
Modeling and Hemofiltration Treatment of Acute Inflammation †

 ,
,
Abstract
:1. Introduction
1.1. Inflammation
1.2. Sepsis
1.3. Mathematical Models of Inflammation
1.4. Endotoxemia: A Model of Sepsis
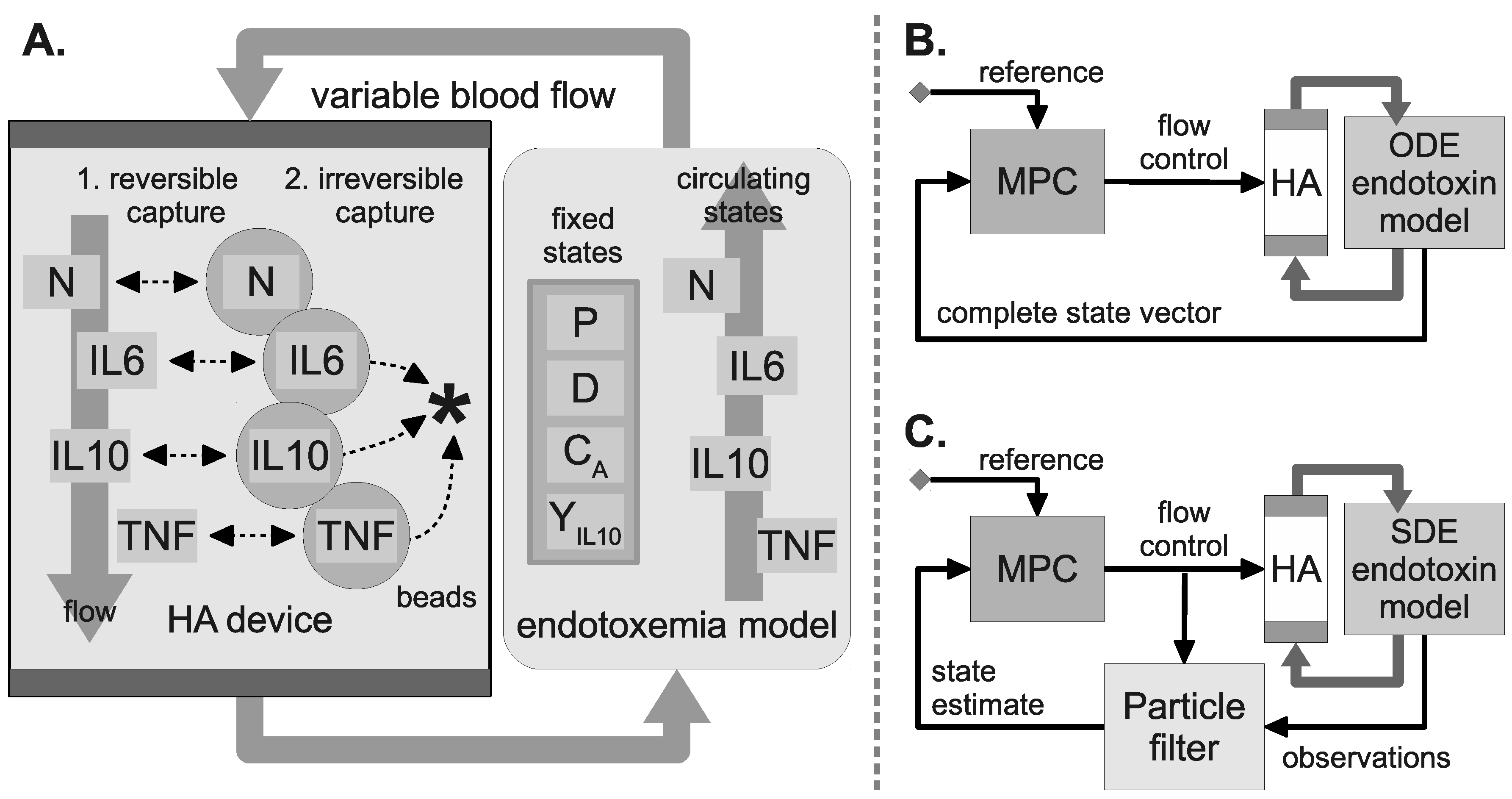
1.5. Hemoadsorption: A Potential Treatment for Sepsis
1.6. Model-Based Treatment
1.6.1. Model Predictive Control
1.6.2. State Estimation: Mapping Diagnostics into Model Representations
1.7. Manuscript Overview
2. Materials and Methods
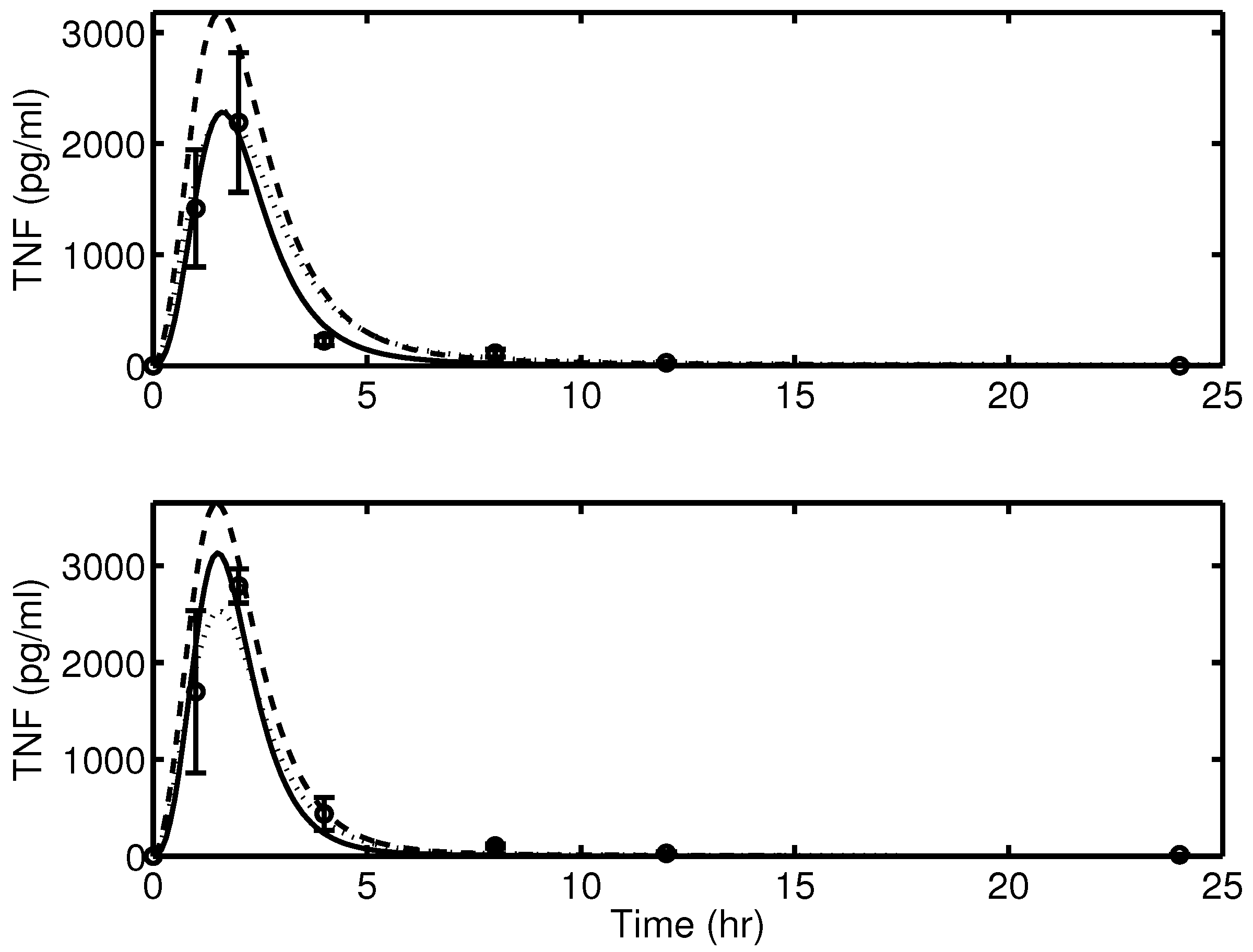
2.1. Experimental Data
2.2. Mathematical Model of Acute Inflammation
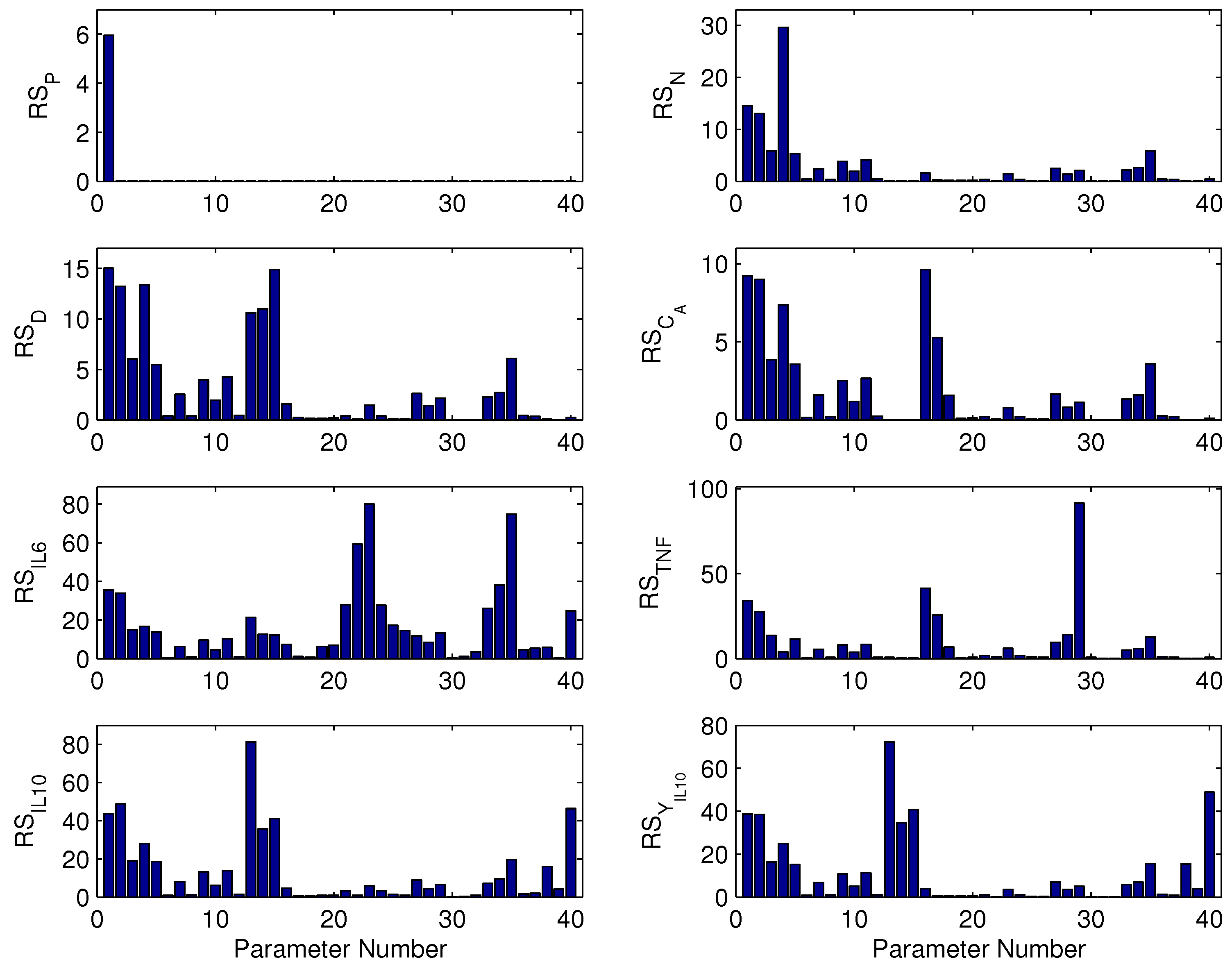
2.3. Parametric Sensitivity by Finite Difference Method
2.4. In Silico Treatment
2.5. Stochastic Endotoxemia Model
2.6. Observation Model
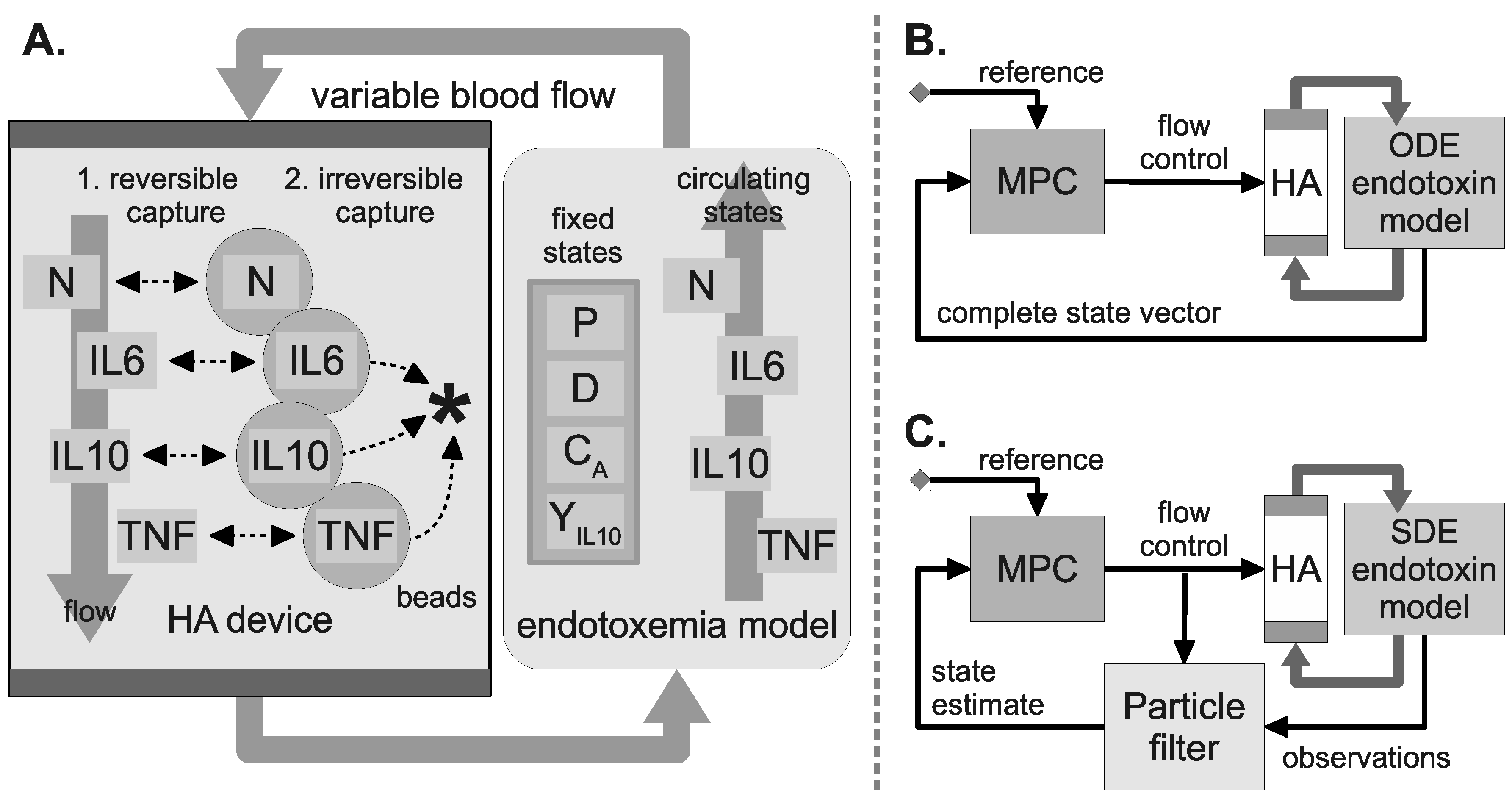
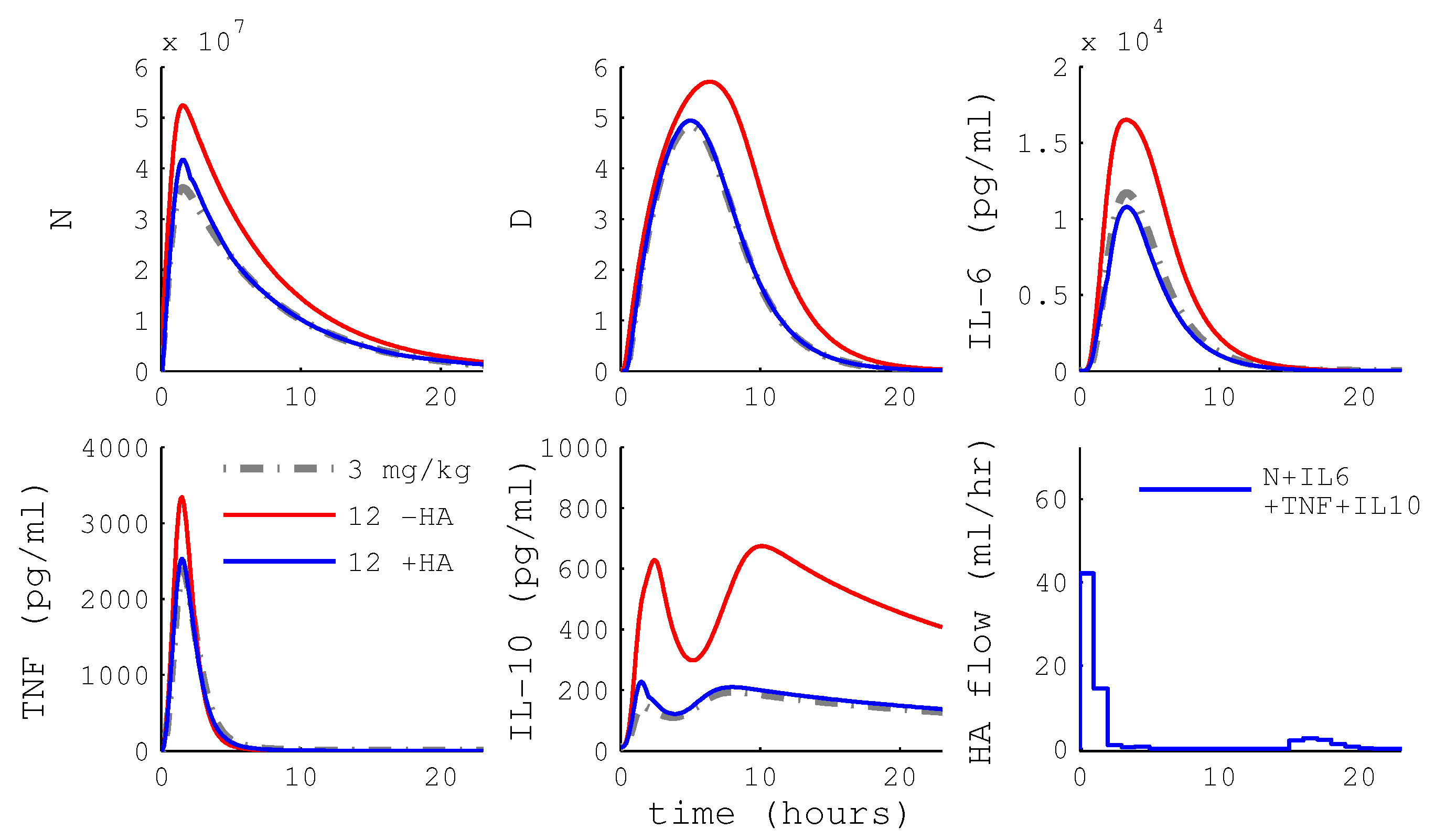
2.7. Hemoadsorption Model
2.8. WBC Capture Model
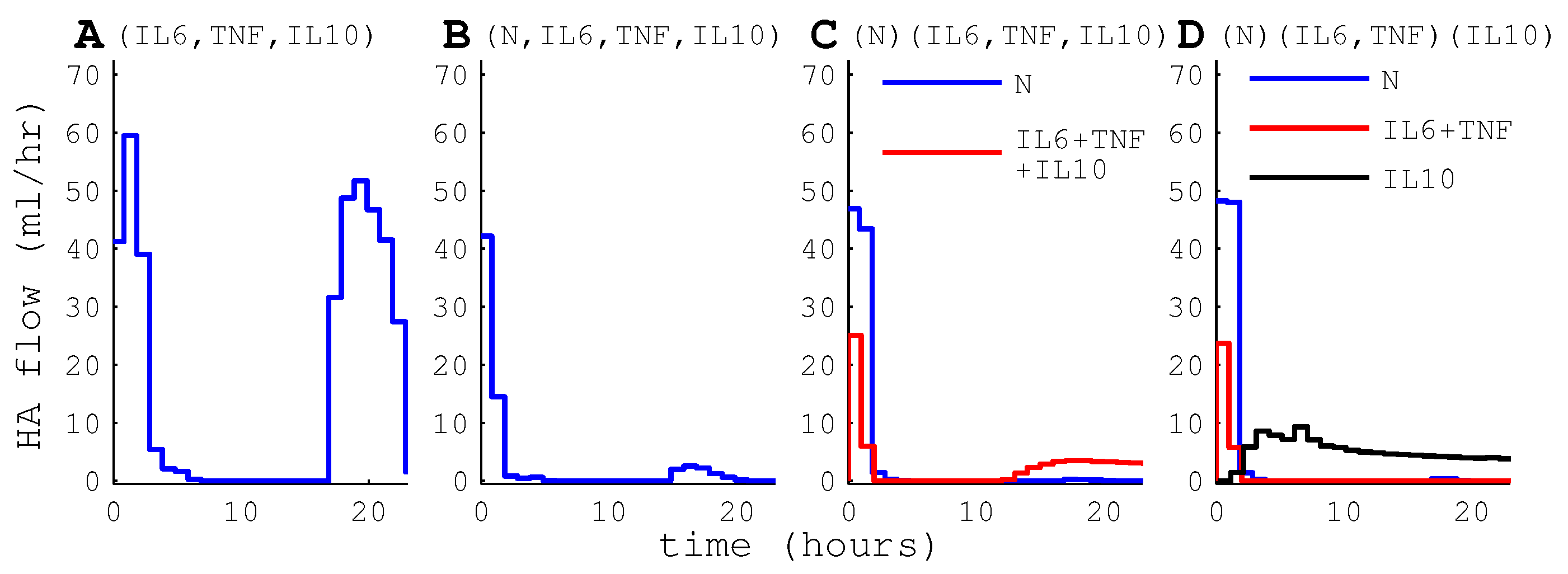
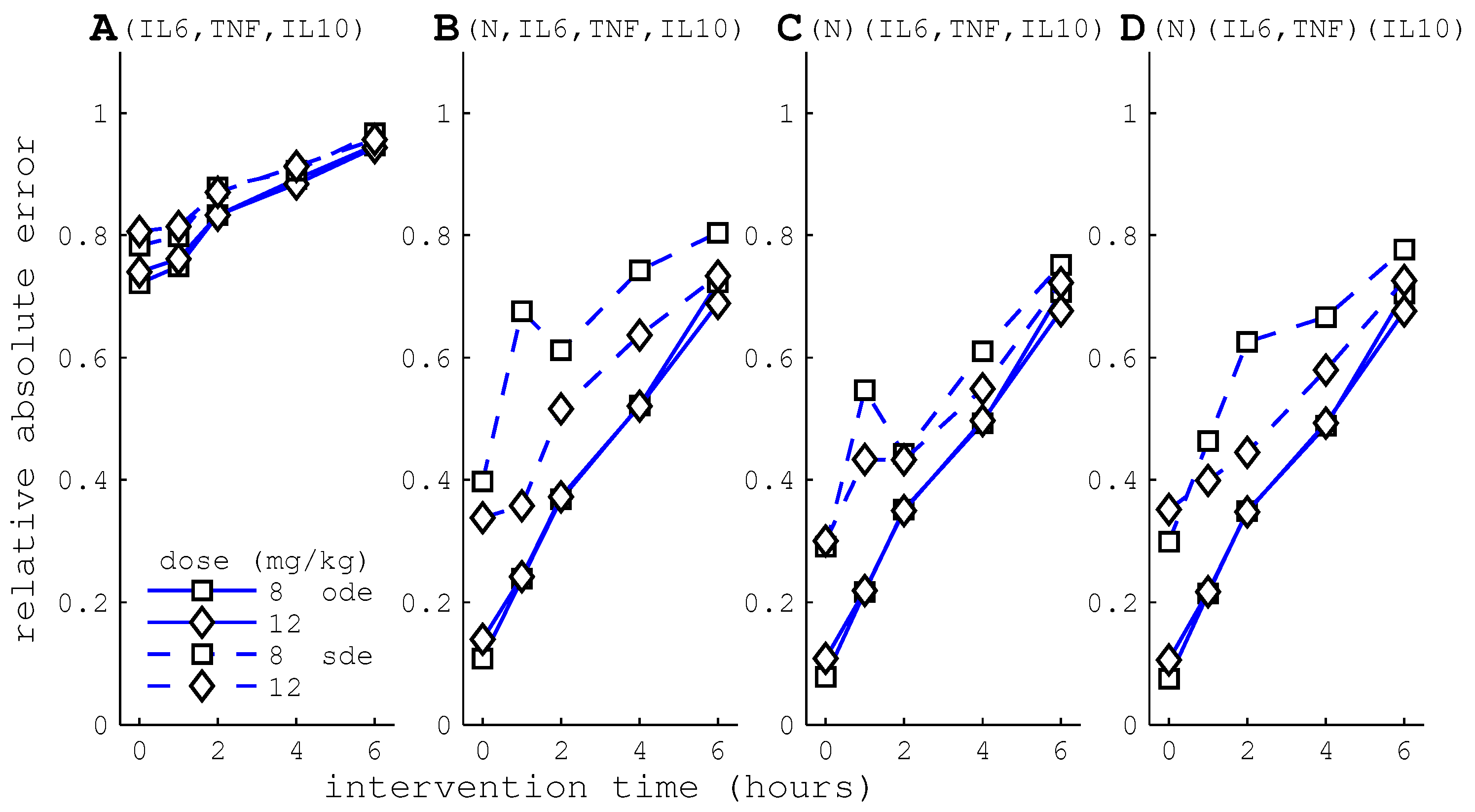
2.9. HA Device Configurations
2.10. Model Predictive Control
2.11. HA Performance Metric
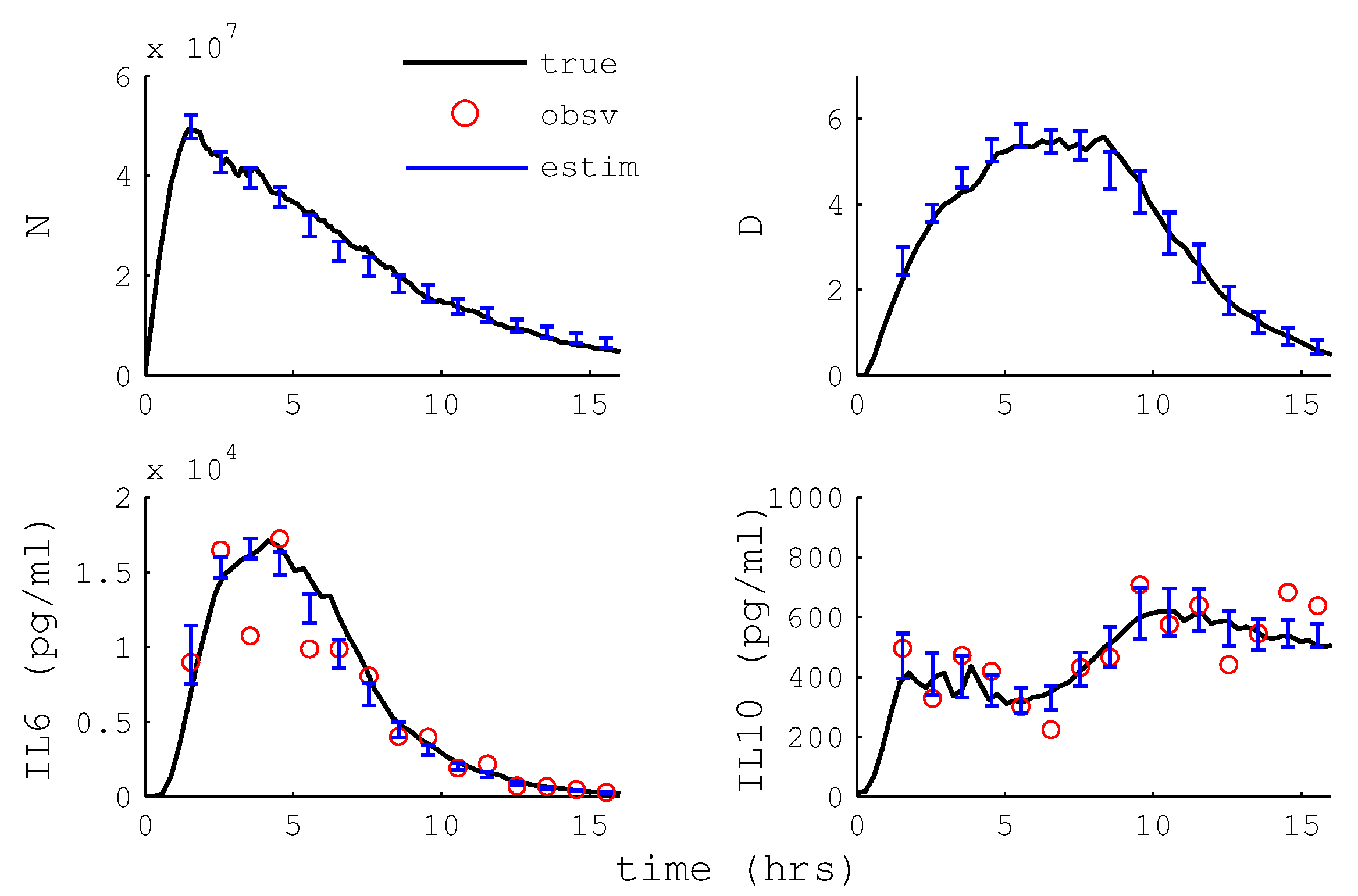
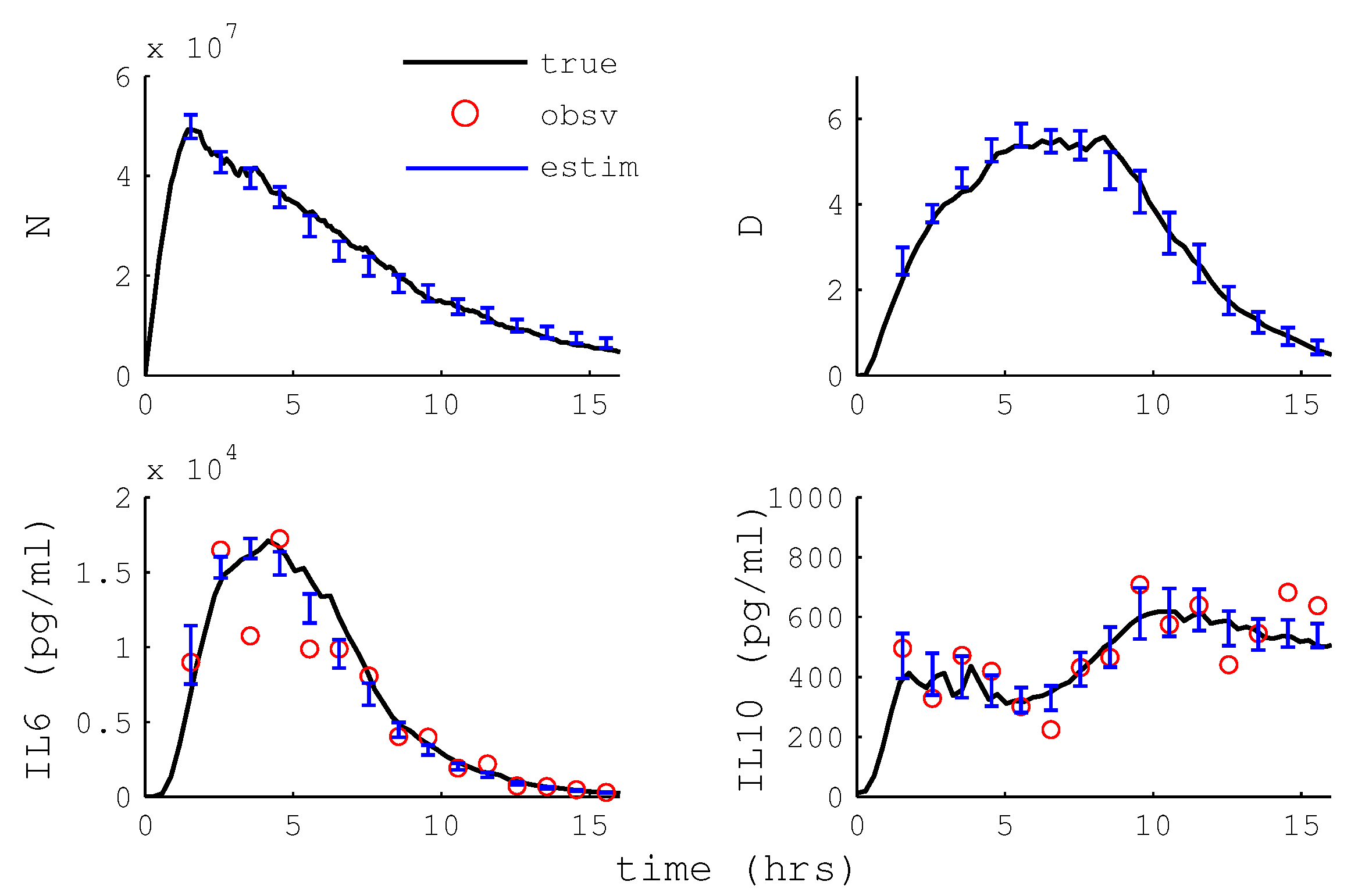
2.12. Particle Filter State Estimation
2.13. State Estimation Performance Metric
3. Results
3.1. Parameter Sensitivity Analysis
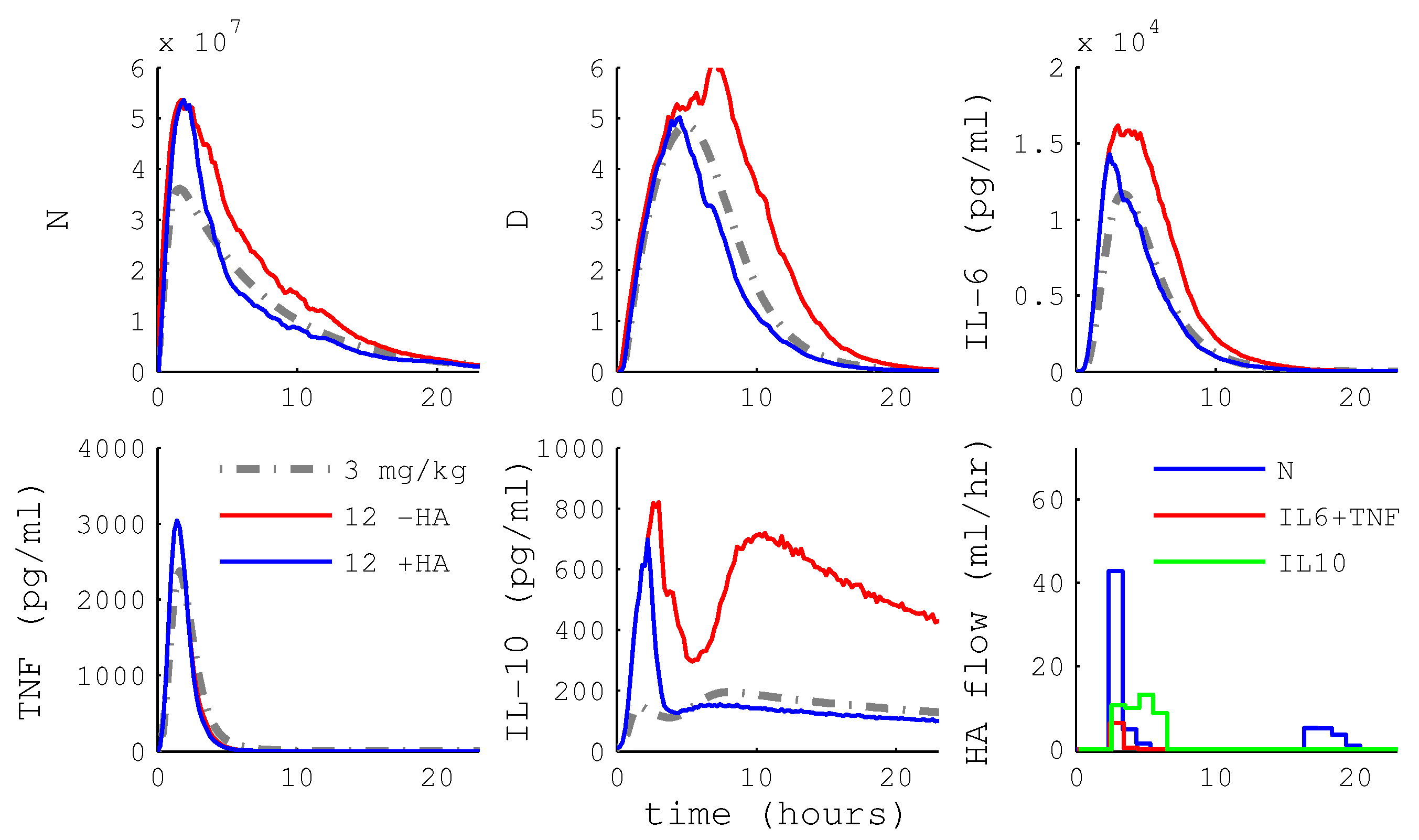
3.2. Controlling the Inflammatory Response
3.2.1. MPC Using HA
3.2.2. HA Efficacy: Cytokine Versus WBC Capture
3.2.3. Differential Capture of Inflammatory Mediators
3.2.4. Particle Filter State Estimation
3.2.5. Hemoadsorption Control with State Estimation
4. Discussion
4.1. Trading Off Biological Fidelity and Model Structure
4.2. Biological Fidelity Challenges Parameter Estimation
4.3. Cell Capture Is Predicted Key to HA Efficacy
4.4. HA Devices with Differential Cytokine and WBC Capture May Have Little Benefit
4.5. MPC Reference Trajectory
4.6. State Estimation
4.7. Real-Time Control of HA
4.8. Limitations
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A. Model Structure Justification
Appendix A.1. Model Selection Technique
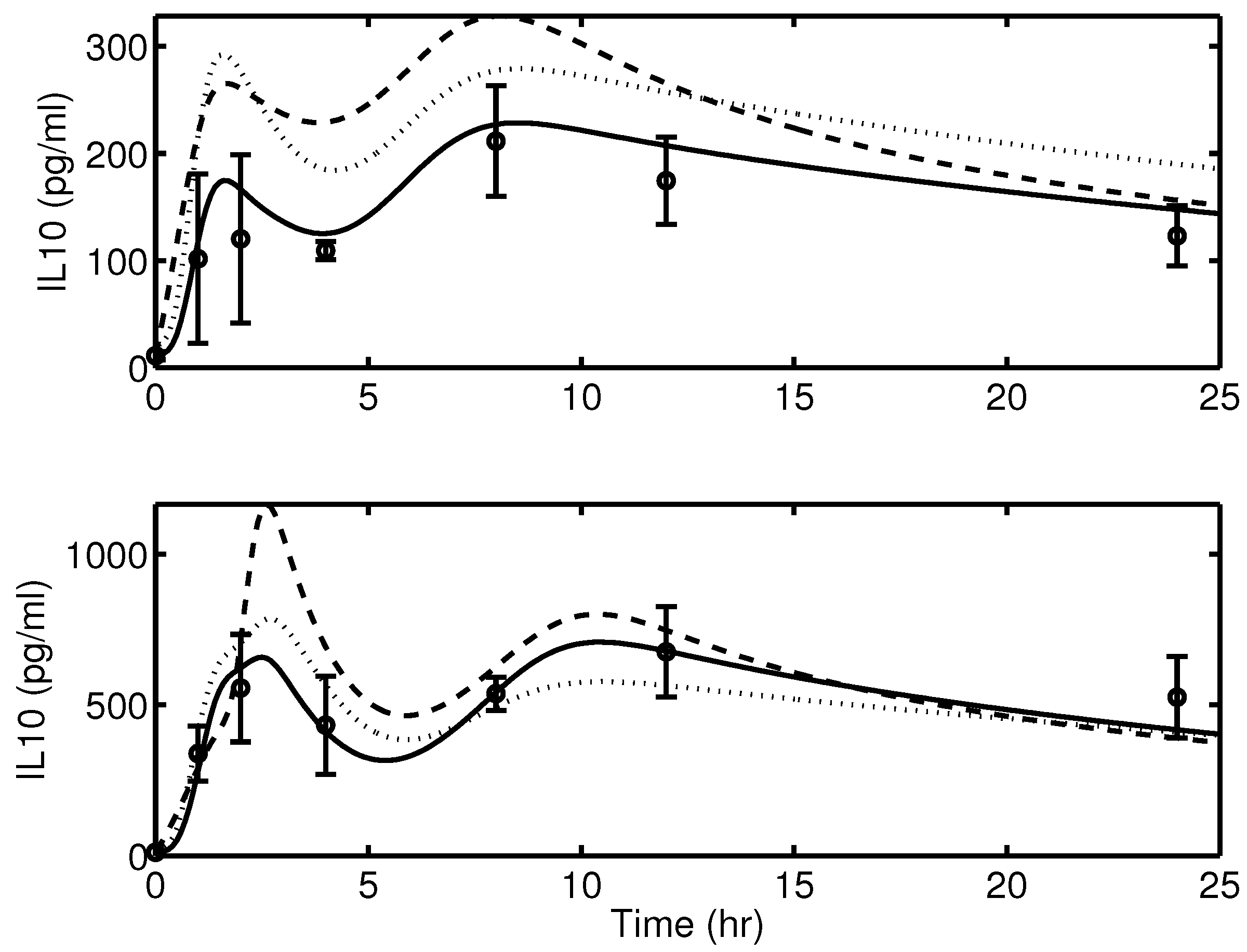
Appendix A.2. Model Selection Comparisons




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model (Equation) | AIC | BIC |
|---|---|---|
| IL-6 (Equation (14)) | 274.1 | 299.7 |
| AV-1 (Equation (A4)) | 303.3 | 328.2 |
| AV-2 (Equation (A5)) | 280.0 | 305.6 |
| TNF (Equation (18)) | 274.1 | 299.7 |
| AV-3 (Equation (A6)) | 282.2 | 307.8 |
| AV-4 (Equation (A7)) | 277.0 | 303.2 |
| IL-10 (Equation (22)) | 274.1 | 299.7 |
| AV-5 (Equation (A8)) | 290.2 | 315.1 |
| AV-6 (Equation (A9)) | 287.4 | 312.4 |
| Effect of D on IL-10 (Equation (12)) | 274.1 | 299.7 |
| AV-7 | 279.4 | 305.0 |
| AV-8 | 281.1 | 306.7 |
References
