Kat2a and Kat2b Acetyltransferase Activity Regulates Craniofacial Cartilage and Bone Differentiation in Zebrafish and Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Zebrafish
2.2. Genotyping of Zebrafish
2.3. Mice and Genotyping
2.4. Zebrafish kat2a and kat2b Mutant Generation
2.5. In Situ Hybridization, Alcian Blue Staining, and Alizarin Red Staining
- kat2a Fw—5′-GAATTCCCTACACCGAACTCT-3′, Rv—5′-ACTGACACTGGGAAGAAA CTA-3’
- kat2b Fw—5′-CACGTCATTGGAAACTC-3’, Rv—5′-CGCCTCATTCTTCTTTAC-3′
2.6. Immunohistochemistry and Western Blotting
2.7. Quantitative PCR
- Dlx1: Fw 5′-CCGGAGGTTCCAACAAACT-3′; Rv 5′-TCTGGAACCCATATCTTGACCT-3′, Probe #13.
- Twist1: Fw 5′-AGCTACGCCTTCTCCGTCT-3′; Rv 5′-TCCTTCTCTGGAAACAATGACA-3′, Probe #58.
- Runx2: Fw 5′-TCCACAAGGACAGAGTCAGATTAC-3′; Rv 5′-TGGCTCAGATAGGAGGGGTA-3′, Probe #60.
- Bmp4: Fw 5′-GAGGAGTTTCGATCACGAAGA-3′; Rv 5’-GCTCTGCCGAGGAGATCA-3′, Probe #89.
- Sox9: Fw 5′-TATCTTCAAGGCGCTGCAA-3′; Rv 5′-TCGGTTTTGGGAGTGGTG-3′, Probe #101.
- Col2a1: Fw 5′-AAGAACCAGACTGCCTCAAC-3′; Rv 5′-CCTTTGGCCCTAATTTTCG-3′, Probe #56.
2.8. Statistical Analysis
3. Results
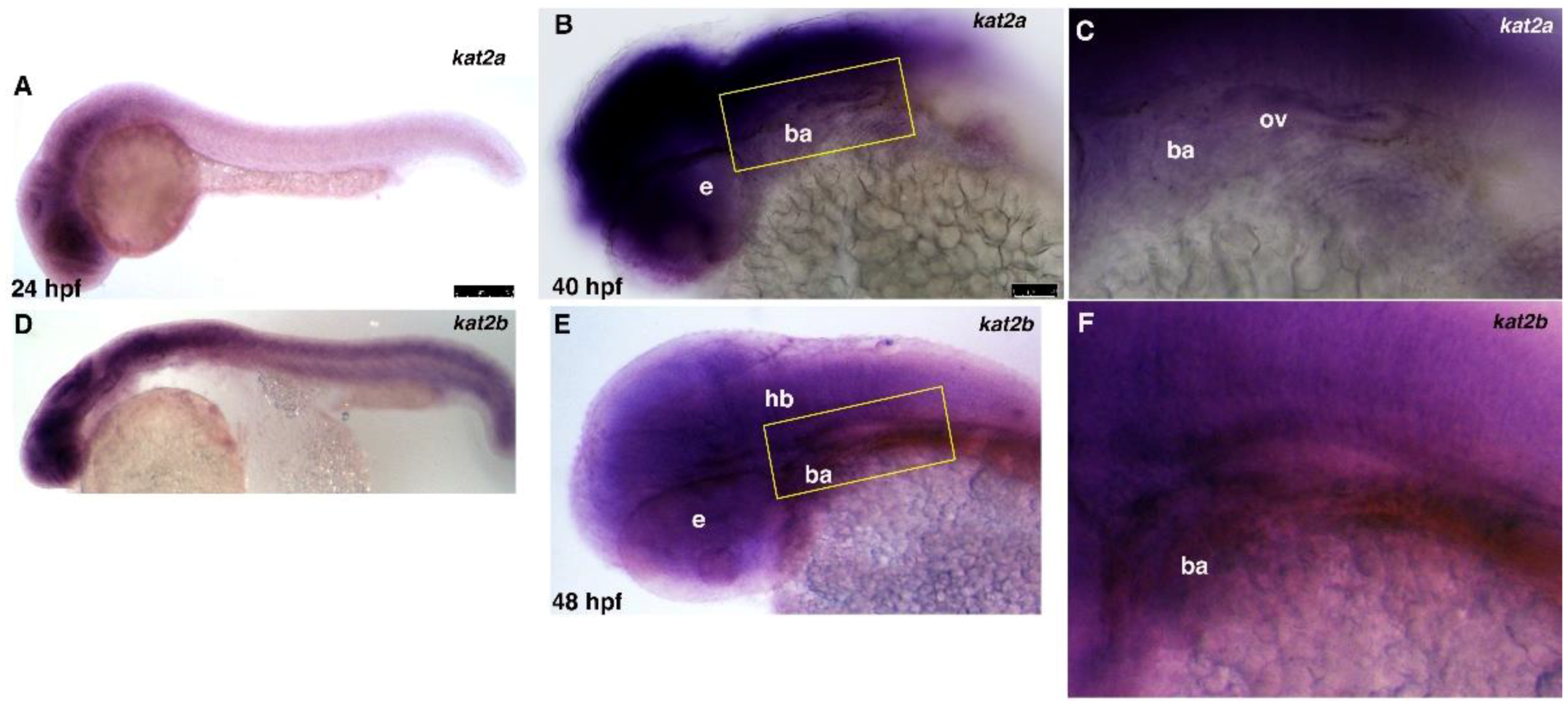
3.1. kat2a and kat2b Are Expressed Broadly during Zebrafish Embryogenesis
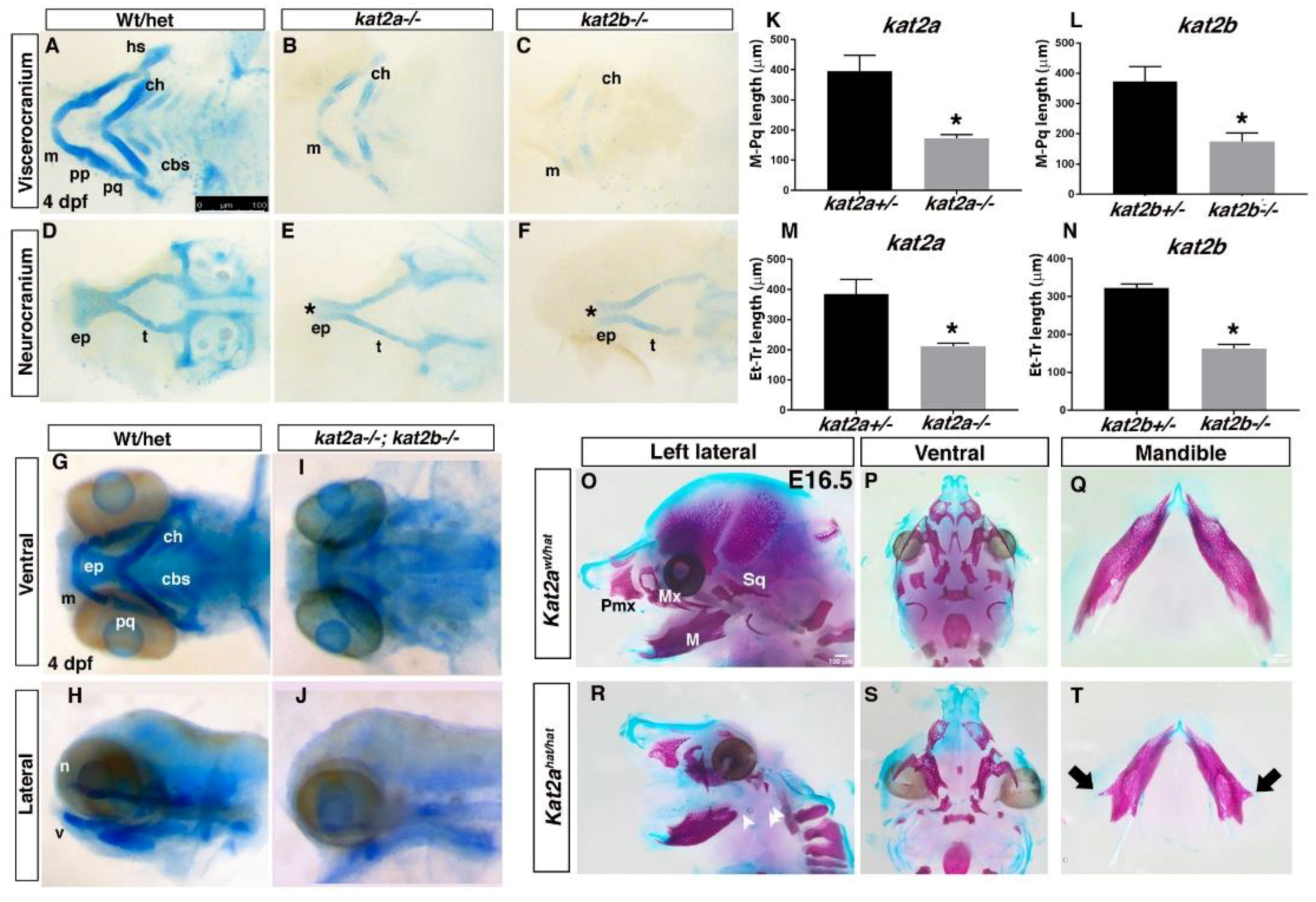
3.2. Loss of kat2a and kat2b in Zebrafish, and Kat2ahat/hat in Mice, Results in Craniofacial Cartilage and Bone Defects
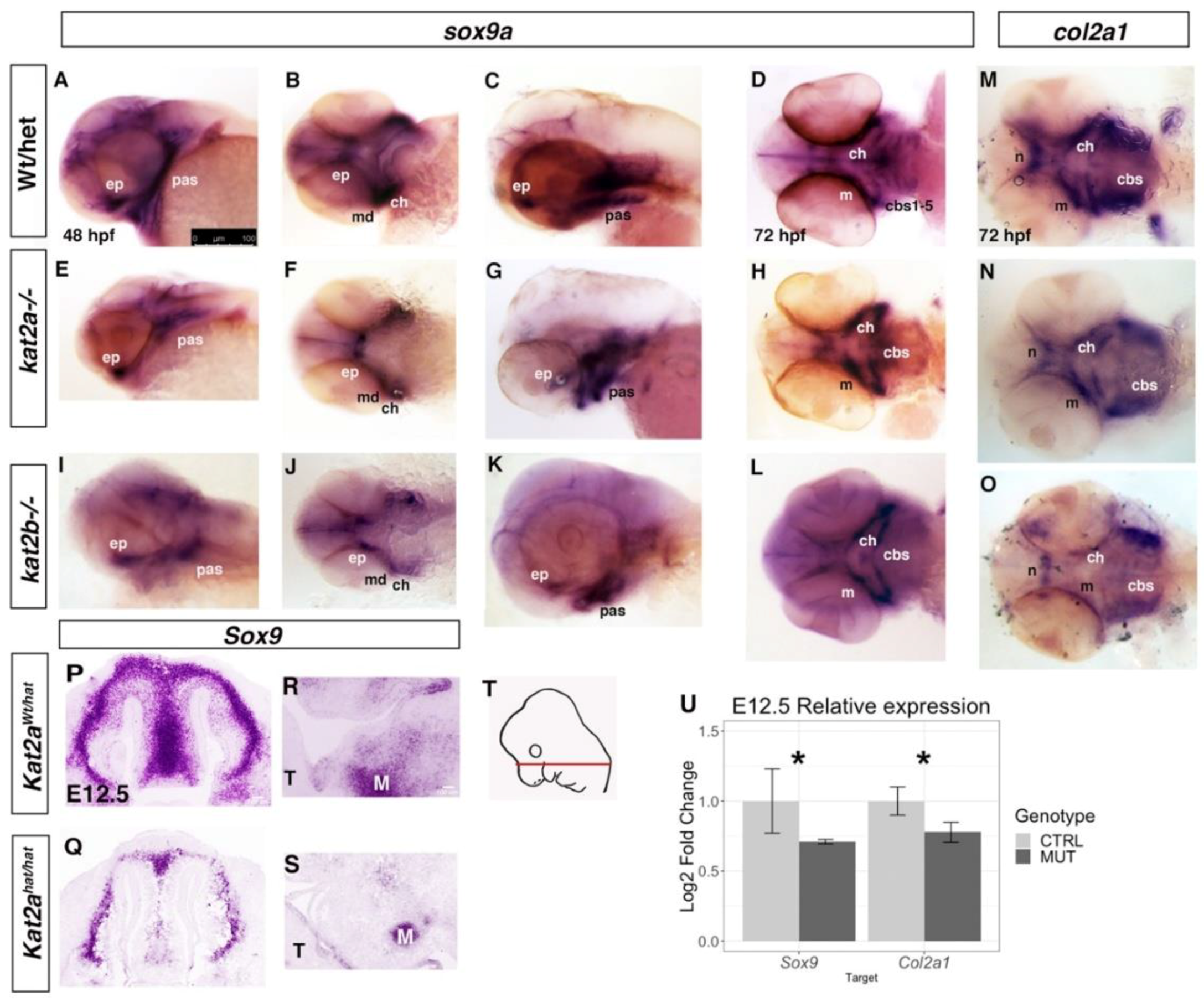
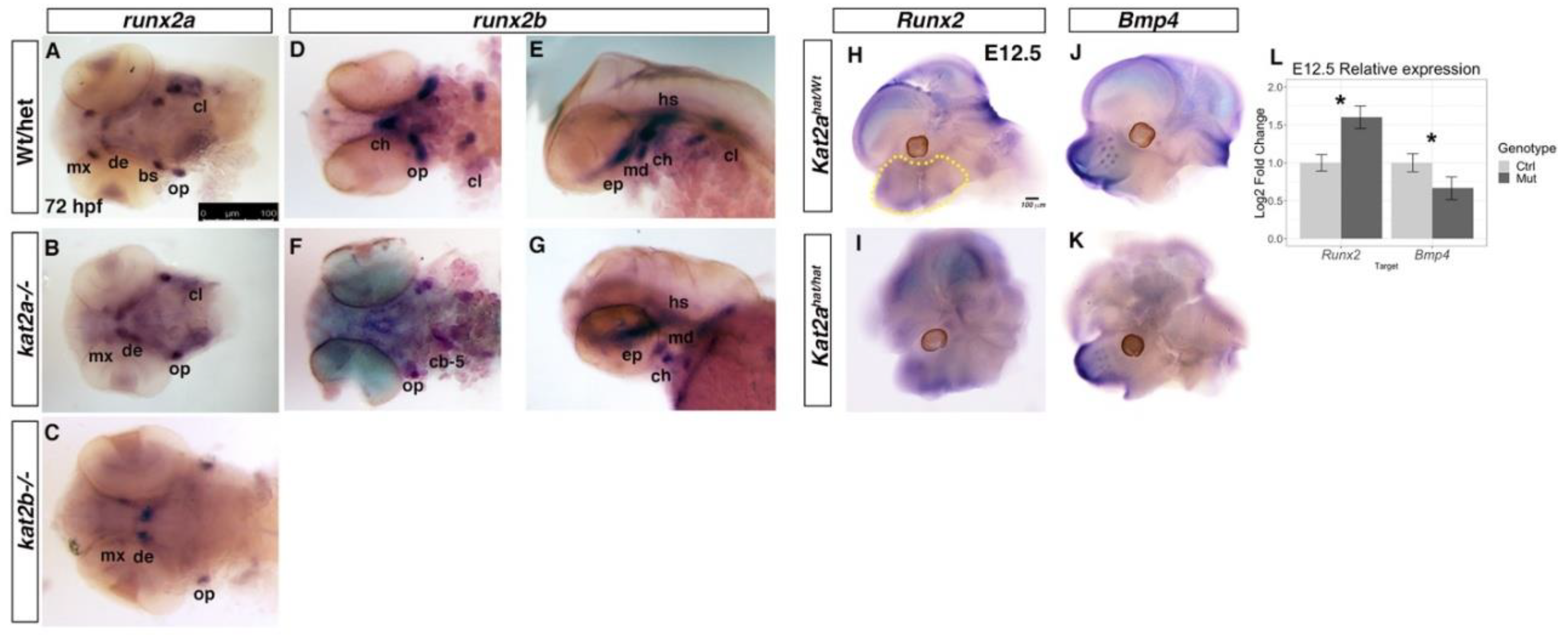
3.3. Cartilage and Bone Markers are Differentially Expressed in kat2a and kat2b Zebrafish and Kat2aHat/Hat Mouse Embryos
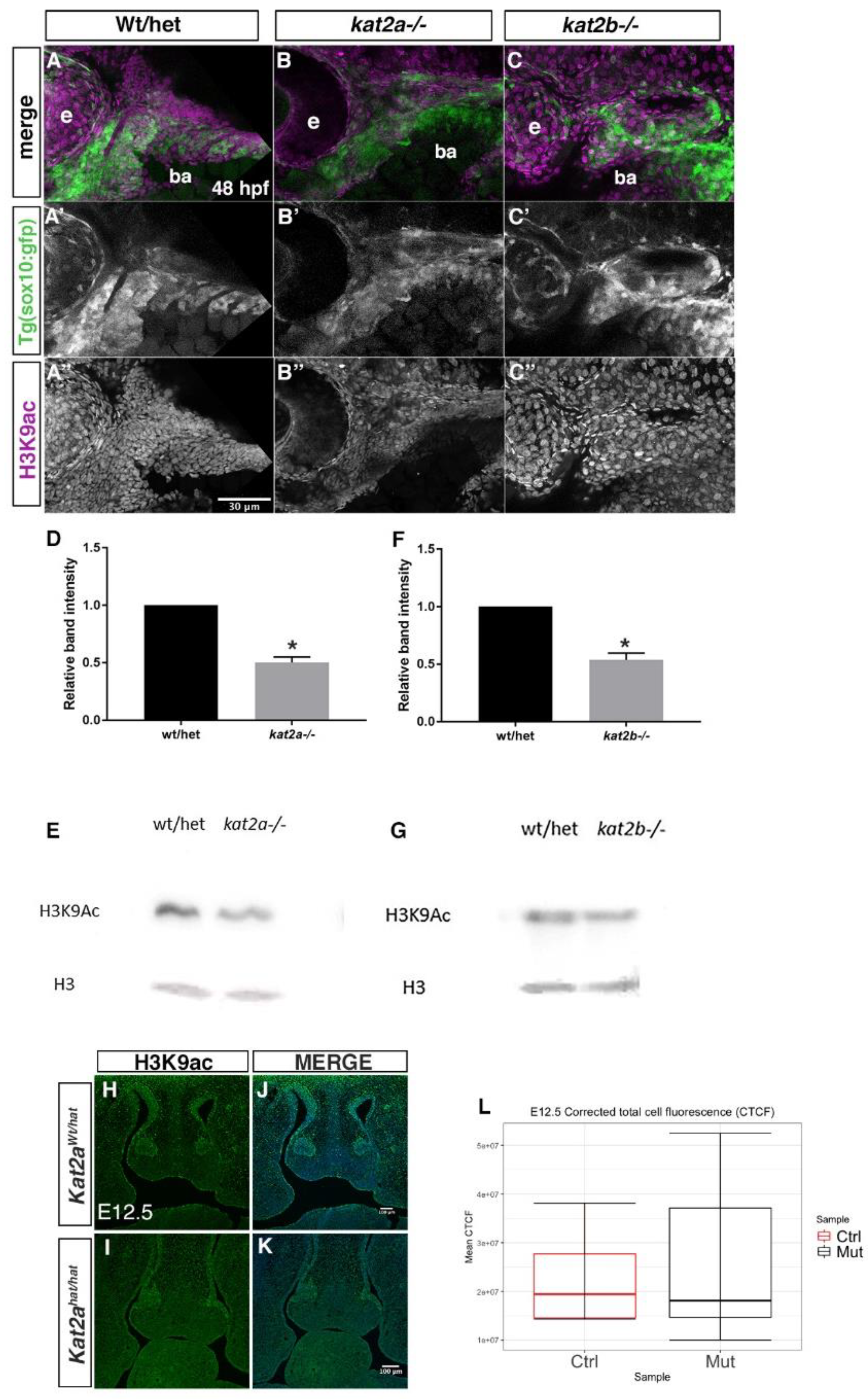
3.4. H3K9ac Is Reduced in Kat2a and Kat2b Mutants
4. Discussion
Supplemental Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chai, Y.; Maxson, R.E., Jr. Recent advances in craniofacial morphogenesis. Dev. Dyn. 2006, 235, 2353–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Douarin, N.M.; Brito, J.M.; Creuzet, S. Role of the neural crest in face and brain development. Brain Res. Rev. 2007, 55, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Le Douarin, N. The Neural Crest; Developmental and Cell Biology Series; Cambridge University Press: Cambridge, UK; New York, NY, USA, 1982; p. 272. [Google Scholar]
- Graham, A. Development of the pharyngeal arches. Am. J. Med. Genet. A 2003, 119A, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.F.; Kimmel, C.B. Segment and cell type lineage restrictions during pharyngeal arch development in the zebrafish embryo. Development 1994, 120, 483–494. [Google Scholar] [PubMed]
- Raible, D.W.; Eisen, J.S. Regulative interactions in zebrafish neural crest. Development 1996, 122, 501–507. [Google Scholar] [PubMed]
