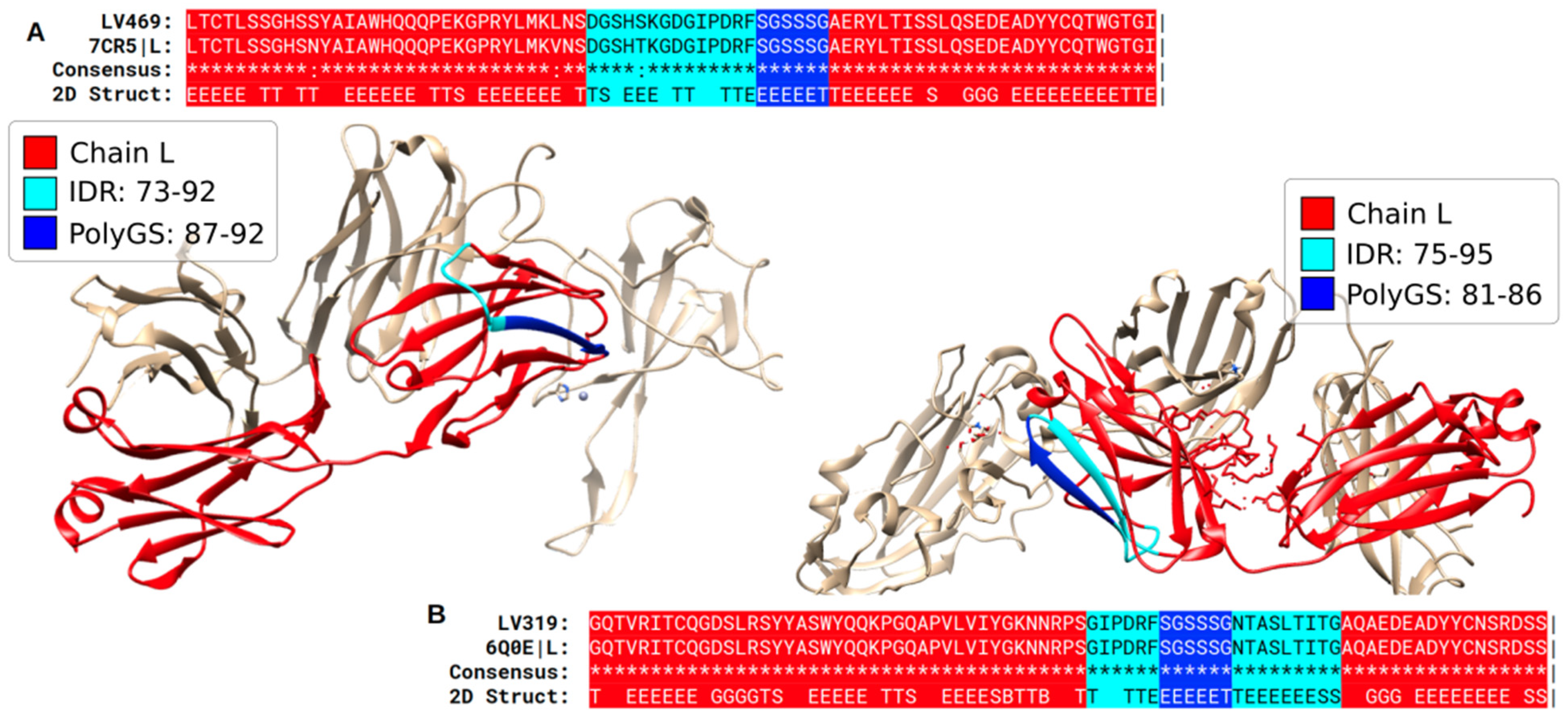
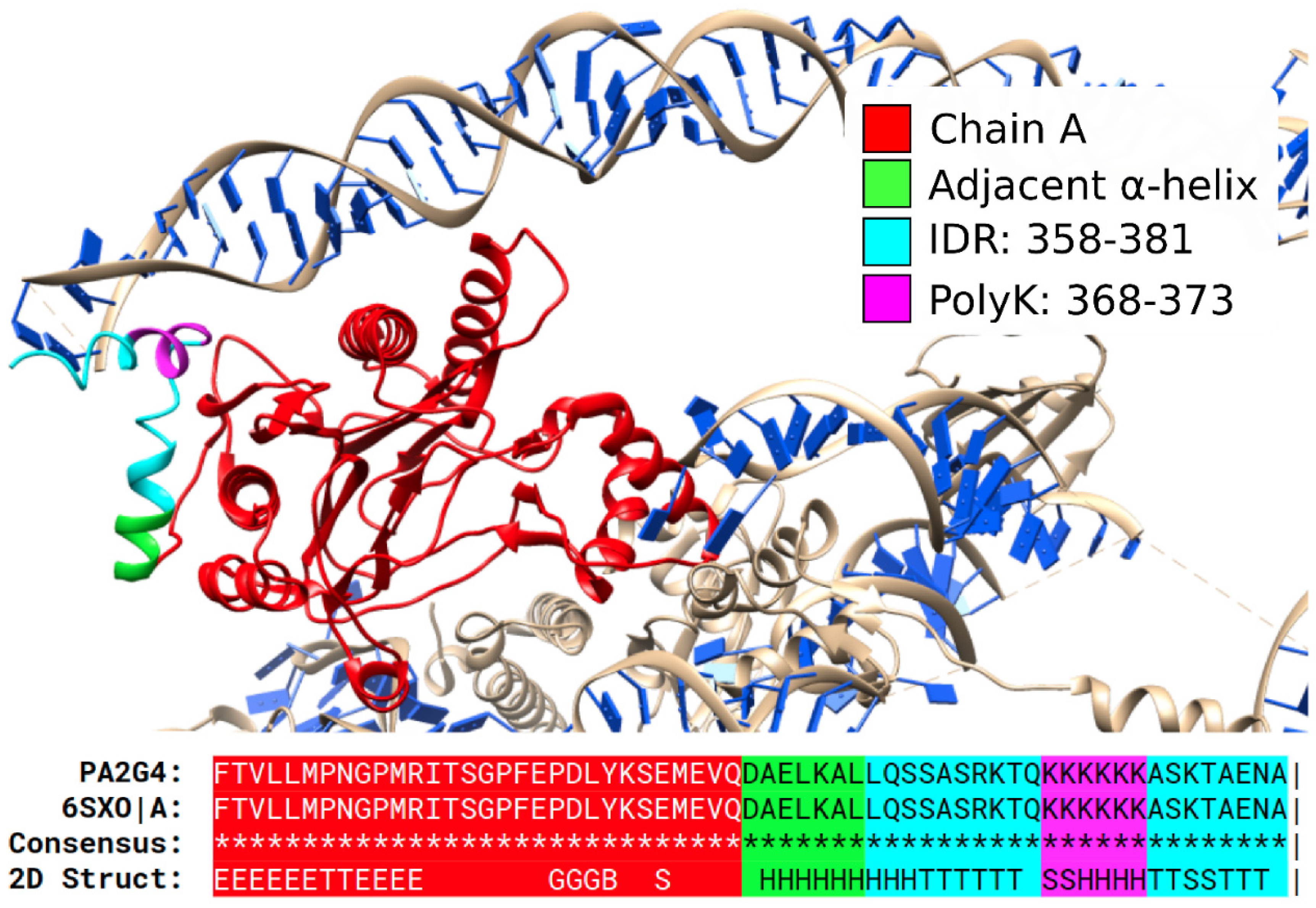
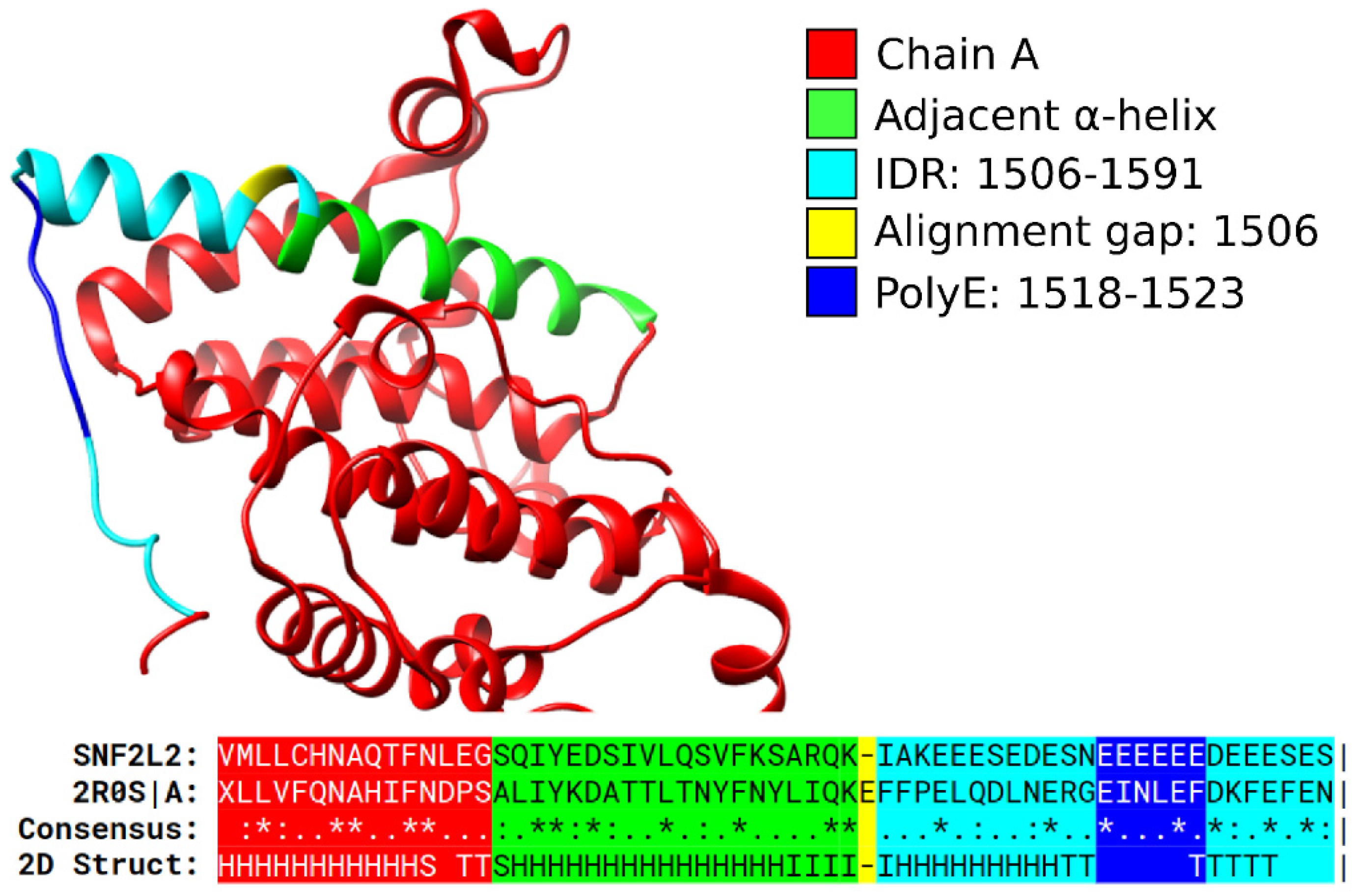
3.1. Specific PolyX and PolyXYs Can Produce Structural Gain in IDRs
3.2. PolyX and PolyXYs Accumulation in PDB-IDRs
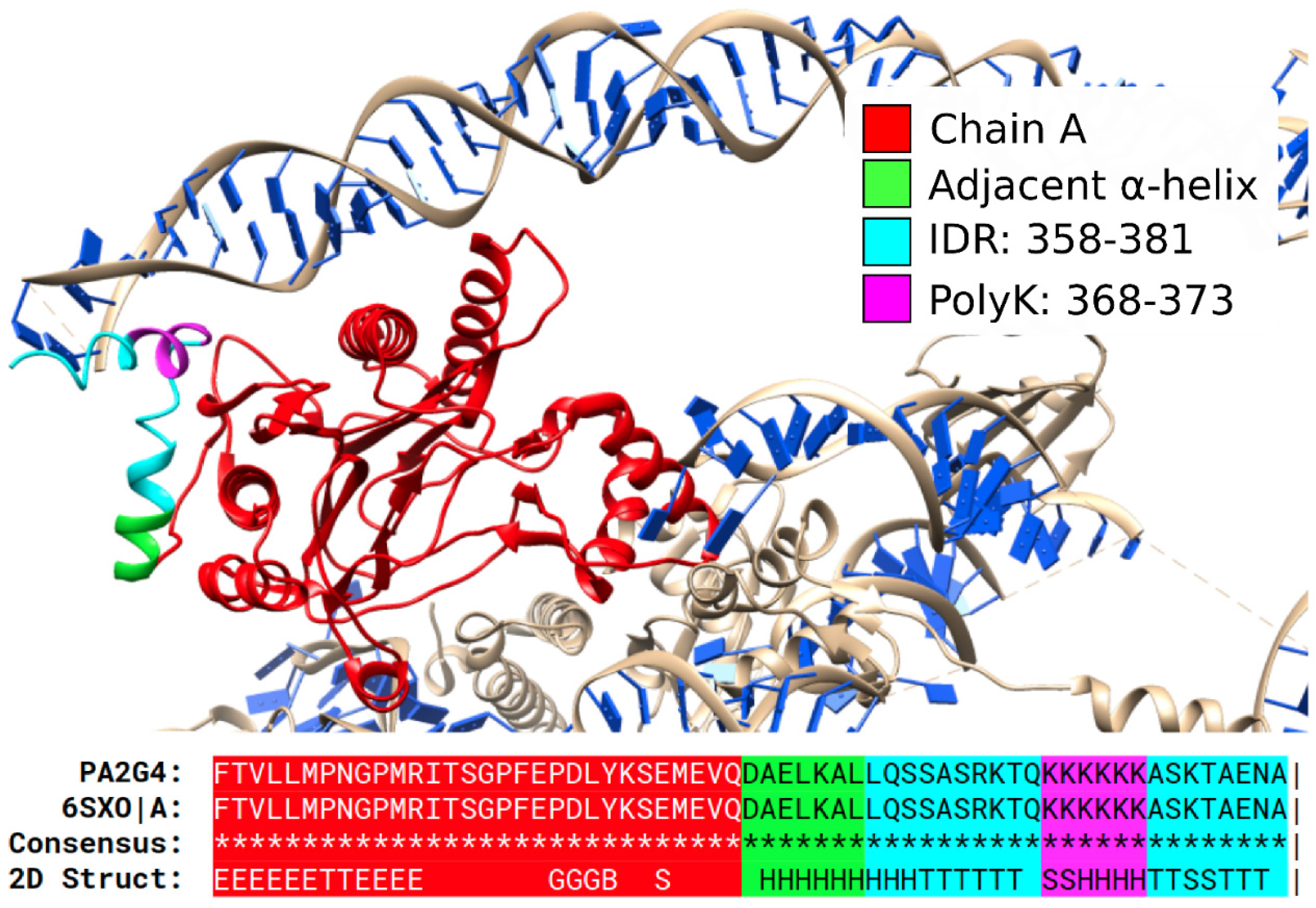
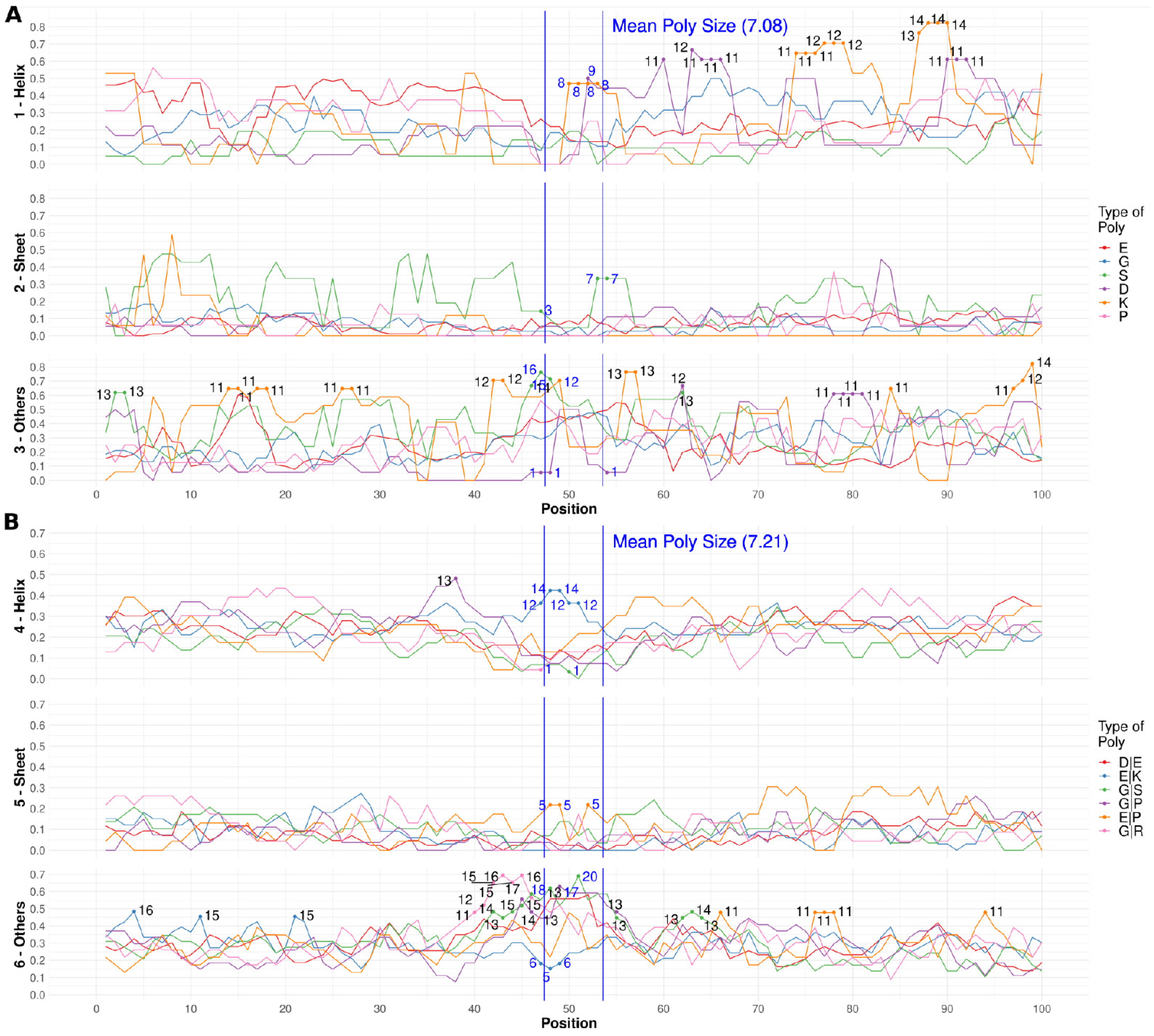
3.3. Secondary Structures from the PDB Associated with PolyX in IDRs
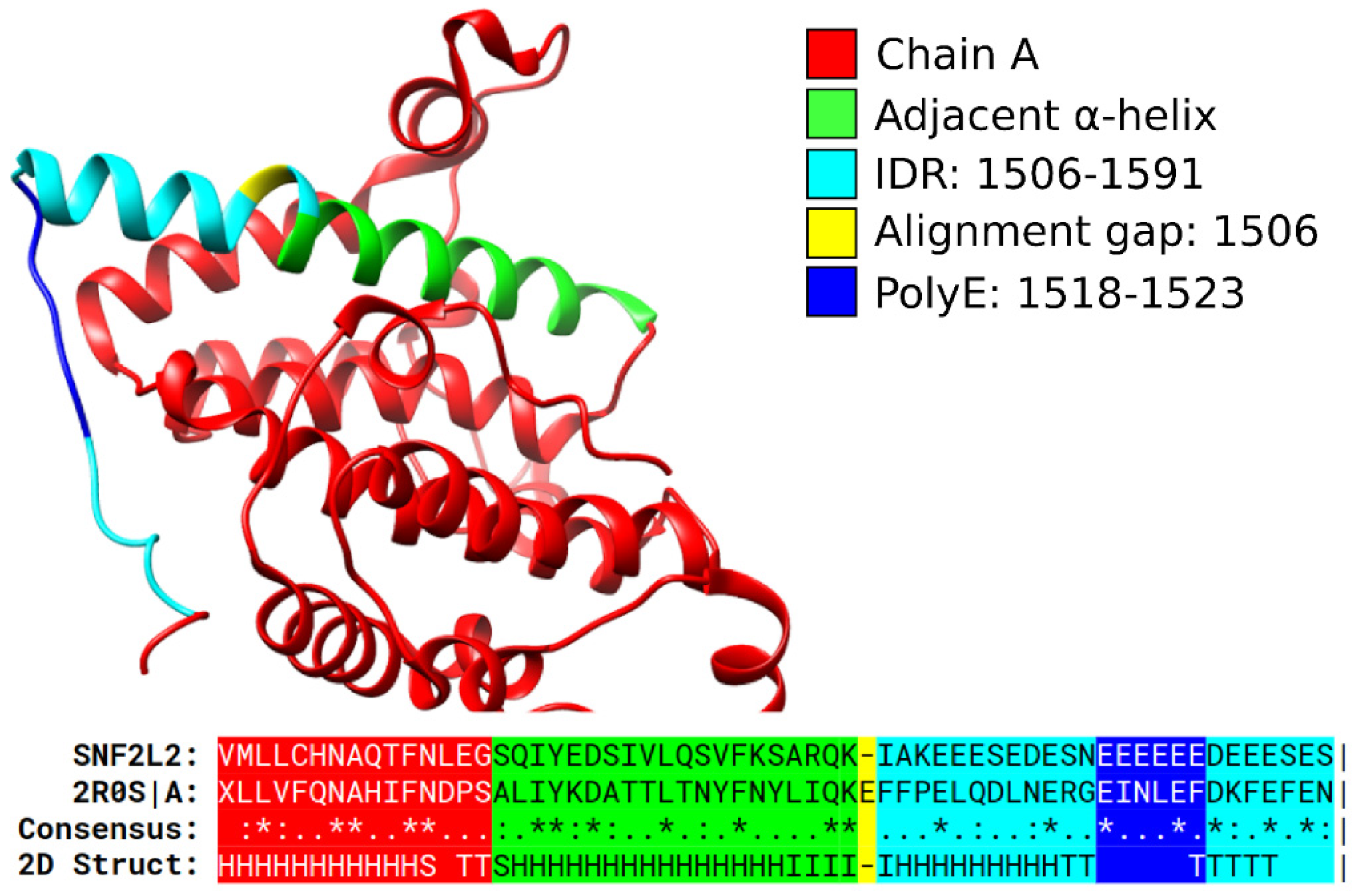
3.4. Secondary Structures from the PDB Associated with PolyXY in IDRs
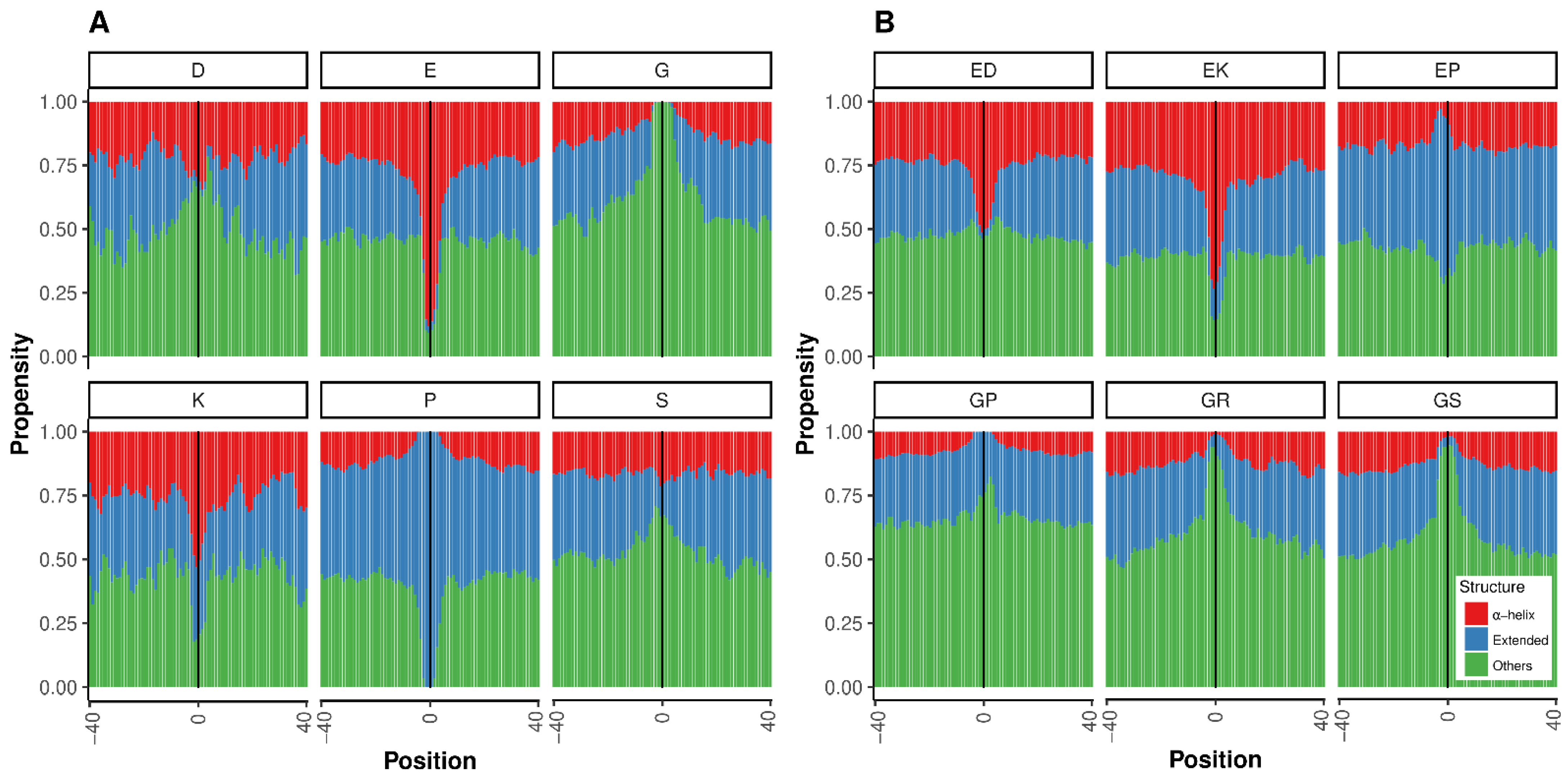
3.5. Calculation of Structural Propensities for PolyX and PolyXY in IDRs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tompa, P. Intrinsically Unstructured Proteins. Trends Biochem. Sci. 2002, 27, 527–533. [Google Scholar] [CrossRef]
- Peng, Z.; Yan, J.; Fan, X.; Mizianty, M.J.; Xue, B.; Wang, K.; Hu, G.; Uversky, V.N.; Kurgan, L. Exceptionally Abundant Exceptions: Comprehensive Characterization of Intrinsic Disorder in All Domains of Life. Cell. Mol. Life Sci. 2015, 72, 137–151. [Google Scholar] [CrossRef]
- Jorda, J.; Xue, B.; Uversky, V.N.; Kajava, A.V. Protein Tandem Repeats—the More Perfect, the Less Structured. FEBS J. 2010, 277, 2673–2682. [Google Scholar] [CrossRef]