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C.A., Jr. Innate immunity. N. Engl. J. Med. 2000, 343, 338–344. [Google Scholar] [CrossRef]
- Cohen, J. The immunopathogenesis of sepsis. Nature 2002, 420, 885–891. [Google Scholar] [CrossRef] [PubMed]
- Abraham, E. Neutrophils and acute lung injury. Crit. Care Med. 2003, 31, S195–S199. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; van der Poll, T. Severe sepsis and septic shock. N. Engl. J. Med. 2013, 369, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, N.; Kobayashi, K. Macrophages in Inflammation. Curr. Drug Targets Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Kong, L.; Fink, M.P.; Weissfeld, L.A.; Yealy, D.M.; Pinsky, M.R.; Fine, J.; Krichevsky, A.; Delude, R.L.; Angus, D.C. Understanding the inflammatory cytokine response in pneumonia and sepsis: Results of the Genetic and Inflammatory Markers of Sepsis (GenIMS) Study. Arch. Intern. Med. 2007, 167, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Bone, R.C.; Balk, R.A.; Cerra, F.B.; Dellinger, R.P.; Fein, A.M.; Knaus, W.A.; Schein, R.M.; Sibbald, W.J. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. Chest 1992, 101, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; Linde-Zwirble, W.T.; Lidicker, J.; Clermont, G.; Carcillo, J.; Pinsky, M.R. Epidemiology of severe sepsis in the United States: analysis of incidence, outcome, and associated costs of care. Crit. Care Med. 2001, 29, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Evans, T. Organ dysfunction during sepsis. Intensive Care Med. 2006, 32, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.P.; Levy, M.M.; Rhodes, A.; Annane, D.; Gerlach, H.; Opal, S.M.; Sevransky, J.E.; Sprung, C.L.; Douglas, I.S.; Jaeschke, R.; et al. Surviving Sepsis Campaign. Crit. Care Med. 2013, 41, 580–637. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.; Levy, M.; Carlet, J.; Bion, J.; Parker, M.; Jaeschke, R.; Reinhart, K.; Angus, D.; Brun-Buisson, C.; Beale, R.; et al. Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock: 2008. Intensive Care Med. 2008, 34, 17–60. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.; Sun, Q.; Dubois, M. Clinical trials of immunomodulatory therapies in severe sepsis and septic shock. Clin. Infect. Dis. 2002, 34, 1084–1093. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Clermont, G.; Vodovotz, Y.; Chow, C.C. The dynamics of acute inflammation. J. Theor. Biol. 2004, 230, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Chow, C.C.; Clermont, G.; Kumar, R.; Lagoa, C.; Tawadrous, Z.; Gallo, D.; Betten, B.; Bartels, J.; Constantine, G.; Fink, M.P.; et al. The acute inflammatory response in diverse shock states. Shock 2005, 24, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Price, P.M.; Yu, F.; Kaldis, P.; Aleem, E.; Nowak, G.; Safirstein, R.L.; Megyesi, J. Dependence of cisplatin-induced cell death in vitro and in vivo on cyclin-dependent kinase 2. J. Am. Soc. Nephrol. 2006, 17, 2434–2442. [Google Scholar] [CrossRef] [PubMed]