- Raible, D.W.; Wood, A.; Hodsdon, W.; Henion, P.D.; Weston, J.A.; Eisen, J.S. Segregation and early dispersal of neural crest cells in the embryonic zebrafish. Dev. Dyn. 1992, 195, 29–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmona-Fontaine, C.; Matthews, H.K.; Kuriyama, S.; Moreno, M.; Dunn, G.A.; Parsons, M.; Stern, C.D.; Mayor, R. Contact inhibition of locomotion in vivo controls neural crest directional migration. Nature 2008, 456, 957–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kague, E.; Gallagher, M.; Burke, S.; Parsons, M.; Franz-Odendaal, T.; Fisher, S. Skeletogenic fate of zebrafish cranial and trunk neural crest. PLoS ONE 2012, 7, e47394. [Google Scholar] [CrossRef] [PubMed]
- Theveneau, E.; Mayor, R. Neural crest delamination and migration: From epithelium-to-mesenchyme transition to collective cell migration. Dev. Biol. 2012, 366, 34–54. [Google Scholar] [CrossRef] [PubMed]
- Seberg, H.E.; Van Otterloo, E.; Cornell, R.A. Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanoma Res. 2017, 30, 454–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisen, J.S.; Weston, J.A. Development of the neural crest in the zebrafish. Dev. Biol. 1993, 159, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Cordero, D.R.; Brugmann, S.; Chu, Y.; Bajpai, R.; Jame, M.; Helms, J.A. Cranial neural crest cells on the move: Their roles in craniofacial development. Am. J. Med. Genet. A 2011, 155A, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Santagati, F.; Rijli, F.M. Cranial neural crest and the building of the vertebrate head. Nat. Rev. Neurosci. 2003, 4, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.C.; Nichols, J.T.; Yan, Y.L.; Leonard, I.F.; BreMiller, R.A.; Amacher, S.L.; Postlethwait, J.H.; Kimmel, C.B. Pharyngeal morphogenesis requires fras1-itga8-dependent epithelial-mesenchymal interaction. Dev. Biol. 2016, 416, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Bronner, M.E.; LeDouarin, N.M. Development and evolution of the neural crest: An overview. Dev. Biol. 2012, 366, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Munoz, W.A.; Trainor, P.A. Neural crest cell evolution: How and when did a neural crest cell become a neural crest cell. Curr. Top. Dev. Biol. 2015, 111, 3–26. [Google Scholar] [PubMed]
- Rothstein, M.; Bhattacharya, D.; Simoes-Costa, M. The molecular basis of neural crest axial identity. Dev. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, S.; Diaz, R.; Trainor, P.A. Signals and switches in Mammalian neural crest cell differentiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008326. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Iseki, S.; Maxson, R.E.; Sucov, H.M.; Morriss-Kay, G.M. Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 2002, 241, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Vivatbutsiri, P.; Morriss-Kay, G.; Saga, Y.; Iseki, S. Cell lineage in mammalian craniofacial mesenchyme. Mech. Dev. 2008, 125, 797–808. [Google Scholar] [CrossRef] [PubMed]