- van der Lee, R.; Buljan, M.; Lang, B.; Weatheritt, R.J.; Daughdrill, G.W.; Dunker, A.K.; Fuxreiter, M.; Gough, J.; Gsponer, J.; Jones, D.T.; et al. Classification of intrinsically disordered regions and proteins. Chem. Rev. 2014, 114, 6589–6631. [Google Scholar] [CrossRef] [PubMed]
- Oldfield, C.J.; Dunker, A.K. Intrinsically Disordered Proteins and Intrinsically Disordered Protein Regions. Annu. Rev. Biochem. 2014, 83, 553–584. [Google Scholar] [CrossRef] [PubMed]
- Babu, M.M. The Contribution of Intrinsically Disordered Regions to Protein Function, Cellular Complexity, and Human Disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompa, P.; Schad, E.; Tantos, A.; Kalmar, L. Intrinsically disordered proteins: Emerging interaction specialists. Curr. Opin. Struct. Biol. 2015, 35, 49–59. [Google Scholar] [CrossRef]
- Mészáros, B.; Tompa, P.; Simon, I.; Dosztányi, Z. Molecular Principles of the Interactions of Disordered Proteins. J. Mol. Biol. 2007, 372, 549–561. [Google Scholar] [CrossRef]
- Bianchi, G.; Longhi, S.; Grandori, R.; Brocca, S. Relevance of Electrostatic Charges in Compactness, Aggregation, and Phase Separation of Intrinsically Disordered Proteins. Int. J. Mol. Sci. 2020, 21, 6208. [Google Scholar] [CrossRef]
- Kastano, K.; Mier, P.; Andrade-Navarro, M.A. The Role of Low Complexity Regions in Protein Interaction Modes: An Illustration in Huntingtin. Int. J. Mol. Sci. 2021, 22, 1727. [Google Scholar] [CrossRef]