- Lagoa, C.E.; Bartels, J.; Baratt, A.; Tseng, G.; Clermont, G.; Fink, M.P.; Billiar, T.R.; Vodovotz, Y. The role of initial trauma in the host’s response to injury and hemorrhage: insights from a correlation of mathematical simulations and hepatic transcriptomic analysis. Shock 2006, 26, 592–600. [Google Scholar] [CrossRef] [PubMed]
- An, G. In silico experiments of existing and hypothetical cytokine-directed clinical trials using agent-based modeling. Crit. Care Med. 2004, 32, 2050–2060. [Google Scholar] [CrossRef] [PubMed]
- Nieman, G.; Brown, D.; Sarkar, J.; Kubiak, B.; Ziraldo, C.; Dutta-Moscato, J.; Vieau, C.; Barclay, D.; Gatto, L.; Maier, K.; et al. A two-compartment mathematical model of endotoxin-induced inflammatory and physiologic alterations in swine. Crit. Care Med. 2012, 40, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Dick, T.E.; Molkov, Y.I.; Nieman, G.; Hsieh, Y.H.; Jacono, F.J.; Doyle, J.; Scheff, J.D.; Calvano, S.E.; Androulakis, I.P.; An, G.; et al. Linking inflammation, cardiorespiratory variability, and neural control in acute inflammation via computational modeling. Front. Physiol. 2012, 3, 222. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Foteinou, P.T.; Calvano, S.E.; Lowry, S.F.; Androulakis, I.P. Agent-based modeling of endotoxin-induced acute inflammatory response in human blood leukocytes. PLoS ONE 2010, 5, e9249. [Google Scholar] [CrossRef] [PubMed]
- Lipniacki, T.; Paszek, P.; Brasier, A.R.; Luxon, B.A.; Kimmel, M. Stochastic regulation in early immune response. Biophys. J. 2006, 90, 725–742. [Google Scholar] [CrossRef] [PubMed]
- Cheong, R.; Hoffmann, A.; Levchenko, A. Understanding NF-kappaB signaling via mathematical modeling. Mol. Syst. Biol. 2008, 4, 192. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Kirschner, D.E. The human immune response to Mycobacterium tuberculosis in lung and lymph node. J. Theor. Biol. 2004, 227, 463–486. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Myers, A.; Flynn, J.L.; Kirschner, D.E. TNF and IL-10 are major factors in modulation of the phagocytic cell environment in lung and lymph node in tuberculosis: A next-generation two-compartmental model. J. Theor. Biol. 2010, 265, 586–598. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Rubin, J.; Clermont, G.; Day, J.; Vodovotz, Y.; Ermentrout, B.G. A reduced mathematical model of the acute inflammatory response: I. Derivation of model and analysis of anti-inflammation. J. Theor. Biol. 2006, 242, 220–236. [Google Scholar] [CrossRef] [PubMed]