- Mork, L.; Crump, G. Zebrafish Craniofacial Development: A Window into Early Patterning. Curr. Top. Dev. Biol. 2015, 115, 235–269. [Google Scholar] [PubMed]
- De Beer, G. The Development of the Vertebrate Skull; Hall, B.K., Hanken, J., Eds.; University of Chicago Press: Chicago, IL, USA, 1985; Volume xivi, p. 554. [Google Scholar]
- Bird, N.C.; Mabee, P.M. Developmental morphology of the axial skeleton of the zebrafish, Danio rerio (Ostariophysi: Cyprinidae). Dev. Dyn. 2003, 228, 337–357. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Miller, C.T.; Kruze, G.; Ullmann, B.; BreMiller, R.A.; Larison, K.D.; Snyder, H.C. The shaping of pharyngeal cartilages during early development of the zebrafish. Dev. Biol. 1998, 203, 245–263. [Google Scholar] [CrossRef] [PubMed]
- Schilling, T.F.; Kimmel, C.B. Musculoskeletal patterning in the pharyngeal segments of the zebrafish embryo. Development 1997, 124, 2945–2960. [Google Scholar] [PubMed]
- Mariotti, M.; Carnovali, M.; Banfi, G. Danio rerio: The Janus of the bone from embryo to scale. Clin. Cases Miner. Bone Metab. 2015, 12, 188–194. [Google Scholar] [CrossRef] [PubMed]
- Cubbage, C.C.; Mabee, P.M. Development of the cranium and paired fins in the zebrafish Danio rerio (Ostariophysi, Cyprinidae). J. Morphol. 1996, 229, 121–160. [Google Scholar] [CrossRef]
- Parada, C.; Chai, Y. Mandible and Tongue Development. Curr. Top. Dev. Biol. 2015, 115, 31–58. [Google Scholar] [PubMed] [Green Version]
- Couly, G.F.; Coltey, P.M.; Le Douarin, N.M. The triple origin of skull in higher vertebrates: A study in quail-chick chimeras. Development 1993, 117, 409–429. [Google Scholar] [PubMed]
- Le Douarin, N.; Kalcheim, C. The Neural Crest, 2nd ed.; Developmental and Cell Biology Series; Cambridge University Press: Cambridge, UK; New York, NY, USA, 1999; Volume xxiii, 445p. [Google Scholar]
- Fraser, S.; Keynes, R.; Lumsden, A. Segmentation in the chick embryo hindbrain is defined by cell lineage restrictions. Nature 1990, 344, 431–435. [Google Scholar] [CrossRef] [PubMed]
- Le Lievre, C.S. Participation of neural crest-derived cells in the genesis of the skull in birds. J. Embryol. Exp. Morphol. 1978, 47, 17–37. [Google Scholar] [PubMed]
- Lumsden, A.; Sprawson, N.; Graham, A. Segmental origin and migration of neural crest cells in the hindbrain region of the chick embryo. Development 1991, 113, 1281–1291. [Google Scholar] [PubMed]
- Osumi-Yamashita, N.; Ninomiya, Y.; Doi, H.; Eto, K. The contribution of both forebrain and midbrain crest cells to the mesenchyme in the frontonasal mass of mouse embryos. Dev. Biol. 1994, 164, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Eberhart, J.K. The midline, oral ectoderm, and the arch-0 problem. Integr. Comp. Biol. 2008, 48, 668–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, N.; Javidan, Y.; Nelson, S.; Carney, T.J.; Kelsh, R.N.; Schilling, T.F. Hedgehog