- Mier, P.; Paladin, L.; Tamana, S.; Petrosian, S.; Hajdu-Soltész, B.; Urbanek, A.; Gruca, A.; Plewczynski, D.; Grynberg, M.; Bernadó, P.; et al. Disentangling the Complexity of Low Complexity Proteins. Brief. Bioinform. 2020, 21, 458–472. [Google Scholar] [CrossRef] [Green Version]
- Kumari, B.; Kumar, R.; Kumar, M. Low Complexity and Disordered Regions of Proteins Have Different Structural and Amino Acid Preferences. Mol. Biosyst. 2015, 11, 585–594. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Huang, J. Musashi-1: An Example of How Polyalanine Tracts Contribute to Self-Association in the Intrinsically Disordered Regions of Rna-Binding Proteins. Int. J. Mol. Sci. 2020, 21, 2289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanek, A.; Popovic, M.; Morató, A.; Estaña, A.; Elena-Real, C.A.; Mier, P.; Fournet, A.; Allemand, F.; Delbecq, S.; Andrade-Navarro, M.A.; et al. Flanking Regions Determine the Structure of the Poly-Glutamine in Huntingtin Through Mechanisms Common Among Glutamine-Rich Human Proteins. Structure 2020, 28, 733–746.e5. [Google Scholar] [CrossRef]
- Ramazzotti, M.; Monsellier, E.; Kamoun, C.; Degl’Innocenti, D.; Melki, R. Polyglutamine repeats are associated to specific sequence biases that are conserved among eukaryotes. PLoS ONE 2012, 7, e30824. [Google Scholar] [CrossRef] [Green Version]