- Daun, S.; Rubin, J.; Vodovotz, Y.; Roy, A.; Parker, R.; Clermont, G. An ensemble of models of the acute inflammatory response to bacterial lipopolysaccharide in rats: Results from parameter space reduction. J. Theor. Biol. 2008, 253, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Fink, M. Animal models of sepsis and its complications. Kidney Int. 2008, 74, 991–993. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Song, M.; Venkataraman, R. Hemoadsorption removes tumor necrosis factor, interleukin-6, and interleukin-10, reduces nuclear factor-kappaB DNA binding, and improves short-term survival in lethal endotoxemia. Crit. Care Med. 2004, 32, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Carter, M.J.; Kellum, J.A. Effects of hemoadsorption on cytokine removal and short-term survival in septic rats. Crit. Care Med. 2008, 36, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- DiLeo, M.; Kellum, J.; Federspiel, W. A Simple Mathematical Model of Cytokine Capture Using a Hemoadsorption Device. Ann. Biomed. Eng. 2009, 37, 222–229. [Google Scholar] [CrossRef] [PubMed]
- DiLeo, M.V.; Fisher, J.D.; Federspiel, W.J. Experimental Validation of a Theoretical Model of Cytokine Capture Using a Hemoadsorption Device. Ann. Biomed. Eng. 2009, 37, 2310–2316. [Google Scholar] [CrossRef] [PubMed]
- Song, S.O.; Song, S.O.K.; Hogg, J.; Peng, Z.Y.; Parker, R.; Kellum, J.a.; Clermont, G. Ensemble models of neutrophil trafficking in severe sepsis. PLoS Comput. Biol. 2012, 8, e1002422. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.S.; Doyle III, F.J. Control–Relevant Modeling in Drug Delivery. Adv. Drug Deliv. Rev. 2001, 48, 211–228. [Google Scholar] [CrossRef]
- Doyle, F.J., III; Seborg, D.; Parker, R.S.; Bequette, B.W.; Jeffrey, A.; Xia, X.; Craig, I.; McAvoy, T.J. A Tutorial on Biomedical Process Control. J. Process Control 2007, 17, 571–594. [Google Scholar] [CrossRef]
- Muske, K.R.; Rawlings, J.B. Model predictive control with linear models. AIChE J. 1993, 39, 262–287. [Google Scholar] [CrossRef]
- Day, J.; Rubin, J.; Clermont, G. Using nonlinear model predictive control to find optimal therapeutic strategies to modulate inflammation. Math. Biosci. Eng. 2010, 7, 739–763. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.B.; Bakshi, B.R. Particle filtering and moving horizon estimation. Comput. Chem. Eng. 2006, 30, 1529–1541. [Google Scholar] [CrossRef]
- Arulampalam, M.; Maskell, S.; Gordon, N.; Clapp, T. A tutorial on particle filters for online nonlinear/non-Gaussian Bayesian tracking. IEEE Trans. Signal Process. 2002, 50, 174–188. [Google Scholar] [CrossRef]