signaling is required for cranial neural crest morphogenesis and chondrogenesis at the midline in the zebrafish skull. Development 2005, 132, 3977–3988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartz, M.E.; Sheehan-Rooney, K.; Dixon, M.J.; Eberhart, J.K. Examination of a palatogenic gene program in zebrafish. Dev. Dyn. 2011, 240, 2204–2220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jugessur, A.; Shi, M.; Gjessing, H.K.; Lie, R.T.; Wilcox, A.J.; Weinberg, C.R.; Christensen, K.; Boyles, A.L.; Daack-Hirsch, S.; Nguyen, T.T.; et al. Maternal genes and facial clefts in offspring: A comprehensive search for genetic associations in two population-based cleft studies from Scandinavia. PLoS ONE 2010, 5, e11493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campeau, P.M.; Lu, J.T.; Dawson, B.C.; Fokkema, I.F.; Robertson, S.P.; Gibbs, R.A.; Lee, B.H. The KAT6B-related disorders genitopatellar syndrome and Ohdo/SBBYS syndrome have distinct clinical features reflecting distinct molecular mechanisms. Hum. Mutat. 2012, 33, 1520–1525. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, P.M.; Neitzel, L.R.; Quintana, A.M.; Geiger, E.A.; Zackai, E.H.; Clouthier, D.E.; Artinger, K.B.; Ming, J.E.; Shaikh, T.H. Kabuki syndrome genes KMT2D and KDM6A: Functional analyses demonstrate critical roles in craniofacial, heart and brain development. Hum. Mol. Genet. 2015, 24, 4443–4453. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Layman, W.S.; Martin, D.M.; Scacheri, P.C. Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am. J. Med. Genet. A 2010, 152A, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xiao, A. Epigenetic regulation in neural crest development. Birth Defects Res. A Clin. Mol. Teratol. 2011, 91, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Rada-Iglesias, A.; Bajpai, R.; Prescott, S.; Brugmann, S.A.; Swigut, T.; Wysocka, J. Epigenomic annotation of enhancers predicts transcriptional regulators of human neural crest. Cell Stem Cell 2012, 11, 633–648. [Google Scholar] [CrossRef] [PubMed]
- Minoux, M.; Holwerda, S.; Vitobello, A.; Kitazawa, T.; Kohler, H.; Stadler, M.B.; Rijli, F.M. Gene bivalency at Polycomb domains regulates cranial neural crest positional identity. Science 2017, 355, eaal2913. [Google Scholar] [CrossRef] [PubMed]
- Bolande, R.P. Neurocristopathy: Its growth and development in 20 years. Pediatr. Pathol. Lab. Med. 1997, 17, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Kirby, R.S.; Mai, C.T. Population-based birth defects surveillance systems in the United States. Birth Defects Res. A Clin. Mol. Teratol. 2006, 76, 835–836. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zang, C.; Rosenfeld, J.A.; Schones, D.E.; Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Peng, W.; Zhang, M.Q.; et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat. Genet. 2008, 40, 897–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agalioti, T.; Chen, G.; Thanos, D. Deciphering the transcriptional histone acetylation code for a human gene. Cell 2002, 111, 381–392. [Google Scholar] [CrossRef]