- Escobedo, A.; Topal, B.; Kunze, M.; Aranda, J.; Chiesa, G.; Mungianu, D.; Bernardo-Seisdedos, G.; Eftekharzadeh, B.; Gairí, M.; Pierattelli, R.; et al. Side chain to main chain hydrogen bonds stabilize a polyglutamine helix in a transcription factor. Nat. Commun. 2019, 10, 2034. [Google Scholar] [CrossRef] [Green Version]
- Delucchi, M.; Schaper, E.; Sachenkova, O.; Elofsson, A.; Anisimova, M. A New Census of Protein Tandem Repeats and Their Relationship with Intrinsic Disorder. Genes 2020, 11, 407. [Google Scholar] [CrossRef] [Green Version]
- Cascarina, S.M.; Elder, M.R.; Ross, E.D. Atypical structural tendencies among low-complexity domains in the Protein Data Bank proteome. PLoS Comput. Biol. 2020, 16, e1007487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piovesan, D.; Necci, M.; Escobedo, N.; Monzon, A.M.; Hatos, A.; Mičetić, I.; Quaglia, F.; Paladin, L.; Ramasamy, P.; Dosztányi, Z.; et al. MobiDB: Intrinsically disordered proteins in 2021. Nucleic Acids Res. 2021, 49, D361–D367. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped Blast and Psi-Blast: A New Generation of Protein Database Search Programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Totzeck, F.; Andrade-Navarro, M.A.; Mier, P. The Protein Structure Context of PolyQ Regions. PLoS ONE 2017, 12, e0170801. [Google Scholar] [CrossRef] [PubMed]