- Freeman, B.D.; Natanson, C. Anti-inflammatory therapies in sepsis and septic shock. Expert Opin. Investig. Drugs 2000, 9, 1651–1663. [Google Scholar] [CrossRef] [PubMed]
- Bellingan, G. Inflammatory cell activation in sepsis. Br. Med. Bull. 1999, 55, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Pretolani, M. Interleukin-10: An Anti-inflammatory Cytokine with Therapeutic Potential. Clin. Exp. Allergy 1999, 29, 1164–1171. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, M.R. Sepsis: A pro- and anti-inflammatory disequilibrium syndrome. Contrib. Nephrol. 2001, 132, 354–366. [Google Scholar] [PubMed]
- Matzinger, P. The danger model: A renewed sense of self. Science 2002, 296, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Giannoudis, P.V.; Smith, R.M.; Perry, S.L.; Windsor, A.J.; Dickson, R.A.; Bellamy, M.C. Immediate IL-10 expression following major orthopaedic trauma: Relationship to anti-inflammatory response and subsequent development of sepsis. Intensive Care Med. 2000, 26, 1076–1081. [Google Scholar] [CrossRef] [PubMed]
- Kamm, K.; Vanderkolk, W.; Lawrence, C.; Jonker, M.; Davis, A.T. The effect of traumatic brain injury upon the concentration and expression of interleukin-1beta and interleukin-10 in the rat. J. Trauma 2006, 60, 152–157. [Google Scholar] [CrossRef] [PubMed]
- Day, J.; Rubin, J.; Vodovotz, Y.; Chow, C.C.; Reynolds, A.; Clermont, G. A reduced mathematical model of the acute inflammatory response II. Capturing scenarios of repeated endotoxin administration. J. Theor. Biol. 2006, 242, 237–256. [Google Scholar] [CrossRef] [PubMed]
- Iversen, M.H.; Hahn, R.G. Acute effects of vitamin A on the kinetics of endotoxin in conscious rabbits. Intensive Care Med. 1999, 25, 1160–1164. [Google Scholar] [CrossRef] [PubMed]
- Warner, A.E.; DeCamp, M.M.; Molina, R.M.; Brain, J.D. Pulmonary removal of circulating endotoxin results in acute lung injury in sheep. Lab. Investig. 1988, 59, 219–230. [Google Scholar] [PubMed]
- Xing, Z.; Gauldie, J.; Cox, G.; Baumann, H.; Jordana, M.; Lei, X.F.; Achong, M.K. IL-6 is an antiinflammatory cytokine required for controlling local or systemic acute inflammatory responses. J. Clin. Investig. 1998, 101, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, I.Y.; Johnson, R.C.; Schmahai, T.J. Preclinical Safety Evaluation of Recombinant Human Interleukin-10. Regul. Toxicol. Pharmacol. 2002, 35, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Parker, R.S. Dynamic Modeling of Exercise Effects on Plasma Glucose and Insulin Levels. J. Diabetes Sci. Technol. 2007, 1, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Yue, H.; Brown, M.; Knowles, J.; Wang, H.; Broomhead, D.S.; Kell, D.B. Insights into the Behavior of Systems Biology Models from Dynamics Sensitivity and Identifiability Analysis: A Case Study of an NF-κB Signalling Pathway. Mol. Biosyst. 2006, 2, 640–649. [Google Scholar] [CrossRef] [PubMed]