- Gates, L.A.; Shi, J.; Rohira, A.D.; Feng, Q.; Zhu, B.; Bedford, M.T.; Sagum, C.A.; Jung, S.Y.; Qin, J.; Tsai, M.J.; et al. Acetylation on histone H3 lysine 9 mediates a switch from transcription initiation to elongation. J. Biol. Chem. 2017, 292, 14456–14472. [Google Scholar] [CrossRef] [PubMed]
- Karmodiya, K.; Krebs, A.R.; Oulad-Abdelghani, M.; Kimura, H.; Tora, L. H3K9 and H3K14 acetylation co-occur at many gene regulatory elements, while H3K14ac marks a subset of inactive inducible promoters in mouse embryonic stem cells. BMC Genom. 2012, 13, 424. [Google Scholar] [CrossRef] [PubMed]
- Vastenhouw, N.L.; Schier, A.F. Bivalent histone modifications in early embryogenesis. Curr. Opin. Cell Biol. 2012, 24, 374–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guenther, M.G.; Levine, S.S.; Boyer, L.A.; Jaenisch, R.; Young, R.A. A chromatin landmark and transcription initiation at most promoters in human cells. Cell 2007, 130, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Hosey, A.M.; Chaturvedi, C.P.; Brand, M. Crosstalk between histone modifications maintains the developmental pattern of gene expression on a tissue-specific locus. Epigenetics 2010, 5, 273–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, Z.; Tora, L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 2007, 26, 5341–5357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Geng, R.; Guo, X.; Yuan, R.; Zhou, X.; Zhong, Y.; Huo, Y.; Zhou, M.; Shen, Q.; Li, Y.; et al. PCAF/GCN5-Mediated Acetylation of RPA1 Promotes Nucleotide Excision Repair. Cell Rep. 2017, 20, 1997–2009. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Yamauchi, J.; Kuwata, T.; Tamura, T.; Yamashita, T.; Bae, N.; Westphal, H.; Ozato, K.; Nakatani, Y. Distinct but overlapping roles of histone acetylase PCAF and of the closely related PCAF-B/GCN5 in mouse embryogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 11303–11306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, Q.; Yu, L.R.; Wang, L.; Zhang, Z.; Kasper, L.H.; Lee, J.E.; Wang, C.; Brindle, P.K.; Dent, S.Y.; Ge, K. Distinct roles of GCN5/PCAF-mediated H3K9ac and CBP/p300-mediated H3K18/27ac in nuclear receptor transactivation. EMBO J. 2011, 30, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Wang, C.; Kuang, X.; Feng, X.; Sartorelli, V.; Ying, H.; Ge, K.; Dent, S.Y. Gcn5 and PCAF regulate PPARgamma and Prdm16 expression to facilitate brown adipogenesis. Mol. Cell. Biol. 2014, 34, 3746–3753. [Google Scholar] [CrossRef] [PubMed]