- Mier, P.; Alanis-Lobato, G.; Andrade-Navarro, M.A. Context characterization of amino acid homorepeats using evolution, position, and order. Proteins 2017, 854, 709–719. [Google Scholar] [CrossRef] [PubMed]
- Estaña, A.; Barozet, A.; Mouhand, A.; Vaisset, M.; Zanon, C.; Fauret, P.; Sibille, N.; Bernado, P.; Cortes, J. Predicting Secondary Structure Propensities in Idps Using Simple Statistics from Three-Residue Fragments. J. Mol. Biol. 2020, 432, 5447–5459. [Google Scholar] [CrossRef]
- Chandonia, J.M.; Fox, N.K.; Brenner, S.E. SCOPe: Classification of large macromolecular structures in the structural classification of proteins-extended database. Nucleic Acids Res. 2019, 47, D475–D481. [Google Scholar] [CrossRef] [Green Version]
- Ozenne, V.; Schneider, R.; Yao, M.; Huang, J.R.; Salmon, L.; Zweckstetter, M.; Jensen, M.R.; Blackledge, M. Mapping the potential energy landscape of intrinsically disordered proteins at amino acid resolution. Am. Chem. Soc. 2012, 134, 15138–15148. [Google Scholar] [CrossRef] [Green Version]
- Estaña, A.; Sibille, N.; Delaforge, E.; Vaisset, M.; Cortés, J.; Bernadó, P. Realistic Ensemble Models of Intrinsically Disordered Proteins Using a Structure-Encoding Coil Database. Structure 2019, 27, 381–391.e2. [Google Scholar] [CrossRef] [Green Version]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Holehouse, A.S.; Das, R.K.; Ahad, J.N.; Richardson, M.O.; Pappu, R.V. CIDER: Resources to Analyze Sequence-Ensemble Relationships of Intrinsically Disordered Proteins. Biophys. J. 2017, 112, 16–21. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, R.K.; Pappu, R.V. Conformations of intrinsically disordered proteins are influenced by linear sequence distributions of oppositely charged residues. Proc. Natl. Acad. Sci. USA 2013, 110, 13392–13397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uversky, V.N. The Alphabet of Intrinsic Disorder: II. Various Roles of Glutamic Acid in Ordered and Intrinsically Disordered Proteins. Intrinsically Disord Proteins 2013, 1, e24684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chou, P.Y.; Fasman, G.D. Prediction of protein conformation. Biochemistry 1974, 13, 222–245. [Google Scholar] [CrossRef] [PubMed]
- Uversky, V.N. The Protein Disorder Cycle. Biophys. Rev. 2021, 13, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Da-tabase: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Wilhelm, P.; Lewandowski, B.; Trapp, N.; Wennemers, H. A crystal structure of an oligoproline PPII-helix, at last. J. Am. Chem. Soc. 2014, 136, 15829–15832. [Google Scholar] [CrossRef] [PubMed]
- Urbanek, A.; Morató, A.; Allemand, F.; Delaforge, E.; Fournet, A.; Popovic, M.; Delbecq, S.; Sibille, N.; Bernadó, P. A General Strategy to Access Structural Information at Atomic Resolution in Polyglutamine Homorepeats. Angew. Chem. Int. Ed. Engl. 2018, 57, 3598–3601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urbanek, A.; Elena-Real, C.A.; Popovic, M.; Morató, A.; Fournet, A.; Allemand, F.; Delbecq, S.; Sibille, N.; Bernadó, P. Site-Specific Isotopic Labeling SSIL: Access to High-Resolution Structural and Dynamic Information in Low-Complexity Proteins. Chembiochem 2020, 21, 769–775. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef] [Green Version]
- Tang, W.S.; Fawzi, N.L.; Mittal, J. Refining All-Atom Protein Force Fields for Polar-Rich, Prion-like, Low-Complexity Intrinsically Disordered Proteins. J. Phys. Chem. B 2020, 124, 9505–9512. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
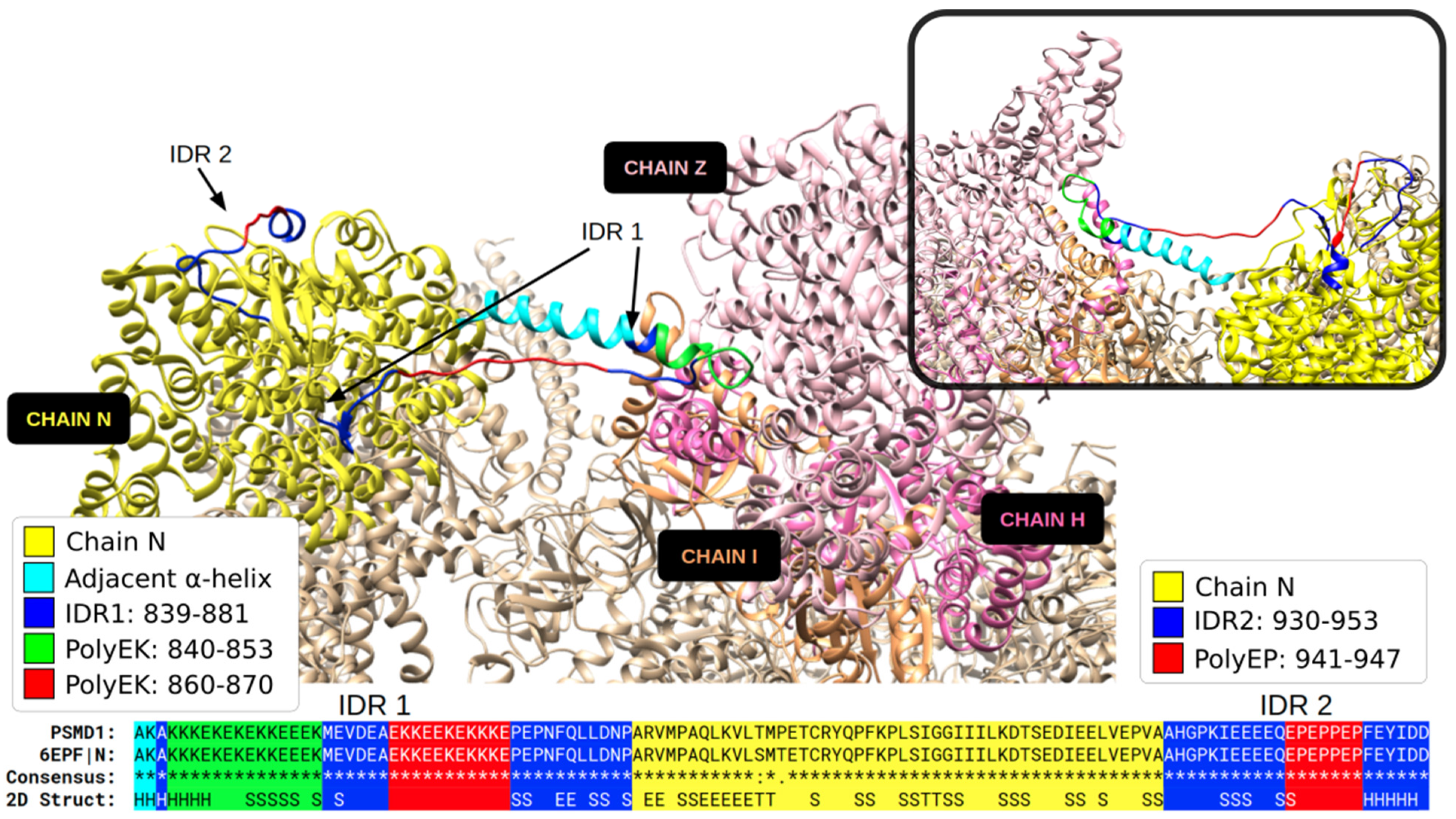{kind=link}
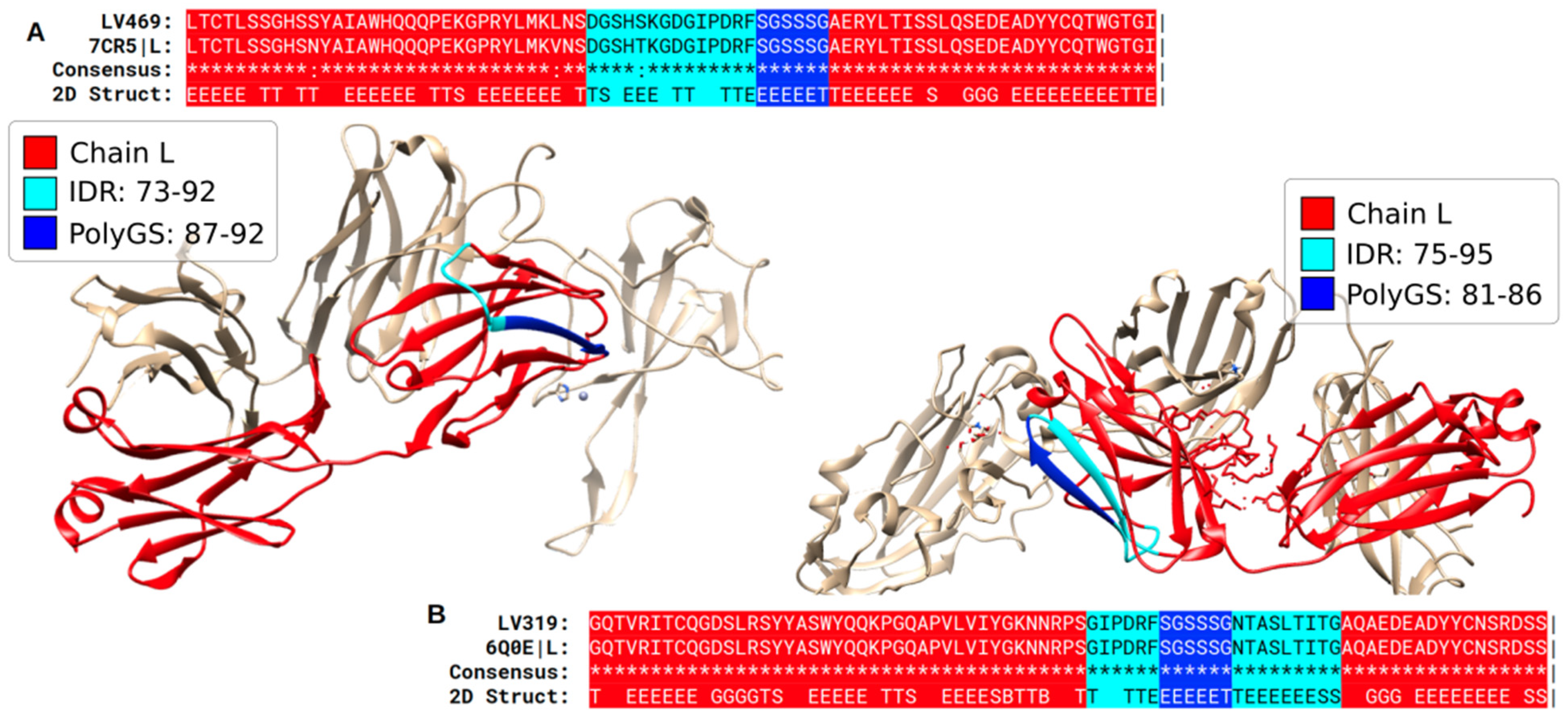{kind=link}
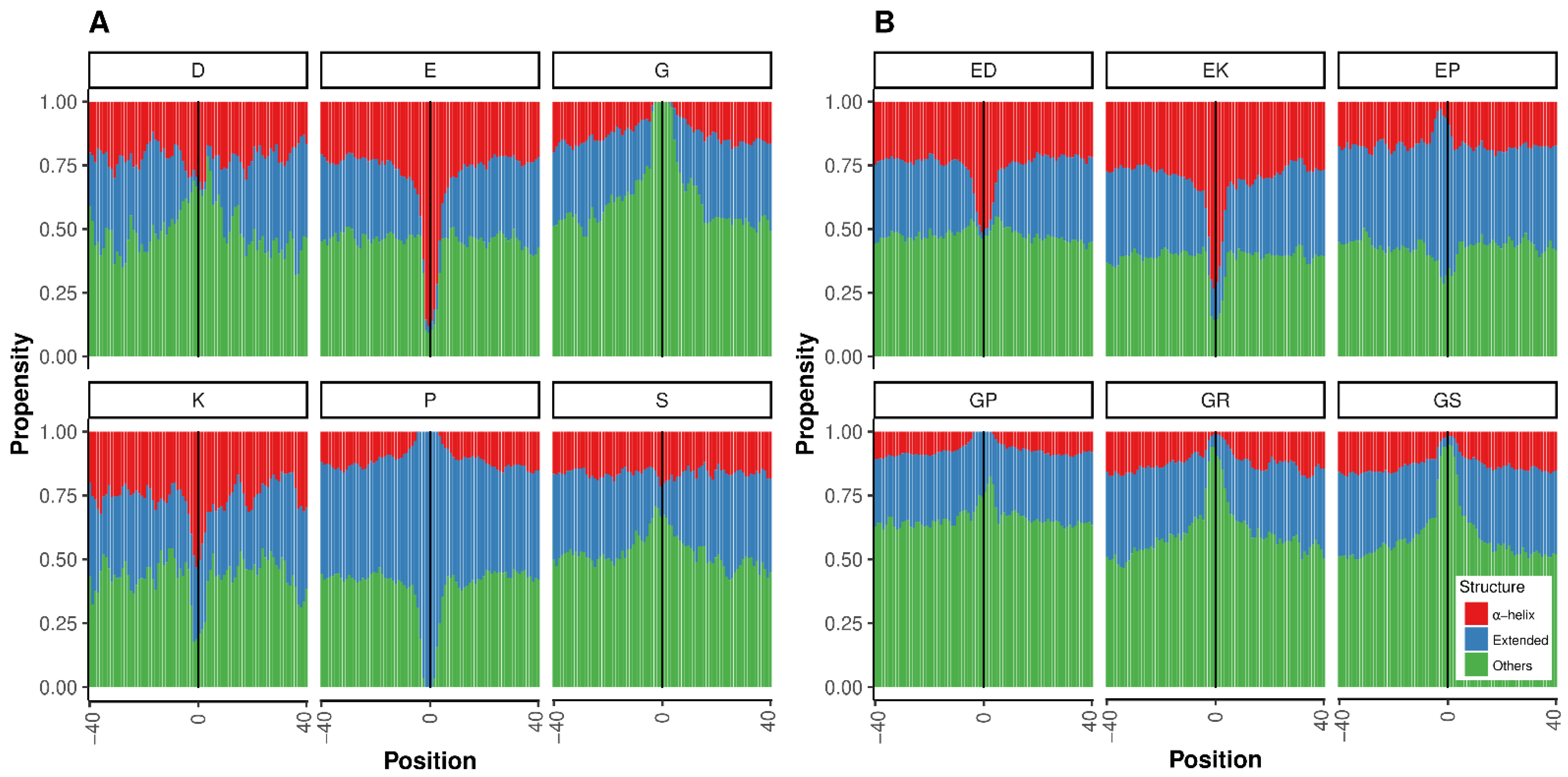{kind=link}
| polyX | Count | PDB Coverage | Rank in PDB-IDRs | Rank in IDRs | Rank in Proteome |
|---|---|---|---|---|---|
| polyE | 91 | 0.68 | 1 | 1 | 1 |
| polyG | 38 | 0.49 | 2 | 4 | 5 |
| polyS | 21 | 0.65 | 3 | 3 | 4 |
| polyD | 18 | 0.49 | 4 | 9 | 11 |
| polyK | 17 | 0.71 | 5 | 6 | 8 |
| polyP | 16 | 0.40 | 6 | 2 | 2 |
| polyXY | Count | PDB Coverage | Rank in PDB-IDRs | Rank in IDRs | Rank in Proteome |
|---|---|---|---|---|---|