- Zak, D.E.; Goyne, G.E.; Schwaber, J.S.; Doyle, F.J., III. Importance of Input Perturbations and Stochastic Gene Expression in the Reverse Engineering of Genetic Regulatory networks: Insights from an Identifiability Analysis of an in silico Network. Genome Res. 2003, 13, 2396–2405. [Google Scholar] [CrossRef] [PubMed]
- Hindmarsh, A.C.; Brown, P.N.; Grant, K.E.; Lee, S.L.; Serban, R.; Shumaker, D.E.; Woodward, C.S. SUNDIALS: Suite of nonlinear and differential/algebraic equation solvers. ACM Trans. Math. Softw. 2005, 31, 363–396. [Google Scholar] [CrossRef]
- Valenti, I.E. Characterization of a Novel Sorbent Polymer for the Treatment of Sepsis. Master’s Thesis, University of Pittsburgh, Pittsburgh, PA, USA, 2010. [Google Scholar]
- Rimmelé, T.; Kellum, J.A. Clinical review: Blood purification for sepsis. Crit. Care 2011, 15, 205. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Wang, H.; Carter, M.; Dileo, M.; Bishop, J.; Zhou, F.; Wen, X.; Rimmelé, T.; Singbartl, K.; Federspiel, W.; et al. Acute Removal of Common Sepsis Mediators does not Explain the Effects of Extracorporeal Blood Purification in Experimental Sepsis. Kidney Int. 2012, 81, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Bishop, C.M. Pattern Recognition and Machine Learning; Springer: New York, NY, USA, 2006; Chapter 13; pp. 363–369. [Google Scholar]
- Audoly, S.; D’Angio, L.; Saccomani, M.P.; Cobelli, C. Global Identifiability of Linear Compartmental Models—A Computer Algebra Algorithm. IEEE Trans. Biomed. Eng. 1998, 45, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Chappell, M.J.; Godfrey, K.R. Structural identifiability of the parameters of a nonlinear batch reactor model. Math. Biosci. 1992, 108, 241–251. [Google Scholar] [CrossRef]
- Ljung, L.; Glad, T. On global identifiability for arbitrary model parametrizations. Automatica 1994, 30, 265–276. [Google Scholar] [CrossRef]
- Pohjanpalo, H. System identifiability based on the power series expansion of the solution. Math. Biosci. 1978, 41, 21–33. [Google Scholar] [CrossRef]
- Audoly, S.; Belu, G.; D’Angio, L.; Saccomani, M.P.; Cobelli, C. Global Identifiability of Nonlinear Models of Biological Systems. IEEE Trans. Biomed. Eng. 2001, 48, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Riedemann, N.C.; Guo, R.; Ward, P.A. Novel strategies for the treatment of sepsis. Nat. Med. 2003, 9, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, P.N.; Dyanov, H.M.; Park, P.; Wang, J.; Newell, K.A.; Quigg, R.J. Acute renal failure in endotoxemia is caused by TNF acting directly on TNF receptor-1 in kidney. J. Immunol. 2002, 168, 5817–5823. [Google Scholar] [CrossRef] [PubMed]