- Koutsogiannouli, E.A.; Wagner, N.; Hader, C.; Pinkerneil, M.; Hoffmann, M.J.; Schulz, W.A. Differential Effects of Histone Acetyltransferase GCN5 or PCAF Knockdown on Urothelial Carcinoma Cells. Int. J. Mol. Sci. 2017, 18, 1449. [Google Scholar] [CrossRef] [PubMed]
- Bondy-Chorney, E.; Denoncourt, A.; Sai, Y.; Downey, M. Non-histone targets of KAT2A and KAT2B implicated in cancer biology. Biochem. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Wilde, J.J.; Siegenthaler, J.A.; Dent, S.Y.; Niswander, L.A. Diencephalic Size Is Restricted by a Novel Interplay Between GCN5 Acetyltransferase Activity and Retinoic Acid Signaling. J. Neurosci. 2017, 37, 2565–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.; Chen, Y.; Underhill, T.M.; Mymryk, J.S.; Torchia, J. The coactivator p/CIP/SRC-3 facilitates retinoic acid receptor signaling via recruitment of GCN5. J. Biol. Chem. 2003, 278, 39402–39412. [Google Scholar] [CrossRef] [PubMed]
- Kurooka, H.; Honjo, T. Functional interaction between the mouse notch1 intracellular region and histone acetyltransferases PCAF and GCN5. J. Biol. Chem. 2000, 275, 17211–17220. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Edmondson, D.G.; Evrard, Y.A.; Wakamiya, M.; Behringer, R.R.; Roth, S.Y. Loss of Gcn5l2 leads to increased apoptosis and mesodermal defects during mouse development. Nat. Genet. 2000, 26, 229–232. [Google Scholar] [CrossRef] [PubMed]
- Bu, P.; Evrard, Y.A.; Lozano, G.; Dent, S.Y. Loss of Gcn5 acetyltransferase activity leads to neural tube closure defects and exencephaly in mouse embryos. Mol. Cell. Biol. 2007, 27, 3405–3416. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.K.; Aparicio-Sanchez, J.J.; Buxton, S.; Ketley, A.; Mohamed, T.; Rutland, C.S.; Loughna, S.; Brook, J.D. Acetylation of TBX5 by KAT2B and KAT2A regulates heart and limb development. J. Mol. Cell. Cardiol. 2018, 114, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Westerfield, M. The Zebrafish Book: A Guide for the Laboratory Use of Zebrafish (Danio Rerio); University of Oregon Press: Eugene, OR, USA, 1995. [Google Scholar]
- Dutton, J.R.; Antonellis, A.; Carney, T.J.; Rodrigues, F.S.; Pavan, W.J.; Ward, A.; Kelsh, R.N. An evolutionarily conserved intronic region controls the spatiotemporal expression of the transcription factor Sox10. BMC Dev. Biol. 2008, 8, 105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Doyle, E.L.; Booher, N.J.; Standage, D.S.; Voytas, D.F.; Brendel, V.P.; Vandyk, J.K.; Bogdanove, A.J. TAL Effector-Nucleotide Targeter (TALE-NT) 2.0: Tools for TAL effector design and target prediction. Nucleic Acids Res. 2012, 40, W117–W122. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed]