| polyDE | 43 | 0.57 | 1 | 3 | 4 |
| polyEK | 33 | 0.47 | 2 | 13 | 10 |
| polyGS | 29 | 0.54 | 3 | 4 | 3 |
| polyGP | 27 | 0.30 | 4 | 1 | 2 |
| polyEP | 23 | 0.69 | 5 | 18 | 23 |
| polyGR | 23 | 0.46 | 6 | 12 | 16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves-Kulik, M.; Mier, P.; Kastano, K.; Cortés, J.; Bernadó, P.; Schmid, F.; Andrade-Navarro, M.A. Low Complexity Induces Structure in Protein Regions Predicted as Intrinsically Disordered. Biomolecules 2022, 12, 1098. https://doi.org/10.3390/biom12081098
Gonçalves-Kulik M, Mier P, Kastano K, Cortés J, Bernadó P, Schmid F, Andrade-Navarro MA. Low Complexity Induces Structure in Protein Regions Predicted as Intrinsically Disordered. Biomolecules. 2022; 12(8):1098. https://doi.org/10.3390/biom12081098
Chicago/Turabian StyleGonçalves-Kulik, Mariane, Pablo Mier, Kristina Kastano, Juan Cortés, Pau Bernadó, Friederike Schmid, and Miguel A. Andrade-Navarro. 2022. "Low Complexity Induces Structure in Protein Regions Predicted as Intrinsically Disordered" Biomolecules 12, no. 8: 1098. https://doi.org/10.3390/biom12081098
APA StyleGonçalves-Kulik, M., Mier, P., Kastano, K., Cortés, J., Bernadó, P., Schmid, F., & Andrade-Navarro, M. A. (2022). Low Complexity Induces Structure in Protein Regions Predicted as Intrinsically Disordered. Biomolecules, 12(8), 1098. https://doi.org/10.3390/biom12081098