- Singbartl, K.; Bockhorn, S.G.; Zarbock, A.; Schmolke, M.; Van Aken, H. T cells modulate neutrophil-dependent acute renal failure during endotoxemia: critical role for CD28. J. Am. Soc. Nephrol. 2005, 16, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Zarbock, A.; Schmolke, M.; Spieker, T.; Jurk, K.; Van Aken, H.; Singbartl, K. Acute uremia but not renal inflammation attenuates aseptic acute lung injury: A critical role for uremic neutrophils. J. Am. Soc. Nephrol. 2006, 17, 3124–3131. [Google Scholar] [CrossRef] [PubMed]
- Schefold, J.C.; Hasper, D.; von Haehling, S.; Meisel, C.; Reinke, P.; Schlosser, H. Interleukin-6 serum level assessment using a new qualitative point-of-care test in sepsis: A comparison with ELISA measurements. Clin. Biochem. 2008, 41, 893–898. [Google Scholar] [CrossRef] [PubMed]
- Dengler, J.; Schefold, J.C.; Graetz, D.; Meisel, C.; Splettstösser, G.; Volk, H.; Schlosser, H. Point-of-care testing for interleukin-6 in cerebro spinal fluid (CSF) after subarachnoid haemorrhage. Med. Sci. Monit. 2008, 14, BR265–BR268. [Google Scholar] [PubMed]
- Day, J.; Rubin, J.; Clermont, G. Using nonlinear model predictive control to find optimal therapeutic strategies to modulate inflammation. Math. Biosci. Eng. 2010, 7, 739–763. [Google Scholar] [CrossRef] [PubMed]
- Alves-Filho, J.C.; de Freitas, A.; Russo, M.; Cunha, F.Q. Toll-like receptor 4 signaling leads to neutrophil migration impairment in polymicrobial sepsis. Crit. Care Med. 2006, 34, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Call, D.R.; Nemzek, J.A.; Ebong, S.J.; Bolgos, G.L.; Newcomb, D.E.; Remick, D.G. Ratio of Local to Systemic Chemokine Concentrations Regulates Neutrophil Recruitment. Am. J. Pathol. 2001, 158, 715–721. [Google Scholar] [CrossRef]
- Akaike, H. A Bayesian Extension of the Minimal AIC Procedures of Autoregressive Model Fitting. Biometrika 1979, 66, 237–242. [Google Scholar] [CrossRef]
- Schwarz, G. Estimating the dimension of a model. Ann. Stat. 1978, 6, 461–464. [Google Scholar] [CrossRef]
- Posada, D.; Buckley, T.R. Model Selection and Model Averaging in Phylogenetics: Advantages of Akaike Information Criterion and Bayesian Approaches over Likelihood Ratio Tests. Syst. Biol. 2004, 53, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Hadley, J.S.; Wang, J.E.; Foster, S.J.; Thiemermann, C.; Hinds, C.J. Peptidoglycan of Staphylococcus aureus upregulates monocyte expression of CD14, Toll-like receptor 2 (TLR2), and TLR4 in human blood: Possible implications for priming of lipopolysaccharide signaling. Infect. Immun. 2005, 73, 7613–7619. [Google Scholar] [CrossRef] [PubMed]