- Bedell, V.M.; Wang, Y.; Campbell, J.M.; Poshusta, T.L.; Starker, C.G.; Krug, R.G., 2nd; Tan, W.; Penheiter, S.G.; Ma, A.C.; Leung, A.Y.; et al. In vivo genome editing using a high-efficiency TALEN system. Nature 2012, 491, 114–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doudna, J.A.; Charpentier, E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Akhmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thisse, B.; Heyer, V.; Lux, A.; Alunni, V.; Degrave, A.; Seiliez, I.; Kirchner, J.; Parkhill, J.P.; Thisse, C. Spatial and temporal expression of the zebrafish genome by large-scale in situ hybridization screening. Methods Cell Biol. 2004, 77, 505–519. [Google Scholar] [PubMed]
- Johnson, C.W.; Hernandez-Lagunas, L.; Feng, W.; Melvin, V.S.; Williams, T.; Artinger, K.B. Vgll2a is required for neural crest cell survival during zebrafish craniofacial development. Dev. Biol. 2011, 357, 269–281. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.L.; Clouthier, D.E.; Artinger, K.B. Redundant roles of PRDM family members in zebrafish craniofacial development. Dev. Dyn. 2013, 242, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.L.; Lee, D.; Luo, R.; Henion, P.D.; Halpern, M.E. Genes dependent on zebrafish cyclops function identified by AFLP differential gene expression screen. Genesis 2000, 26, 86–97. [Google Scholar] [CrossRef]
- Walker, M.B.; Kimmel, C.B. A two-color acid-free cartilage and bone stain for zebrafish larvae. Biotech. Histochem. 2007, 82, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Manley, N.R.; Condie, B.G. Whole mount in situ hybridization of E8.5 to E11.5 mouse embryos. J. Vis. Exp. 2011, 2797. [Google Scholar] [CrossRef] [PubMed]
- Asp, J.; Abramsson, A.; Betsholtz, C. Nonradioactive in situ hybridization on frozen sections and whole mounts. Methods Mol. Biol. 2006, 326, 89–102. [Google Scholar] [PubMed]
- McLeod, M.J. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology 1980, 22, 299–301. [Google Scholar] [CrossRef] [PubMed]
- Barske, L.; Askary, A.; Zuniga, E.; Balczerski, B.; Bump, P.; Nichols, J.T.; Crump, J.G. Competition between Jagged-Notch and Endothelin1 Signaling Selectively Restricts Cartilage Formation in the Zebrafish Upper Face. PLoS Genet. 2016, 12, e1005967. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, Y.; Jin, C.; Zhang, M.; Tang, F.; Zhou, Y. Histone Acetyltransferase GCN5 Regulates Osteogenic Differentiation of Mesenchymal Stem Cells by Inhibiting NF-kappaB. J. Bone Miner. Res. 2016, 31, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Sauka-Spengler, T.; Bronner-Fraser, M. Evolution of the neural crest viewed from a gene regulatory perspective. Genesis 2008, 46, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malatesta, M.; Steinhauer, C.; Mohammad, F.; Pandey, D.P.; Squatrito, M.; Helin, K. Histone acetyltransferase PCAF is required for Hedgehog-Gli-dependent transcription and cancer cell proliferation. Cancer Res. 2013, 73, 6323–6333. [Google Scholar] [CrossRef] [PubMed]
- Gai, X.; Tu, K.; Li, C.; Lu, Z.; Roberts, L.R.; Zheng, X. Histone acetyltransferase PCAF accelerates apoptosis by repressing a GLI1/BCL2/BAX axis in hepatocellular carcinoma. Cell Death Dis. 2015, 6, e1712. [Google Scholar] [CrossRef] [PubMed]