- Rittirsch, D.; Hoesel, L.M.; Ward, P.A. The disconnect between animal models of sepsis and human sepsis. J. Leukoc. Biol. 2007, 81, 137–143. [Google Scholar] [CrossRef] [PubMed]




| IL-6 | TNF | IL-10 | N | |
|---|---|---|---|---|
| 0.62 | 0.188 | 0.682 | 0.177 | |
| 0.013 | 0.015 | 0.072 | 0.010 | |
| 0.006 | 0.007 | 0.022 | 0 |
| HA | Specificity | ||
|---|---|---|---|
| Configuration | Column 1 | Column 2 | Column 3 |
| A | TNF, IL-6, IL-10 | - | - |
| B | N, TNF, IL-6, IL-10 | - | - |
| C | N | TNF, IL-6, IL-10 | - |
| D | N | TNF, IL-6 | IL-10 |
| No. | Parameter | No. | Parameter | No. | Parameter | No. | Parameter |
|---|---|---|---|---|---|---|---|
| 1 | 11 | 21 | 31 | ||||
| 2 | 12 | 22 | 32 | ||||
| 3 | 13 | 23 | 33 | ||||
| 4 | 14 | 24 | 34 | ||||
| 5 | 15 | 25 | 35 | ||||
| 6 | 16 | 26 | 36 | ||||
| 7 | 17 | 27 | 37 | ||||
| 8 | 18 | 28 | 38 | ||||
| 9 | 19 | 29 | 39 | ||||
| 10 | 20 | 30 | 40 |
| Parameter Groups | Parameters |
|---|---|
| , , , , , , , , , , | |
| , , | |
| , , | |
| , , , , , , , | |
| , , , | |
| , , , , , , | |
| , , |
| States | Relative Sensitivity (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| 100 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| 13.8 | 64 | 0.2 | 1.9 | 2.8 | 5.7 | 11 | 0.5 | |
| 11.8 | 41.0 | 28.7 | 1.6 | 2.4 | 4.9 | 9.4 | 0.3 | |
| 13.0 | 45.8 | 0.1 | 23.3 | 2.3 | 5.1 | 10.0 | 0.2 | |
| 5.4 | 17.0 | 7.0 | 1.4 | 36.3 | 5.1 | 23.3 | 4.6 | |
| 9.8 | 23.9 | 0.3 | 21.2 | 3.9 | 33.3 | 7.3 | 0.2 | |
| 8.5 | 31.0 | 30.8 | 1.1 | 3.5 | 3.9 | 8.0 | 12.9 | |
| 8.7 | 29.6 | 33.2 | 1.1 | 1.6 | 3.6 | 6.9 | 15.3 | |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parker, R.S.; Hogg, J.S.; Roy, A.; Kellum, J.A.; Rimmelé, T.; Daun-Gruhn, S.; Fedorchak, M.V.; Valenti, I.E.; Federspiel, W.J.; Rubin, J.; et al. Modeling and Hemofiltration Treatment of Acute Inflammation. Processes 2016, 4, 38. https://doi.org/10.3390/pr4040038
Parker RS, Hogg JS, Roy A, Kellum JA, Rimmelé T, Daun-Gruhn S, Fedorchak MV, Valenti IE, Federspiel WJ, Rubin J, et al. Modeling and Hemofiltration Treatment of Acute Inflammation. Processes. 2016; 4(4):38. https://doi.org/10.3390/pr4040038
Chicago/Turabian StyleParker, Robert S., Justin S. Hogg, Anirban Roy, John A. Kellum, Thomas Rimmelé, Silvia Daun-Gruhn, Morgan V. Fedorchak, Isabella E. Valenti, William J. Federspiel, Jonathan Rubin, and et al. 2016. "Modeling and Hemofiltration Treatment of Acute Inflammation" Processes 4, no. 4: 38. https://doi.org/10.3390/pr4040038
APA StyleParker, R. S., Hogg, J. S., Roy, A., Kellum, J. A., Rimmelé, T., Daun-Gruhn, S., Fedorchak, M. V., Valenti, I. E., Federspiel, W. J., Rubin, J., Vodovotz, Y., Lagoa, C., & Clermont, G. (2016). Modeling and Hemofiltration Treatment of Acute Inflammation. Processes, 4(4), 38. https://doi.org/10.3390/pr4040038