- Brewster, R.; Lee, J.; Ruiz i Altaba, A. Gli/Zic factors pattern the neural plate by defining domains of cell differentiation. Nature 1998, 393, 579–583. [Google Scholar] [CrossRef] [PubMed]
- Marine, J.C.; Bellefroid, E.J.; Pendeville, H.; Martial, J.A.; Pieler, T. A role for Xenopus Gli-type zinc finger proteins in the early embryonic patterning of mesoderm and neuroectoderm. Mech. Dev. 1997, 63, 211–225. [Google Scholar] [CrossRef]
- Figueiredo, A.L.; Maczkowiak, F.; Borday, C.; Pla, P.; Sittewelle, M.; Pegoraro, C.; Monsoro-Burq, A.H. PFKFB4 control of AKT signaling is essential for premigratory and migratory neural crest formation. Development 2017, 144, 4183–4194. [Google Scholar] [CrossRef] [PubMed]
- Fantauzzo, K.A.; Soriano, P. PI3K-mediated PDGFRalpha signaling regulates survival and proliferation in skeletal development through p53-dependent intracellular pathways. Genes Dev. 2014, 28, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Kubota, Y.; Mitsukawa, N.; Uchida, M.; Uchida, Y.; Akita, S.; Hasegawa, M.; Satoh, K. Low-level mesodermal somatic mutation mosaicism: Late-onset craniofacial and cervical spinal hyperostoses. Am. J. Med. Genet. A 2014, 164A, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Sundaresan, N.R.; Pillai, V.B.; Wolfgeher, D.; Samant, S.; Vasudevan, P.; Parekh, V.; Raghuraman, H.; Cunningham, J.M.; Gupta, M.; Gupta, M.P. The deacetylase SIRT1 promotes membrane localization and activation of Akt and PDK1 during tumorigenesis and cardiac hypertrophy. Sci. Signal. 2011, 4, ra46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Sun, G.; Wang, Z.; Wan, Y.; Guo, J.; Shi, L. PCAF-mediated Akt1 acetylation enhances the proliferation of human glioblastoma cells. Tumour Biol. 2015, 36, 1455–1462. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sen, R.; Pezoa, S.A.; Carpio Shull, L.; Hernandez-Lagunas, L.; Niswander, L.A.; Artinger, K.B. Kat2a and Kat2b Acetyltransferase Activity Regulates Craniofacial Cartilage and Bone Differentiation in Zebrafish and Mice. J. Dev. Biol. 2018, 6, 27. https://doi.org/10.3390/jdb6040027
Sen R, Pezoa SA, Carpio Shull L, Hernandez-Lagunas L, Niswander LA, Artinger KB. Kat2a and Kat2b Acetyltransferase Activity Regulates Craniofacial Cartilage and Bone Differentiation in Zebrafish and Mice. Journal of Developmental Biology. 2018; 6(4):27. https://doi.org/10.3390/jdb6040027
Chicago/Turabian StyleSen, Rwik, Sofia A. Pezoa, Lomeli Carpio Shull, Laura Hernandez-Lagunas, Lee A. Niswander, and Kristin Bruk Artinger. 2018. "Kat2a and Kat2b Acetyltransferase Activity Regulates Craniofacial Cartilage and Bone Differentiation in Zebrafish and Mice" Journal of Developmental Biology 6, no. 4: 27. https://doi.org/10.3390/jdb6040027